Abstract
Porous carbon materials have gained increasing attention in catalysis applications due to their tailorable surface properties, large specific surface area, excellent thermal stability, and low cost. Even though porous carbon materials have been employed for thermal-catalytic dry reforming of methane (DRM), the structure–function relationship, especially the critical factor affecting catalytic performance, is still under debate. Herein, various porous carbon-based samples with disparate pore structures and surface properties are prepared by alkali (K2CO3) etching and the following CO2 activation of low-cost petroleum pitch. Detailed characterization clarifies that the quinone/ketone carbonyl functional groups on the carbon surface are the key active sites for DRM. Density functional theory (DFT) calculations also show that the C=O group have the lowest transition state energy barrier for CH4* cleavage to CH3* (2.15 eV). Furthermore, the cooperative interplay between the specific surface area and quinone/ketone carbonyl is essential to boost the cleavage of C-H and C-O bonds, guaranteeing enhanced DRM catalytic performance. The MC-600-800 catalyst exhibited an initial CH4 conversion of 51% and a reaction rate of 12.6 mmolCH4 gcat.−1 h−1 at 800 °C, CH4:CO2:N2= 1:1:8, and GHSV = 6000 mL gcat.−1 h−1. Our work could pave the way for the rational design of metal-free carbon-based DRM catalysts and shed new light on the high value-added utilization of heavy oils.
1. Introduction
Methane is an ideal resource for hydrogen or syngas (CO + H2) production via catalytic technology such as steam reforming of methane (SRM), partial oxidation of methane (POM), dry reforming of methane (DRM), and catalytic methane decomposition (CMD) [1,2,3,4]. Among all the catalytic methane transformation strategies, DRM, utilizing two greenhouse gases (CH4 + CO2) to produce high-quality syngas (CO/H2), possesses the integrated functions of greenhouse gas elimination and valuable chemical supply. The DRM product, namely syngas with a molar ratio of 1, can be employed as an ideal feedstock for liquid fuels or light olefin production via Fischer–Tropsch synthesis technology [5,6]. Even though DRM is gaining more and more attention from industry and academia due to its large-scale application potential and environmental benefits, the absence of stable and effective catalysts leads to an absence of commercially feasible technology [7]. Therefore, the current interest in DRM is mainly focused on the development of suitable catalysts and optimizing their catalytic stability. Various transition metals such as Ni, Co, and Pd have been explored as active catalysts for DRM, but most of them suffer from high cost or rapid deactivation because of sintering at high reaction temperatures as well as carbon deposition. Therefore, the search for DRM catalysts with excellent catalytic performance and appropriate economic cost for large-scale utilization is highly urgent [8].
Metal-free carbon-based catalysts stand out among various candidates regarding high-temperature sintering/carbon deposition-resistance and low cost [9]. Firstly, the pure carbon-based catalysts without doping from metallic active sites could effectively avoid the undesirable active sites sintering, especially under high reaction temperatures. Furthermore, deactivation by carbon deposition is not problematic for DRM, as the deposited carbon can act as a new catalyst for the subsequent DRM reaction, as reported in other literature, thus improving the activity in the later stage [10]. So far, many researchers have evaluated the DRM performance of various carbon materials, such as activated carbon (AC), carbon black (CB), carbon nanotubes (CNT), ordered mesoporous carbon materials, etc. Li et al. [11] found that coal char could exhibit DRM catalytic activity, and its specific surface area and ash content are two key factors affecting the DRM performance. Petroleum asphalt-based carbon materials also showed appealing activity and stability for DRM [12]. It is worth noting that the application of coal char and petroleum asphalt-based carbon materials in DRM are attractive due to the realization of the value-added utilization of low-cost resources. Suelves et al. [13] reported a linear relationship between methane conversion and the number of oxygen-containing functional groups on the carbon surface by measuring the desorption of CO and CO2 during thermal treatment. Even though tremendous progress has been made in carbon-based DRM catalyst exploration and active site identification, the structure–function relationship of the carbon-based catalysts has not been well defined, leading to difficulties in understanding the catalytic mechanism and to further enhance DRM catalytic performance.
To tackle the above-mentioned obstacles, this study aims at optimizing the content of the oxygen-containing functional groups and architectural properties of the petroleum pitch-derived porous carbon materials by tuning the K2CO3 activation temperature and the subsequent CO2 activation atmosphere. Additionally, the DRM reaction is used as a model to investigate the influence of oxygen-containing functional groups on the catalyst surface in activating C-H bonds. Three discrete oxygen-containing functional groups can be identified by deconvolution of the O1s peak of X-ray photoelectron spectroscopy (XPS): C=O group, O=C-O group, and C-OH/C-O-C group. Based on experimental and characterization results, the role of the quinone/ketone carbonyl oxygen-containing functional groups (C=O) in DRM is identified. Density functional theory (DFT) calculations also show that C=O groups play a crucial role in promoting the cleavage of CH4. Furthermore, CO2 activation can increase the specific microporous surface area and the accessibility of reactants to the catalytic interface without losing the number of essential active sites. This would further enhance the DRM performance of the carbon-based catalyst. This study contributes to the in-depth understanding of how specific oxygen-containing functional groups affect the activity of porous carbon materials during catalytic processes, which not only enriches the understanding of the activity nature of carbon-based catalysts in DRM reactions but also provides an essential theoretical basis for the design of novel and efficient carbon-based catalysts.
2. Results and Discussion
2.1. Preparation and Structural Characterization of K2CO3-Activated Catalysts
The porous carbon materials catalysts were prepared by the K2CO3 chemical activation method. The preparation scheme is shown in Figure 1. According to the research literature, the mechanism of preparing porous carbon materials by activation with K2CO3 at high temperatures includes the following chemical reactions (Equations (1)–(4)) [14]:
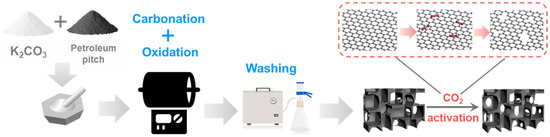
Figure 1.
Schematic illustration of the preparation procedure of metal-free catalyst (MC-T, T denotes the CO2 activation temperature, °C).
The XRD patterns of the MC-T catalysts are displayed in Figure 2a. All samples exhibited two broad peaks at 24° and 43°, which can be attributed to the disorder graphite structure of crystal planes (002) and (100) (0.34 nm and 0.21 nm interplanar distances, respectively) [15,16]. The (002) peak is a standard characteristic of the interlayer stacking structure of the conjugated aromatic system, which indicates the graphite-like structure of the porous carbon materials catalysts. Compared with the standard diffraction peak position (26°), the diffraction peak shifted to a lower angle. According to the Bragg diffraction formula [17] (d = nλ/2sin θ, where d is the inter planar spacing, 2θ is the diffraction peak angle, n = 1, λ = 1.5406 Å), the activation of K2CO3 increased the carbon layer spacing. With the increase with activation temperature during the preparation process, the diffraction peak widened, and the intensity decreased, indicating an increase in the amorphous degree of the porous carbon materials.
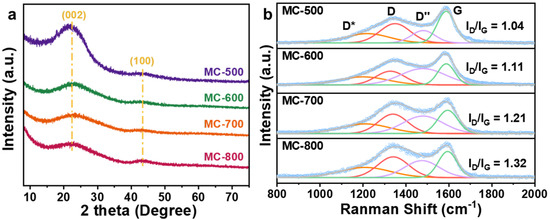
Figure 2.
(a) XRD patterns and (b) Raman spectra of MC-500/600/700/800 catalysts.
