
Carbon-Supported Fe-Based Catalyst for Thermal-Catalytic CO2 Hydrogenation into C2+ Alcohols: The Effect of Carbon Support Porosity on Catalytic Performance

 ,
, 
Abstract
1. Introduction
2. Results and Discussion
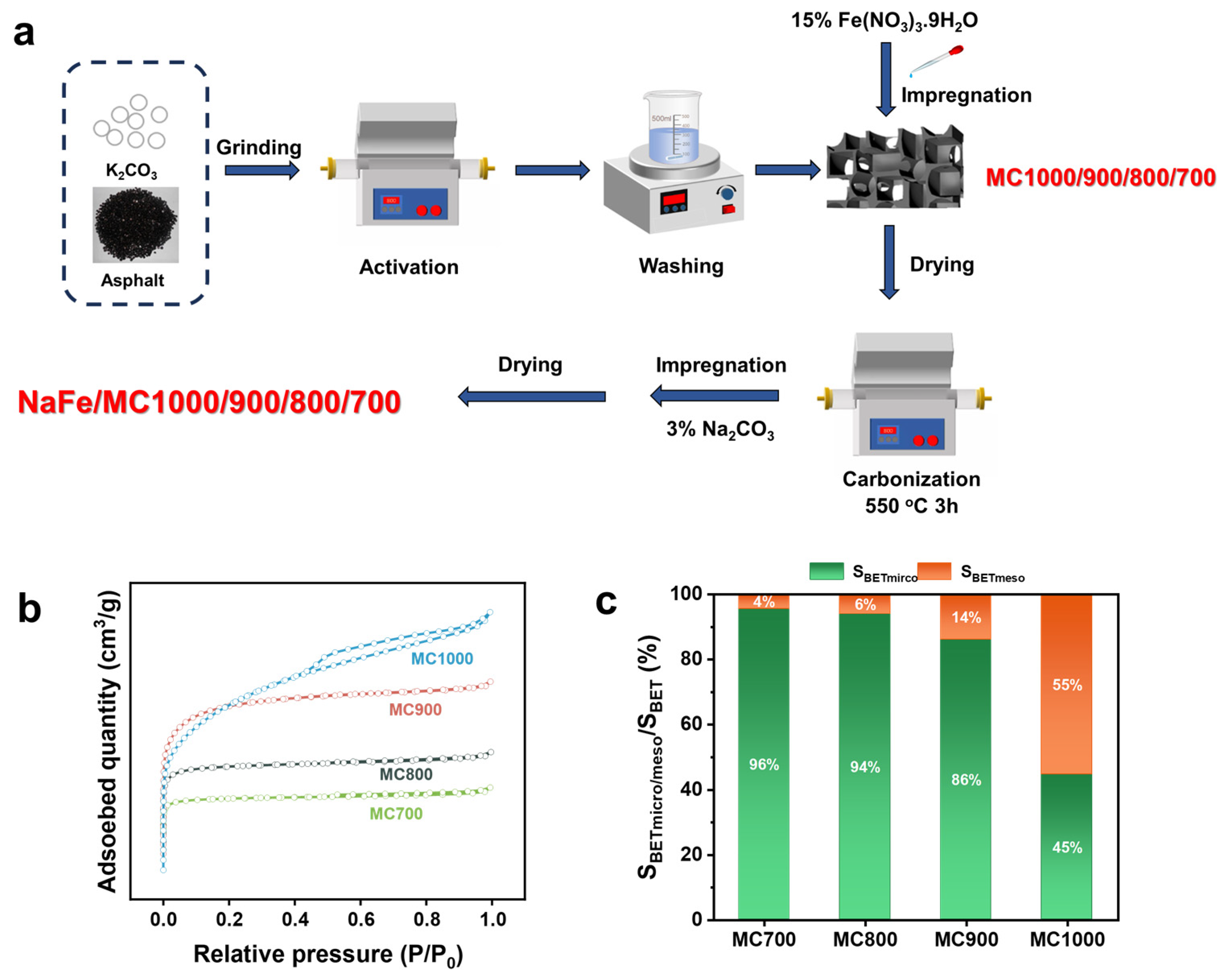
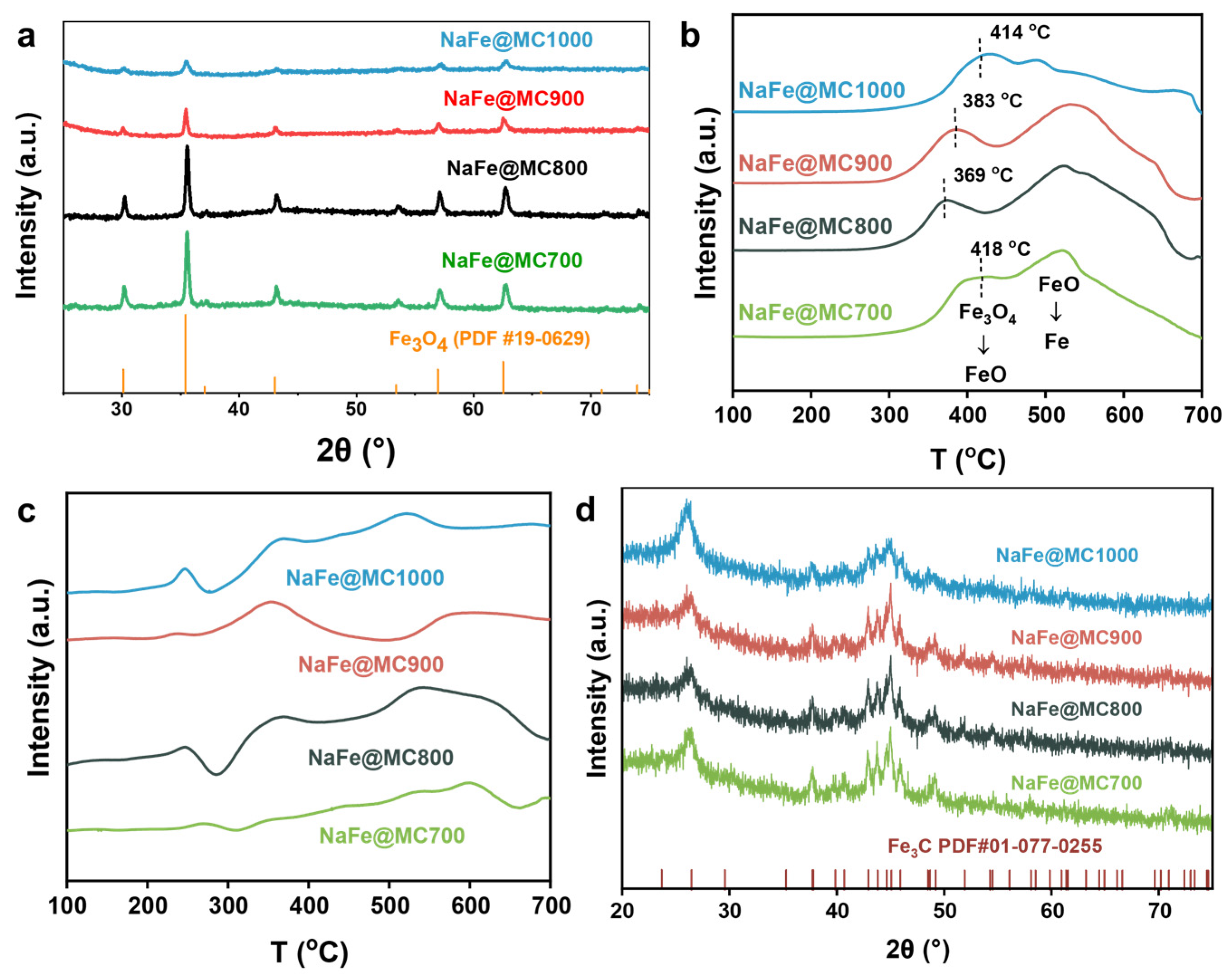
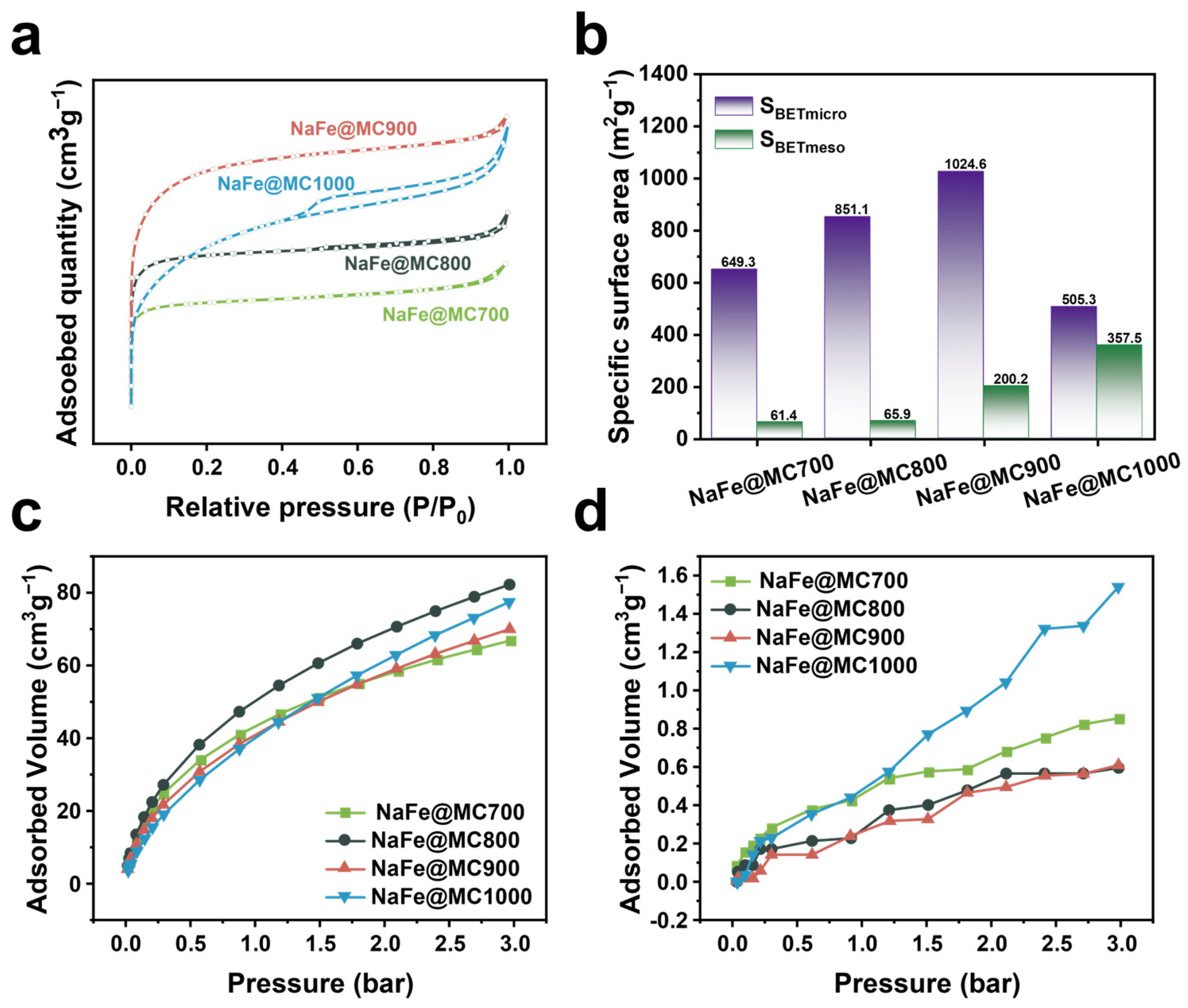
2.1. Structural Characterization of Catalysts
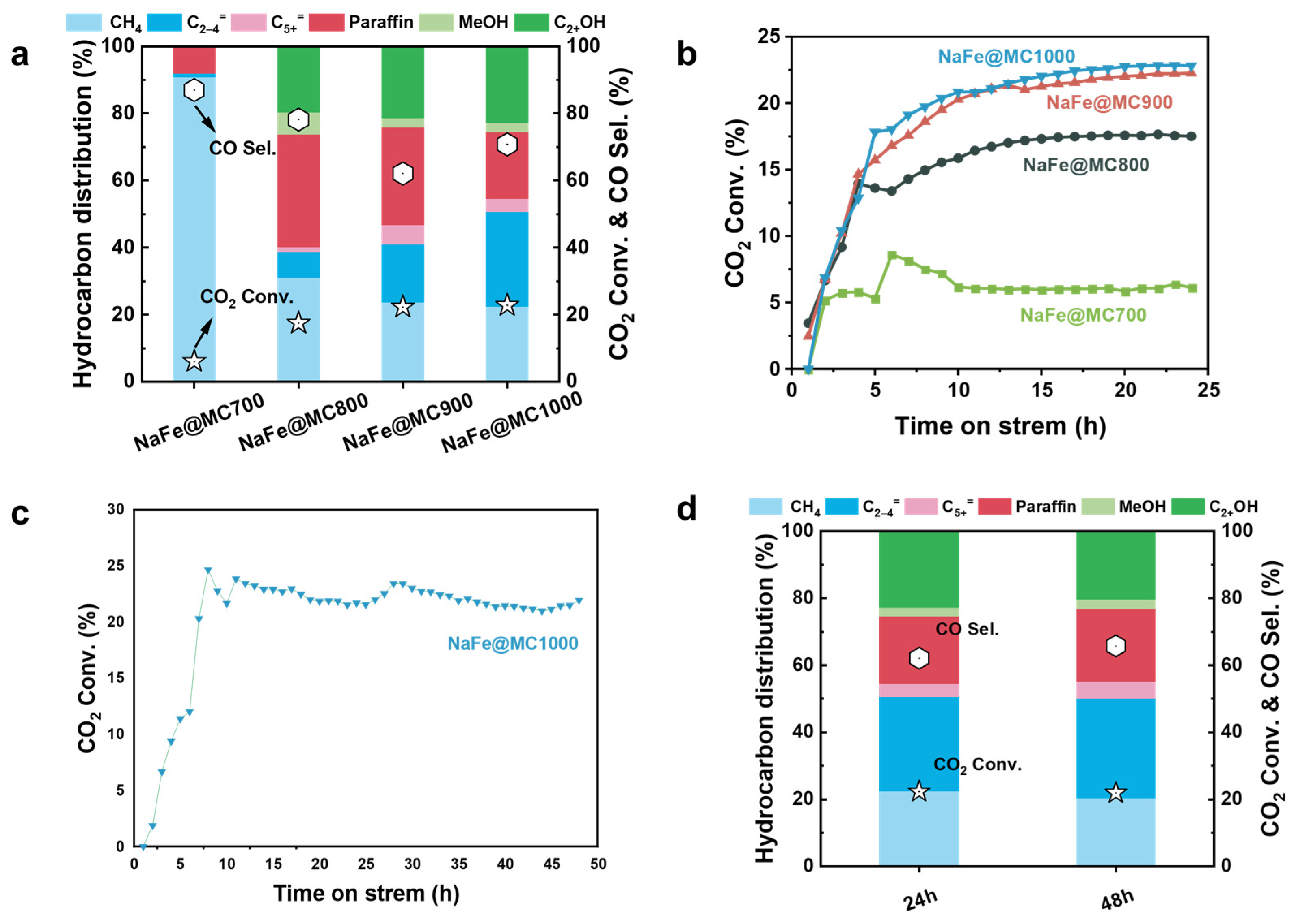
2.2. Catalytic Performance
2.3. Influence of Catalyst Pore Structure on Catalytic Performance