Figure 2b shows the Raman spectra and the fitted compositions of the MC-T catalysts. In addition, the D peak (near 1350 cm−1) and G peak (near 1580 cm−1) can be attributed to the defective carbon and graphitic carbon, respectively [18]. In addition, there are also D* peaks located near 1200 cm−1 and D″ peaks near 1450 cm−1, attributed to the vibration of C-C and C=C bonds of polyenic compounds, respectively, which is typical for annealed and activated organic structures [19,20]. Notably, The ID/IG values increased as the carbonization temperature increased (1.04, 1.11, 1.21, 1.32 for MC-500/600/700/800/, respectively), indicating that the defect degree of the catalysts increased with the increase in carbonization temperature. The imperfect structures, such as defects, edges, and vacancies, could lead to the localization of π electrons and promote the generation of oxygen-containing groups on the surface, therefore altering the catalytic performance.
The effect of the K2CO3 activation temperature on the catalyst morphology was further monitored by scanning electron microscope (SEM) (Figure 3). The SEM images revealed that all the porous carbon-based catalysts presented a three-dimensional lamellar porous structure. With the increase in activation temperature, the carbon flakes became more dispersed. This structural change was conducive to the exposure of catalytically active sites and mass transfer behavior during the reaction process, thus accelerating the diffusion and adsorption of the reactants and improving their conversion rate.

Figure 3.
SEM images of (a) MC-500, (b) MC-600, (c) MC-700, and (d) MC-800 catalysts.
2.2. Structure–Function Relationship of the K2CO3-Activated Catalysts
The DRM reaction of K2CO3-activated catalysts was performed at 800 °C (Figure 4). The conversion of CO2 and CH4 on MC-600 was much higher than that of MC-500/700/800, reaching a maximum of 64.32% and 50.66%, respectively. The corresponding initial reaction rates were also the highest, reaching 10.39 mmolCH4 gcat.−1 h−1 and 16.08 mmolCO2 gcat.−1 h−1, respectively. The highest conversion may result from more oxygen-containing functional groups on MC-600. However, all catalysts had poor stability, with the CO2 and CH4 conversion decreasing significantly during the 8 h test. This phenomenon may be attributed to carbon deposition during the reaction process, preventing the reactants from reaching the active site [21]. Notably, as illustrated in Figure 4, the CO2 conversion consistently surpassed the CH4 conversion, indicating that the reverse water-gas shift reaction (RWGS: CO2 + H2 → CO + H2O) occurred to a certain extent. For the MC-600/700/800 catalysts, the molar ratio of H2/CO was between 0.23 and 0.46 during the catalytic performance test, below the theoretical value of 1.0. Moreover, the H2/CO ratio gradually declined while the reaction time evolved, indicating that the RWGS reaction intensified [22]. Conversely, the H2/CO ratio from the MC-500 catalyst exceeded 1.0, potentially due to the thermal decomposition of the catalyst under a high reaction temperature.
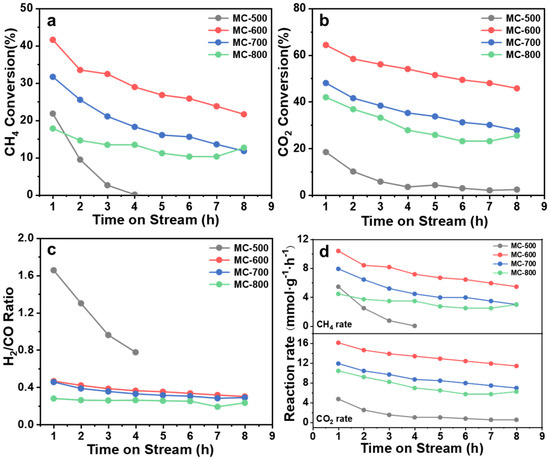
Figure 4.
(a) CH4 conversion, (b) CO2 conversion, (c) H2/CO ratio and (d) Reaction rate over reaction time of MC-500/600/700/800 catalysts. Reaction conditions: GHSV = 6000 mL gcat. −1 h−1; P = 1 atm; CH4: CO2 = 1:1; temperature: 800 °C.
Several researchers have reported that oxygen-containing functional groups can be employed as active sites of metal-free carbon catalysts, especially in dehydrogenation reactions [23,24]. Oxygen-containing functional groups could tailor the electronic properties of the porous carbon materials catalysts, thus optimizing the relevant catalytic reactions. Typical oxygen-containing functional groups, such as carboxyl (O=C-O), quinone/ketone carbonyl (C=O), and hydroxyl (C-OH) on carbon materials are shown in Figure 5a. According to the X-ray photoelectron spectroscopy (XPS) results shown in Figure 5b, the porous carbon materials catalyst was mainly composed of C (284.3 eV) and O (531.9 eV) elements. At the same time, a trace amount of N with XPS binding energy at 399.9 eV was also detected in the porous carbon materials, which can be derived from the petroleum pitch precursor [25]. Notably, the MC-500 samples also contained trace amounts of S (S 2p, 165 eV; S 2s, 225 eV), which may be attributed to the incomplete decomposition of the S-containing groups in the petroleum pitch precursor under lower calcination temperatures [26,27]. Moreover, with the increase in activation temperature, the ratio of N/C content (0.09, 0.05, 0.02, 0.01 for MC-500/600/700/800/, respectively) and the ratio of O/C content (0.48, 0.31, 0.24, 0.17 for MC-500/600/700/800/, respectively) decreased gradually, which indicates that the N- and O-containing groups could be decomposed at high temperatures. The oxygen-containing functional groups were further characterized by deconvolution of the XPS O 1s spectra (Figure 6). The peaks can be attributed to C=O (531.0–531.8 eV), O=C-O (532.2–532.8 eV), C-OH/C-O-C (533.5 eV), and adsorbed water (535.0–536.2 eV), respectively [28]. The relative concentration of oxygen-containing groups was quantified using the peak area fitting method (Table 1). The MC-600 catalyst possessed the highest content of the C=O group, which is consistent with the results of the C 1s XPS analysis (Figure 7 and Table 2) [29]. Considering the superior CH4 conversion of the MC-600 catalyst, the C=O group may be the essential active site for C-H bond cleavage during DRM reaction [30]. As reported in other literature, C-H bonding in CH4 molecule could activate the C=O group on the porous carbon materials surface [31]. Therefore, it is reasonable to believe that the C=O active sites on porous carbon materials could deliver the C-H bond cleavage function via the C=O → C-OH → C=O pathway. In addition, O=C-O, C-OH, and other acidic oxygen-containing groups have been widely reported as the active sites of cracking side reactions [32]. Typically, a lower activation temperature is favorable for the formation of oxygen-containing functional groups. However, as the activation temperature increases, oxygen atoms are inclined to be removed and converted to gaseous oxides [33]. Therefore, 600 °C is the optimum activation temperature with the most active sites for C-H bond cleavage and the least active sites for cracking side reactions.
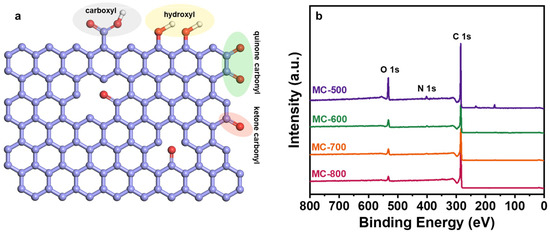
Figure 5.
(a) Oxygen-containing functional groups on the carbon surface and (b) XPS survey spectra of MC-500/600/700/800 catalysts.
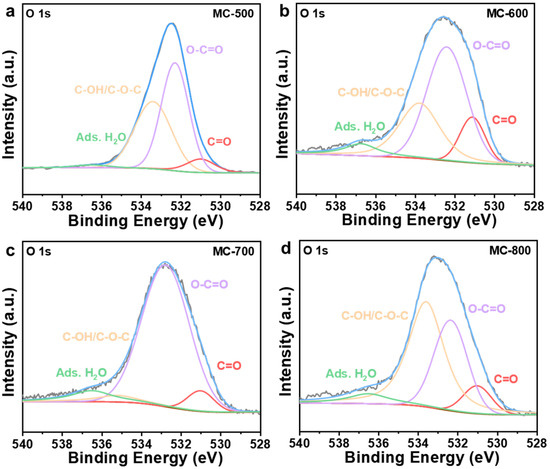
Figure 6.
High-resolution O 1s XPS spectra of (a) MC-500, (b) MC-600, (c) MC-700, and (d) MC-800 catalysts.

Table 1.
Surface elements and oxygen-containing groups content of MC-500/600/700/800 catalysts.
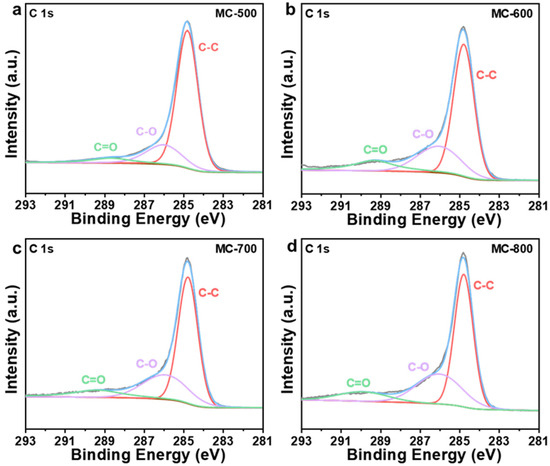
Figure 7.
High-resolution C 1s XPS spectra of (a) MC-500, (b) MC-600, (c) MC-700, and (d) MC-800 catalysts.
Table 2.
C-C, C-O and C=O bonds content of MC-500/600/700/800 catalysts.
N2 adsorption/desorption isotherms were performed to analyze the specific surface area and pore size of the porous carbon materials (Figure 8). All of the catalysts displayed typical I isotherms, indicating that the porous carbon materials catalysts prepared by chemical K2CO3 activation were mainly microporous structures and had a narrow pore size distribution centered at 1.2 nm [34]. As expected, with the increase in activation temperature, the N2 adsorption capacity of the catalyst increased obviously, indicating that the increase in activation temperature could significantly enhance the pore structure of the catalyst, which can be attributed to the stronger etching capability of K2CO3 under high temperatures. As listed in Table 3, the porous carbon materials catalysts exhibited markedly different total specific surface areas (SBET), ranging from 214 m2 g−1 to 1748 m2 g−1. With the increase in activation temperature, the SBET and the total pore volume (Vt) increased mainly in the microporous specific surface area (Smir) and microporous volume (Vmir). In contrast, the pore size (DAVE) first increased and then decreased. This could be attributed to the increased microporous collapse and reduced pore size when the calcination temperature exceeded 700 °C [35]. Notably, no positive correlation between the architectural properties and the DRM catalytic performance was detected from the K2CO3-activated catalysts. However, theoretically, advanced structure properties, such as high specific surface area and large pore size, are favorable to speed up the transportation of reactant molecules and increase the contact opportunity to the active sites, thus boosting catalytic performance. Therefore, increasing the specific surface area of the MC-T series catalysts by further activation methods without decreasing the concentration of the quinone/ketone carbonyl groups seems to be essential to further enhance the DRM catalytic performance of porous carbon-based catalysts.
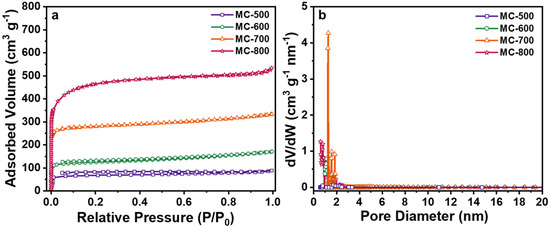
Figure 8.
(a) N2 adsorption/desorption isotherms and (b) Pore size distribution curves of MC-500/600/700/800 catalysts.
Table 3.
Pore structure parameters of MC-500/600/700/800 catalysts.
2.3. Preparation and Structural Characterization of CO2-Activated Catalysts
To address the fast deactivation and low H2/CO ratio of the MC-T series catalysts in DRM, we proposed a secondary activation strategy with CO2 as the activation agent. An MC-600 catalyst with a superior DRM performance was employed as the CO2 activation precursor. This strategy is expected to significantly increase the specific surface area and defectivity of the catalyst through carbon gasification reaction (CO2 + C → 2CO). Furthermore, the oxygen-rich activation environment is also beneficial for the formation of oxygen-containing functional groups, especially the quinone/ketone carbonyl groups [36]. In this section, we prepared three CO2-activated catalysts by changing the CO2 activation temperature.
Figure 9a shows the XRD patterns of MC-600 and MC-600-X (X represents the CO2 activation temperature). All of the CO2-activated catalysts showed two diffraction peaks at about 24° and 43°, which correspond to the (002) and (100) crystal planes of graphitic carbon, respectively [15]. Compared with the MC-600 catalyst, the diffraction peaks of the three modified catalysts broadened and weakened with the increase in activation temperature, indicating the increase in amorphization of the porous carbon materials catalysts [37]. The Raman spectra of the catalysts after CO2 activation are shown in Figure 9b. The ID/IG of MC-600-600, MC-600-700, and MC-600-800 were 1.23, 1.29, and 1.49, respectively, which indicates that the CO2 activation temperature has a significant effect on the defective degree of the carbon-based catalysts. The higher the CO2 activation temperature, the more intense the reaction between CO2 and C, resulting in a higher defective degree of the catalyst [38], which is consistent with the XRD results. The defective structure could provide more anchored sites for oxygen-containing functional groups, thus enhancing the adsorption and activation of CO2/CH4 molecules as well as the DRM catalytic performance.
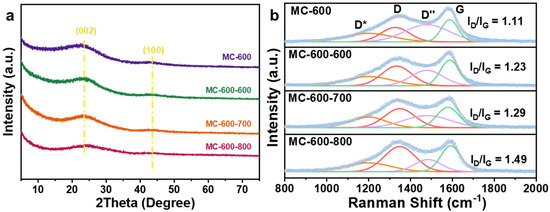
Figure 9.
(a) XRD patterns and (b) Raman spectra of MC-600 and MC-600-600/700/800 catalysts.
The microstructures of the catalysts before and after CO2 activations were observed by SEM. As shown in Figure 10, all catalysts showed a porous structure. Compared with the unmodified porous carbon materials catalyst MC-600, the surface of the MC-600-T series was rougher, and more pores were formed in the carbon. It has been widely accepted that the advanced architecture induced by CO2 activation is conducive to the enhanced transportation of reactant gases and the following DRM reaction [39].

Figure 10.
SEM images of (a) MC-600, (b) MC-600-600, (c) MC-600-700, and (d) MC-600-800 catalysts.
2.4. Structure–Function Relationship of CO2-Activated Catalysts
The DRM performance of the CO2-activated catalysts was tested at 800 °C to investigate the effect of CO2 activation on catalyst performance (Figure 11). In general, CO2 activation effectively improved the CH4 conversion but had no significant effect on the CO2 conversion. The initial CH4 conversion of MC-600-600, MC-600-700, and MC-600-800 catalysts was 45.0%, 48.4%, and 50.7%, respectively, and the deactivation rate was lower than that of the MC-600 catalyst. It should be noted that the catalytic performance of the MC-600-600 was only slightly improved, and the enhanced DRM catalytic performance was more obvious for the MC-600-700 and MC-600-800 catalysts. The initial reaction rates reached 12.12 mmolCH4 gcat.−1 h−1 and 12.62 mmolCH4 gcat.−1 h−1, respectively (Figure 11d). Compared with other carbon-based metal-free catalysts, it has certain advantages under similar reaction conditions (800 °C, CH4/CO2/N2 = 1/1/8) (Table 4). This catalytic performance variation trend could be attributed to the architecture properties and surface quinone/ketone carbonyl group concentration on the porous carbon materials catalysts. Moreover, the CO2 activation step improved the H2/CO ratio significantly. The initial H2/CO ratio of MC-600-600/700/800 was 0.48, 0.53, and 0.57, respectively, which are all higher than that of the MC-600 catalyst. The CO2 activation might increase the number of pores and specific surface area of the catalysts, thus enhancing the adsorption of reactants.
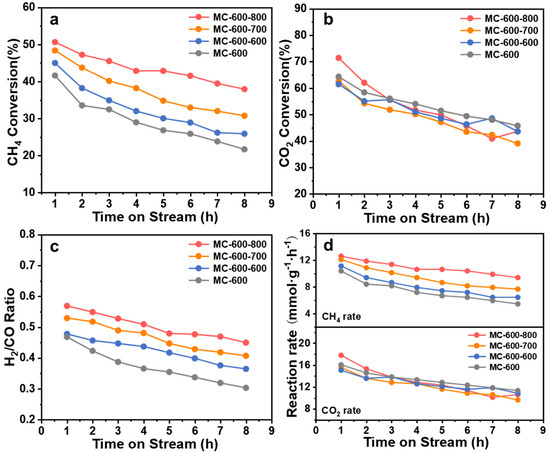
Figure 11.
(a) CH4 conversion, (b) CO2 conversion, (c) H2/CO ratio and (d) Reaction rate over reaction time of MC-600 and MC-600-600/700/800 catalysts. Reaction conditions: GHSV = 6000 mL gcat −1 h−1; P = 1 atm; CH4: CO2 = 1:1; temperature: 800 °C.
Table 4.
Comparison of the DRM performance obtained in this work with other literature a.
XPS spectra were performed to investigate the effect of CO2 activation on the properties of oxygen-containing functional groups (Figure 12). After secondary activation, the catalysts were still predominantly composed of C (284.3 eV) and O (O 1s, 531.9 eV; O 2s 23.3 eV) elements [43], with a trace amount of N (399.9 eV). S (S 2p, 165 eV; S 2s, 225 eV) was also detected in the carbon-based catalyst, which can be attributed to the properties of petroleum pitch feedstock [26,27]. The N/C ratios of the three catalysts (0.03, 0.04, 0.04 for MC-600-600/700/800/, respectively) remained basically the same as the secondary activation temperature increased. In contrast, the O/C ratios of both MC-600-700 and MC-600-800 catalysts decreased compared to those of the MC-600-600 catalyst (0.32, 0.18, 0.24 for MC-600-600/700/800/, respectively). The concentration of oxygen-containing groups analyzed by the deconvolution of the O 1s XPS spectra are listed in Table 5. The relative content of the C=O functional group gradually increased as the activation temperature increased. This trend further clarified the effect of C=O on the catalytic activity of DRM [30], which is consistent with the C 1s XPS results (Figure 13, Table 6). Notably, the relative C=O contents of the MC-600-600/700/800 catalysts were lower than those of the MC-600 catalyst. The high conversions of CH4 and CO2 observed in these catalysts may be attributed to secondary activation, which resulted in a larger specific surface area and enhanced contact between the reactive gases and active sites.
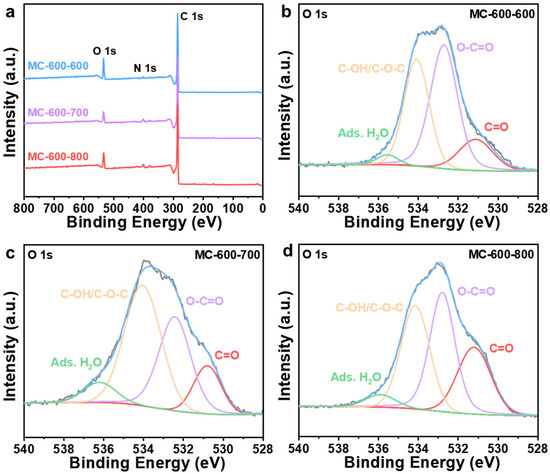
Figure 12.
(a) XPS survey spectra of MC-600-600/700/800 catalysts. High-resolution O 1s XPS spectra of (b) MC-600-600, (c) MC-600-700, and (d) MC-600-800 catalysts.
Table 5.
Surface elements and oxygen-containing groups content of MC-600-600/700/800 catalysts.
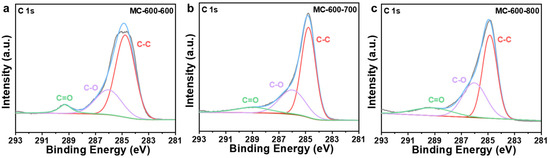
Figure 13.
High-resolution C 1s XPS spectra of (a) MC-600-600, (b) MC-600-700, and (c) MC-600-800 catalysts.
Table 6.
C-C, C-O, and C=O bond content of MC-600-600/700/800 catalysts.
The functional groups on the surface of the MC-600-800 catalyst were monitored by Fourier Transform Infrared (FT-IR) analysis (Figure 14a). In the spectra, the stretching vibration of the -OH group was at around 3400 cm−1, the C-H stretching vibration peak on the benzene ring was around 3000 cm−1, the characteristic peak at around 2300 cm−1 could be attributed to atmospheric CO2, and the bands in the region of around 1400 cm−1 confirmed the existence of C-O or C-H bonds [44]. The typical peaks at around 1000–1300 cm−1 are usually the overlapping effect of C-C/C-O/C-H bonds, while the typical peaks at around 1600–1700 cm−1 can be attributed to the stretching vibration of the C=O group, which corresponds well with the XPS characterization results [44,45,46].
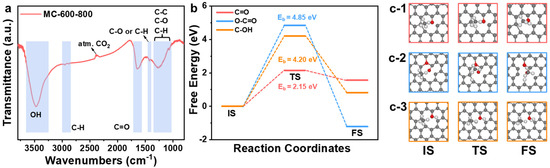
Figure 14.
(a) FT-IR spectrum of MC-600-800 catalyst. (b) Energy profiles of CH4* to CH3* on three models. Energetically favorable configurations of CH4* to CH3* on graphene containing C=O group (c-1), graphene containing O-C=O group (c-2), and graphene containing C-OH group (c-3). Key: transition state (TS), Eb denotes the transition state energy barrier, with the C atom in grey, O atom in red, and H atom in white.
Considering that O-C=O also contributes to the C=O concentration, to further elucidate the effect of different oxygen-containing functional groups on the C-H bond cleavage capacity, graphene cell structures containing different oxygen-containing functional groups were constructed using Materials Studio 2018 software. This study explored the activation process of methane on these oxygen-containing functional group sites (Figure 14b,c). Density functional theory (DFT) results showed that the transition state energy barrier (2.15 eV) for the cleavage of CH4* to CH3* on the graphene-containing C=O groups was the lowest, which was much lower than that of graphene-containing O-C=O groups and C-OH groups (4.85 eV and 4.20 eV, respectively). This finding further confirmed the critical role of C=O groups in the activation of C-H bonds.
Thus, although the total content of C=O and O-C=O groups in the MC-600-600 and MC-600-800 samples is similar (61.73% and 62.42%, respectively), the C=O groups predominate in the MC-600-800 sample treated at higher temperatures. Considering that in the CH4 cracking reaction, the transition state barrier for C=O groups is significantly lower than that for O-C=O groups, this characteristic likely constitutes a critical factor in the superior performance of MC-600-800 over MC-600-600.
The oxygen-containing functional groups in the catalysts after the reaction were characterized by deconvolution of the XPS O 1s spectra (Figure 15 and Table 7). After a reaction of 8 h, the relative contents of the C=O group in MC-600, MC-600-600, MC-600-700, and MC-600-800 were 9.30%, 11.60%, 12.41%, and 24.42%, respectively. Compared to the fresh counterparts, the decrease in the relative content of the C=O group in MC-600-800 throughout the reaction was unnoticeable at only 0.46%. The relative content of C=O groups in MC-600-600 and MC-600-700 decreased by 2.55% and 1.72%, respectively, during the reaction. Conversely, the MC-600 with poor stability delivered obvious C=O group degradation during the DRM reaction (from 12.90% to 9.30%). Therefore, it is reasonable to believe that the stability of the CO2 activated samples is closely related to the content of the C=O group (Figure 11a). This finding also implies that CO2 activation can improve catalyst stability by delaying the degradation of the active groups.
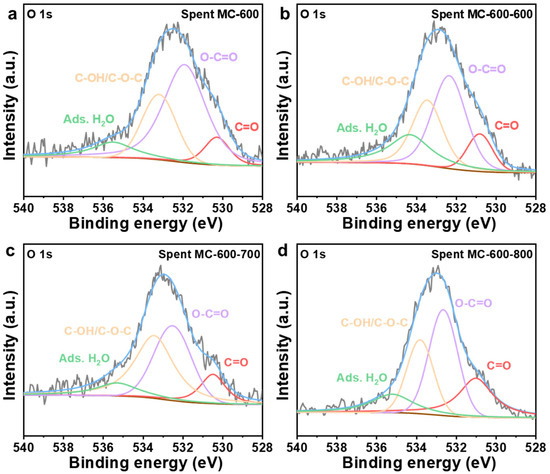
Figure 15.
High-resolution O 1s XPS spectra of (a) spent MC-600, (b) spent MC-600-600, (c) spent MC-600-700, and (d) spent MC-600-800 catalysts.
Table 7.
Surface elements and oxygen-containing groups content of spent MC-600-600/700/800 catalysts.
In order to compare the changes in pore structure and specific surface area of the porous carbon materials catalysts before and after CO2 activation, N2 adsorption-desorption tests were carried out (Figure 16, Table 8). From the N2 adsorption/desorption isotherms, as shown in Figure 12a, the adsorption curve had a steep upward slope at a relative pressure (P/P0) of about 0.1, which confirms the hysteretic type I N2 adsorption behavior [34]. The adsorption curves showed that the CO2-activated catalysts possessed microporous structures, indicating that the CO2 modification did not change the microporous properties of the MC-600 catalyst but enriched the number of micropores, as the Vt and Vmir were increased accordingly, and the VAVE was reduced accordingly. With the secondary activation temperature increase, the SBET of the three catalysts were 398, 500, and 917 m2 g−1, and the Smir were 390, 494, and 897 m2 g−1, respectively. The SBET of the CO2-activated catalysts were all increased compared with that of the unmodified MC-600 catalyst, with the highest increase of about 2.4 times. The pore size distribution, as shown in Figure 15b, also confirmed that the CO2-activated catalysts possessed more micropores than the MC-600 catalyst.
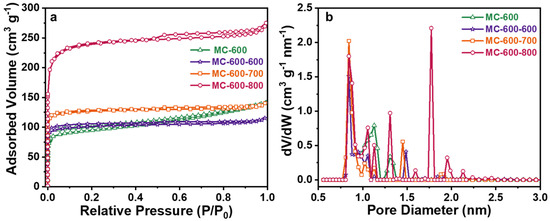
Figure 16.
(a) N2 adsorption/desorption isotherms and (b) pore size distribution curves of MC-600 and MC-600-600/700/800 catalysts.
Table 8.
Pore structure parameters of MC-600 and MC-600-600/700/800 catalysts.
Our analyses indicate that the relative content of C=O groups and the specific surface area of the catalyst together determine the catalytic performance of the catalyst. DFT calculations also showed that C=O groups were essential in activating C-H bonds. Fortunately, due to the higher processing temperature, the O-C=O group is less stable than the C=O group, and the C=O group dominates the MC-600-800, providing it with a lower C-H cleavage energy barrier. At the same time, it has a large specific surface area, which together guarantees the excellent catalytic performance of the MC-600-800 catalyst, which coincides with the core argument of this work.
The adsorption behavior of reactant molecules in the pore structure of the catalysts is also crucial for the DRM performance. The impact of competitive adsorption of CO2 and CH4 molecules in the narrow pore channels on the DRM performance is inevitable. During the DRM reaction, the ideal stoichiometric ratio of CO2 to CH4 is 1, so the closer the molar content of CO2 and CH4 molecules on the porous carbon material surfaces, the more favorable for the reaction. We investigated the adsorption capacity of the MC-600 and MC-600-800 catalysts for CH4 and CO2 molecules by measuring the CH4/CO2 physisorption ratio. As shown in Figure 17, a lower gas adsorption ratio was observed on the MC-600-800 catalysts, indicating well-balanced competitive adsorption of CH4 and CO2 molecules on the active sites, which well explains their superior DRM catalytic performance.
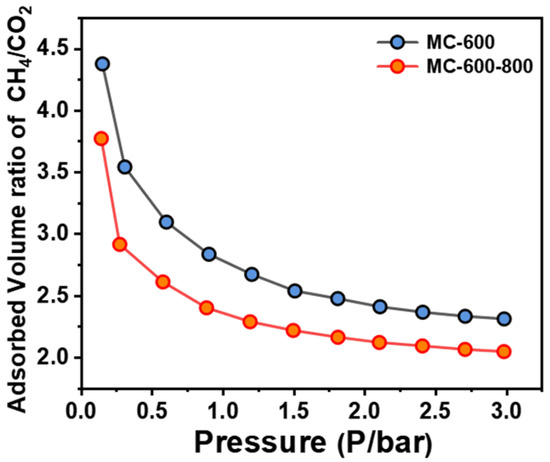
Figure 17.
Competitive adsorption isotherms of CH4 and CO2 on MC-600 and MC-600-800 catalysts.
3. Materials and Methods
3.1. Materials
Petroleum pitch (Sinopec Jiujiang Branch), anhydrous potassium carbonate (K2CO3, analytical grade), and sodium chloride (NaCl, analytical grade) were purchased from Sinopharm Chemical Reagent Group Co., Ltd., Shanghai, China. All reagents were used directly without further purification.
3.2. Catalyst Preparation
3.2.1. Preparation of K2CO3-Activated Catalysts
Firstly, petroleum pitch (1.0 g) and K2CO3 (4.0 g) were fully grounded and mixed. Subsequently, the mixture was pyrolyzed at 500/600/700/800 °C for 2 h with a heating rate of 5 °C ⋅min−1 in a tube furnace under a N2 atmosphere. After cooling down to room temperature, the obtained product was washed with deionized water repeatedly to remove residues then collected by vacuum filtration. Finally, the obtained sample was dried in an oven at 60 °C overnight. The obtained porous carbon materials catalysts were named MC-T (T = 500, 600, 700, and 800 °C), where T represents the temperature during the calcination.
3.2.2. Preparation of CO2-Activated Catalysts
The MC-600 sample prepared by K2CO3 activation of petroleum pitch was heated at 600/700/800 °C for 1 h with a heating rate of 5 °C min−1 in a tubular furnace under a CO2 atmosphere. The modified porous carbon materials catalysts were denoted as MC-600-600/700/800, respectively.
3.3. Catalytic Activity Evaluation
Dry reforming of methane was conducted in a quartz tube reactor with an inner diameter of 6 mm under atmospheric pressure. Before loading, the catalyst was crushed and sieved to 20–40 mesh. The quartz sand and catalyst particles were mixed and placed in the middle of the quartz tube reactor with quartz wool. Before the reaction, the temperature was raised to 600 °C at 5 °C min−1 in a N2 atmosphere and kept for 1 h [47] to remove impurities on the catalyst surface. At the beginning of the reaction, the catalytic performance of 10 Vol % CH4, 10 Vol %CO2, 2 Vol %N2 [48] (internal standard to calculate the conversion of CH4 and CO2), and 78 Vol % Ar were tested. The outlet gas was determined by a gas chromatograph (GC, Huifen HF-900, Purchased from Shandong Huifen Instrument Co., Ltd., Zaozhuang, China.) equipped with a thermal conductivity detector (TCD) and a TDX-01 column. The CH4 conversion, CO2 conversion, and H2/CO ratio were calculated according to the following equations (Equations (5)–(7)):
where Fi,in and Fi,out represent the molar flow rate (mol s−1) of component i of the inlet and outlet.
3.4. Catalyst Characterization
The X-ray diffraction (XRD) patterns were evaluated using an X′ Pert Pro MPD diffractometer with Cu Kα radiation source, the scanning range was 5–75°, and the scanning rate was 10° min−1. The Raman spectra were conducted on a Renishaw RM2000 employing a 512 nm laser (filtered to 2.5 mW) to test the defective structure of the catalysts. The laser beam was focused on the sample surface by a 50× microscope objective. The diffraction grating and exposure time were set to 1800 l/mm and 0.1 s, respectively. The morphology and microstructure of the catalysts were observed by a scanning electron microscopy (SEM, FEI Nova nanoSEM 450) with an electron acceleration voltage of 10 kV; The specific surface area and pore structure parameters were obtained by analyzing the N2 adsorption/desorption isotherms, which were recorded using a Micromeritics ASAP 2020 analyzer (Micromeritics Instrument (Shanghai) Ltd., China.). Before characterization, the catalyst was degassed at 300 °C for 6 h. The specific surface area was calculated by the BET (Brunauer-EmmettTeller) equation, and other pore structure parameters were calculated by the NLDFT (Delocalized Density Function Theory) theory. The surface chemical properties, especially the type and content of oxygen-containing functional groups on the surface, were evaluated on an Escalab 250XI X-ray photoelectron spectroscopy equipped with a monochromatic Al Kα (1486.6 eV) radiation source. The obtained XPS spectra were fitted with XPS peak software (XPSPEAK 4.1) to analyze all chemical compositions. The binding energy was calibrated using the C 1s peak at 284.8 eV [49].
3.5. Computational Details
Materials Studio 2018 software was used to construct three graphene cell structures containing different oxygen-containing functional groups, denoted as C=O, O-C=O, and C-OH (all structures contain around 76 atoms). All structures have periodic boundary conditions.
All the computational calculations were carried out in the framework of the density functional theory (DFT) with the projector augmented plane-wave method, as implemented in the Vienna ab initio simulation package (VASP) [50,51]. The generalized gradient approximation proposed by Perdew, Burke, and Ernzerhof was employed for the exchange-correlation potential [52,53]. The cutoff energy for plane wave was set to 400 eV. Electron smearing was used via the Methfessel-Paxton technique with a smearing width consistent to σ = 0.05 eV. The Monkhorst-Pack meshes with a grid of (1 × 1 × 1) k-points were applied for all calculations. All structures were optimized until the forces on the atoms were less than 0.03 eV/Å. The transition state structure was optimized until the forces on the atoms became converged to 0.05 eV/Å. DFT-D3 functional was used to describe the van der Waals interaction [54]. All transition states were searched by combining the climbing image nudged elastic band (CI-NEB) method with the dimer method, and the stretching frequencies of the saddle points were analyzed to ensure a transition state with only one imaginary frequency [55,56].
The reaction energy (ΔE) and barrier (Eb) were calculated via the following equations (Equations (8) and (9)):
where EIS, ETS, and EFS are the total energy of the corresponding initial state (IS), transitional state (TS), and final state (FS), respectively.
4. Conclusions
Firstly, a series of petroleum asphalt-based derived porous carbon materials with abundant porous structures and surface oxygen-containing groups were successfully prepared using a simple K2CO3 and CO2 activation method and applied in metal-free catalysts for a DRM reaction. For the DRM reaction, the structure–function relationship analysis and DFT calculation showed that the quinone/ketone carbonyl (C=O) groups with the lowest CH4* cleavage energy barrier (2.15 eV) played critical roles in boosting the DRM reaction performance. A higher relative content of surface C=O groups could provide more active sites, thus promoting the dehydrogenation reaction of CH4 more effectively. When the K2CO3 activation temperature exceeds 700 °C, the C=O groups begin to decompose, and the catalyst activity decreases. By following with the secondary CO2 activation of the catalyst with the CO2 process at 800 °C, the architectural properties of the porous carbon-based catalyst were developed with the concentration of quinone/ketone carbonyl (C=O) groups well-maintained. In addition, the CO2 activation also effectively attenuated the competitive adsorption of CH4 and CO2 on the catalyst surface, which is necessary to boost the DRM performance. Benefiting from the cooperative interplay between the advanced architectural properties and the sufficient quinone/ketone carbonyl (C=O) groups, superior DRM performance was achieved. Surface structure of the catalyst and the content of C=O groups were significantly changed, and the contact area between the reacting gas and the active sites was increased, thus further enhancing the catalytic activity. In addition, the secondary activation also effectively attenuated the competitive adsorption of CH4 and CO2, which was necessary for enhancing the DRM performance of the metal-free carbon-based materials. The stepwise activation method proposed in this work provides a new novel strategy for modulating the surface structure and surface-active groups of carbon materials and for rationally designing metal-free porous materials as carbon-based DRM catalysts.
Author Contributions
Conceptualization, K.H. and Y.W.; methodology, K.H. and Y.S.; validation, K.H., Y.S., H.J. and Z.C.; formal analysis, K.H., Y.S., H.J. and S.L.; investigation, K.H., H.F. and L.L.; resources, Y.W. and M.W.; writing—original draft preparation, K.H.; writing—review and editing, Y.W. and M.W. visualization, K.H., Y.S., H.J. and S.C.; supervision, Y.W. and M.W.; project administration, Y.W. and M.W. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Key Research and Development Program of China (2023YFB4104500, 2023YFB4104502), the National Natural Science Foundation of China (22108310), and the Key Laboratory for Green Chemical Technology of the Ministry of Education.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors on request.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| AC | Activated carbon |
| CMD | Catalytic methane decomposition |
| CNT | Carbon nanotube |
| CB | Carbon black |
| DRM | Dry reforming of methane |
| DFT | Density-functional theory |
| FT-IR | Fourier transform infrared |
| GHSV | gas hourly space velocity |
| NLDFT | Non localized density functional theory |
| POM | Partial oxidation of methane |
| RWGS | Reverse water gas shift reaction |
| SRM | Steam reforming of methane |
| SEM | Scanning electron microscopy |
| XPS | X-ray photoelectron spectroscopy |
| XRD | X-ray diffraction |
References
- Ronald, M.; Isa, Y. Decomposition of methane to carbon and hydrogen: A catalytic perspective. Energy Technol. 2019, 7, 1800593. [Google Scholar]
- Li, L.; MD Dostagir, N.H.; Shrotri, A.; Fukuoka, A.; Kobayashi, H. Partial oxidation of methane to syngas via formate intermediate found for a ruthenium-rhenium bimetallic catalyst. ACS Catal. 2021, 11, 3782–3789. [Google Scholar] [CrossRef]
- Palmer, C.; Upham, D.; Smart, S.; Gordon, M.; Metiu, H.; McFarland, E. Dry reforming of methane catalysed by molten metal alloys. Nat. Catal. 2020, 3, 83–89. [Google Scholar] [CrossRef]
- Zhang, J.; Li, X.; Chen, H.; Qi, M.; Zhang, G.; Hu, H.; Ma, X. Hydrogen production by catalytic methane decomposition: Carbon materials as catalysts or catalyst supports. Int. J. Hydrogen Energy 2017, 42, 19755–19775. [Google Scholar] [CrossRef]
- Li, Z.; Lin, Q.; Li, M.; Cao, J.; Liu, F.; Pan, H.; Wang, Z.; Kawi, S. Recent advances in process and catalyst for CO2 reforming of methane. Renew. Sustain. Energy Rev. 2020, 134, 110312. [Google Scholar] [CrossRef]
- Wang, W.; Zeng, C.; Tsubaki, N. Recent advancements and perspectives of the CO2 hydrogenation reaction. Green Carbon 2023, 1, 133–145. [Google Scholar] [CrossRef]
- Li, L.; Yan, K.; Chen, J.; Feng, T.; Wang, F.; Wang, J.; Song, Z.; Ma, C. Fe-rich biomass derived char for microwave-assisted methane reforming with carbon dioxide. Sci. Total Environ. 2019, 657, 1357–1367. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, J.; Tsubaki, N. Clever nanomaterials fabrication techniques encounter sustainable C1 catalysis. Acc. Chem. Res. 2023, 56, 2341–2353. [Google Scholar] [CrossRef]
- Liu, X.; Dai, L. Carbon-based metal-free catalysts. Nat. Rev. Mater. 2016, 1, 16064. [Google Scholar] [CrossRef]
- Moliner, R.; Suelves, I.; Lázaro, M.; Moreno, O. Thermocatalytic decomposition of methane over activated carbons: Influence of textural properties and surface chemistry. Int. J. Hydrogen Energy 2005, 30, 293–300. [Google Scholar] [CrossRef]
- Li, Y.; Xiao, R.; Jin, B.; Zhang, H. Experimental study of the reforming of methane with carbon dioxide over coal char. Int. J. Chem. React. Eng. 2008, 6, A16. [Google Scholar] [CrossRef]
- Samuel, J.; Ok, S. Petroleum asphaltene-based activated nanoporous carbon for CO2 capture and H2 storage. Ind. Eng. Chem. Res. 2023, 62, 9939–9950. [Google Scholar] [CrossRef]
- Suelves, I.; Pinilla, J.; Lázaro, M.; Moliner, R. Carbonaceous materials as catalysts for decomposition of methane. Chem. Eng. J. 2008, 140, 432–438. [Google Scholar] [CrossRef]
- Zhang, F.; Liu, T.; Hou, G.; Kou, T.; Guan, R.; Li, Y. Hierarchically porous carbon foams for electric double layer capacitors. Nano Res. 2016, 9, 2875–2888. [Google Scholar] [CrossRef]
- Xu, C.; Gu, Q.; Li, S.; Ma, L.; Zhou, Y.; Zhang, X.; Jiang, C.; Pham-Huu, C.; Liu, Y. Heteroatom-doped monolithic carbocatalysts with improved sulfur selectivity and impurity tolerance for H2S selective oxidation. ACS Catal. 2021, 11, 8591–8604. [Google Scholar] [CrossRef]
- Kim, Y.S.; Hanif, M.A.; Song, H.; Kim, S.; Cho, Y.; Ryu, S.K.; Kim, H.G. Wood-Derived graphite: A sustainable and cost-effective material for the wide range of industrial applications. Crystals 2024, 14, 309. [Google Scholar] [CrossRef]
- Pope, C. X-Ray Diffraction and the bragg equation. J. Chem. Educ. 1997, 74, 129–131. [Google Scholar] [CrossRef]
- Guo, J.; Lou, H.; Zheng, X. The deposition of coke from methane on a Ni/MgAl2O4 catalyst. Carbon 2007, 45, 1314–1321. [Google Scholar] [CrossRef]
- Dobele, G.; Volperts, A.; Plavniece, A.; Zhurinsh, A.; Upskuviene, D.; Balciunaite, A.; Niaura, G.; Colmenares-Rausseo, L.C.; Tamasauskaite-Tamasiunaite, L.; Norkus, E. Thermochemical Activation of Wood with NaOH, KOH and H3PO4 for the Synthesis of Nitrogen-Doped Nanoporous Carbon for Oxygen Reduction Reaction. Molecules 2024, 29, 2238. [Google Scholar] [CrossRef] [PubMed]
- Gnawali, C.L.; Shrestha, L.K.; Hill, J.P.; Renzhi, M.; Ariga, K.; Adhikari, M.P.; Rajbhandari, R.; Pokharal, B.P. Nanoporous activated carbon material from Terminalia chebula seed for supercapacitor application. C 2023, 9, 109. [Google Scholar] [CrossRef]
- Han, C.; Zhu, X.; Chen, B.; Wang, X. A strategy of constructing the Ni@ silicalite-1 catalyst structure with high activity and resistance to sintering and carbon deposition for dry reforming of methane. Fuel 2024, 355, 129548. [Google Scholar] [CrossRef]
- Hussain, I.; Jalil, A.; Mamat, C.; Siang, T.; Rahman, A.; Azami, M.; Adnan, R. New insights on the effect of the H2/CO ratio for enhancement of CO methanation over metal-free fibrous silica ZSM-5: Thermodynamic and mechanistic studies. Energy Convers. Manag. 2019, 199, 112056. [Google Scholar] [CrossRef]
- Tang, S.; Cao, Z. Site-dependent catalytic activity of graphene oxides towards oxidative dehydrogenation of propane. Phys. Chem. Chem. Phys. 2012, 14, 16558–16565. [Google Scholar] [CrossRef]
- Zhang, G.; Qu, J.; Su, A.; Zhang, Y.; Xu, Y. Towards understanding the carbon catalyzed CO2 reforming of methane to syngas. J. Ind. Eng. Chem. 2015, 21, 311–317. [Google Scholar] [CrossRef][Green Version]
- Song, Y.; Liu, G.; Yuan, Z. N-, P-and B-doped mesoporous carbons for direct dehydrogenation of propane. RSC Adv. 2016, 6, 94636–94642. [Google Scholar] [CrossRef]
- O’Connor, E.; Long, R.D.; Cherkaoui, K.; Thomas, K.; Chalvet, F.; Povey, I.M.; Pemble, M.E.; Hurley, P.K.; Brennan, B.; Hughes, G.; et al. In situ H2S passivation of In0.53Ga0.47As/InP metal-oxide-semiconductor capacitors with atomic-layer deposited HfO2 gate dielectric. Appl. Phys. Lett. 2008, 92, 022902. [Google Scholar] [CrossRef]
- Bae, E.; Hun Kang, Y.; Jang, K.S.; Yun Cho, S. Enhancement of thermoelectric properties of PEDOT: PSS and tellurium-PEDOT: PSS hybrid composites by simple chemical treatment. Sci. Rep. 2016, 6, 18805. [Google Scholar]
- Hu, Z.; Zhao, H.; Chen, C.; Yuan, Z. Castanea mollissima shell-derived porous carbons as metal-free catalysts for highly efficient dehydrogenation of propane to propylene. Catal. Today 2018, 316, 214–222. [Google Scholar] [CrossRef]
- Dolgov, A.; Lopaev, D.; Lee, C.J.; Zoethout, E.; Medvedev, V.; Yakushev, O.; Bijkerk, F. Characterization of carbon contamination under ion and hot atom bombardment in a tin-plasma extreme ultraviolet light source. Appl. Surf. Sci. 2015, 353, 708–713. [Google Scholar] [CrossRef]
- Zhang, J.; Su, D.; Blume, R.; Schlögl, R.; Wang, R.; Yang, X.; Gajovic’, A. Surface chemistry and catalytic reactivity of a nanodiamond in the steam-free dehydrogenation of ethylbenzene. Angew. Chem. Int. Ed. 2010, 122, 8822–8826. [Google Scholar] [CrossRef]
- Zheng, A.; Chu, Y.; Li, S.; Su, D.; Deng, F. Insight into the activation of light alkanes over surface-modified carbon nanotubes from theoretical calculations. Carbon 2014, 77, 122–129. [Google Scholar] [CrossRef]
- Zhou, J.; Sui, Z.; Zhu, J.; Li, P.; Chen, D.; Dai, Y.; Yuan, W. Characterization of surface oxygen complexes on carbon nanofibers by TPD, XPS and FT-IR. Carbon 2007, 45, 785–796. [Google Scholar] [CrossRef]
- Phothong, K.; Tangsathitkulchai, C.; Lawtae, P. The analysis of pore development and formation of surface functional groups in bamboo-based activated carbon during CO2 activation. Molecules 2021, 26, 5641. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Yin, J.; Zhu, X.; Guo, X.; Hu, P.; Yan, X.; Lang, W.; Guo, Y. P-modified microporous carbon nanospheres for direct propane dehydrogenation reactions. Carbon 2019, 152, 855–864. [Google Scholar] [CrossRef]
- Sakina, F.; Baker, R. Metal-and halogen-free synthesis of ordered mesoporous carbon materials. Microporous Mesoporous Mater. 2019, 289, 109622. [Google Scholar] [CrossRef]
- Kim, S.; Jeong, S.; Park, K.; Lee, K.; Kim, H. Effect of oxygen-containing functional groups in metal-free carbon catalysts on the decomposition of methane. Catal. Commun. 2021, 148, 106167. [Google Scholar] [CrossRef]
- Pal, N.; Bhattacharyya, A. Probing the “universal” amorphization of crystalline sulfur in its mixture with ultrahigh surface area porous carbon. J. Phys. Chem. C. 2023, 127, 5713–5719. [Google Scholar] [CrossRef]
- Ashok, J.; Pati, S.; Hongmanorom, P.; Zhang, T.; Chen, J.; Kawi, S. A review of recent catalyst advances in CO2 methanation processes. Catal. Today 2020, 356, 471–489. [Google Scholar] [CrossRef]
- He, T.; Sun, Z.; Wu, J.; Xu, Z.; Zhang, D.; Han, D. Catalytic performance of coal char for the methane reforming process. Chem. Eng. Technol. 2015, 38, 68–74. [Google Scholar] [CrossRef]
- Haghighi, M.; Sun, Z.; Wu, J.; Bromly, J.; Wee, H.; Ng, E.; Wang, Y.; Zhang, D. On the reaction mechanism of CO2 reforming of methane over a bed of coal char. Proc. Combust. Inst. 2007, 31, 1983–1990. [Google Scholar] [CrossRef]
- Tan, Y.; Wang, S.; Li, L.; Meng, B.; Chen, J.; Yang, Z.; Yan, K.; Qin, X. Application of microwave heating for methane dry reforming catalyzed by activated carbon. Chem. Eng. Process 2019, 145, 107662. [Google Scholar] [CrossRef]
- Li, L.; Wang, H.; Jiang, X.; Song, Z.; Zhao, X.; Ma, C. Microwave-enhanced methane combined reforming by CO2 and H2O into syngas production on biomass-derived char. Fuel 2016, 185, 692–700. [Google Scholar] [CrossRef]
- Atuchin, V.V.; Kesler, V.G.; Sapozhnikov, V.K.; Yakovenchuk, V.N. X-ray photoelectron spectrometry and binding energies of Be 1s and O 1s core levels in clinobarylite, BaBe2Si2O7, from Khibiny massif, Kola peninsula. Mater. Charact. 2008, 59, 1329–1334. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, S.; Zheng, Y.; Luo, Z.; Cen, K. Mechanism study of wood lignin pyrolysis by using TG-FTIR analysis. J. Anal. Appl. Pyrol 2008, 82, 170–177. [Google Scholar] [CrossRef]
- Zhang, M.; Bai, Y.; Men, X.; Song, X.; Lv, P.; Wang, J.; Su, W.; Lu, G.; Yu, G. Evolution of oxygen-containing functional groups on coal char surface during gasification in H2O/CO2. J. Energy Inst. 2024, 114, 101622. [Google Scholar] [CrossRef]
- Streletskiy, O.A.; Zavidovskiy, I.A.; Nuriahmetov, I.F.; Khaidarov, A.A.; Pavlikov, A.V.; Minnebaev, K.F. The field-effect transistor based on a polyyne-polyene structure obtained via PVDC dehydrochlorination. J. Compos. Sci 2023, 7, 264. [Google Scholar] [CrossRef]
- Cheng, Z.; Wang, Y.; Jin, D.; Liu, J.; Wang, W.; Gu, Y.; Ni, W.; Feng, Z.; Wu, M. Petroleum pitch-derived porous carbon as a metal-free catalyst for direct propane dehydrogenation to propylene. Catal. Today 2023, 410, 164–174. [Google Scholar] [CrossRef]
- Wu, Z.; Yang, B.; Miao, S.; Liu, W.; Xie, J.; Lee, S.; JPellin, M.; Xiao, D.; Su, D.; Ma, D. Lattice strained Ni-Co alloy as a high-performance catalyst for catalytic dry reforming of methane. ACS Catal. 2019, 9, 2693–2700. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, W.; He, R.; Li, M.; Zhang, J.; Cao, F.; Liu, J.; Lin, S.; Gao, X.; Yang, G.; et al. Carbon-based electron buffer layer on ZnOx-Fe5C2-Fe3O4 boosts ethanol synthesis from CO2 hydrogenation. Angew. Chem. 2023, 135, e202311786. [Google Scholar]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- White, J.A.; Bird, D.M. Implementation of gradient-corrected exchange-correlation potentials in Car-Parrinello total-energy calculations. Phys. Rev. B 1994, 50, 4954. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Henkelman, G.; Jónsson, H. Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points. J. Chem. Phys. 2000, 113, 9978–9985. [Google Scholar] [CrossRef]
- Henkelman, G.; Jónsson, H. A dimer method for finding saddle points on high dimensional potential surfaces using only first derivatives. J. Chem. Phys. 1999, 111, 7010–7022. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).