3. Materials and Methods
3.1. Materials
3.2. Catalyst Preparation
3.3. Catalyst Characterization
3.4. Catalytic Evaluation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gao, P.; Li, S.; Bu, X.; Dang, S.; Liu, Z.; Wang, H.; Zhong, L.; Qiu, M.; Yang, C.; Cai, J.; et al. Direct conversion of CO2 into liquid fuels with high selectivity over a bifunctional catalyst. Nat. Chem. 2017, 9, 1019–1024. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, J.; Qu, Y.; Liu, H.; Tang, C.; Miao, S.; Feng, Z.; An, H.; Li, C. Highly selective conversion of carbon dioxide to lower olefins. ACS Catal. 2017, 7, 8544–8548. [Google Scholar] [CrossRef]
- Wei, J.; Ge, Q.; Yao, R.; Wen, Z.; Fang, C.; Guo, L.; Xu, H.; Sun, J. Directly converting CO2 into a gasoline fuel. Nat. Commun. 2017, 8, 15174. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, M.; Zhou, C.; Zhou, W.; Cheng, K.; Kang, J.; Zhang, Q.; Deng, W.; Wang, Y. Selective transformation of carbon dioxide into lower olefins with a bifunctional catalyst composed of ZnGa2O4 and SAPO-34. Chem. Commun. 2018, 54, 140–143. [Google Scholar] [CrossRef]
- Ni, Y.; Chen, Z.; Fu, Y.; Liu, Y.; Zhu, W.; Liu, Z. Selective conversion of CO2 and H2 into aromatics. Nat. Commun. 2018, 9, 3457. [Google Scholar] [CrossRef]
- Wei, J.; Yao, R.; Ge, Q.; Wen, Z.; Ji, X.; Fang, C.; Zhang, J.; Xu, H.; Sun, J. Catalytic hydrogenation of CO2 to isoparaffins over Fe-based multifunctional catalysts. ACS Catal. 2018, 8, 9958–9967. [Google Scholar] [CrossRef]
- Li, Z.; Qu, Y.; Wang, J.; Liu, H.; Li, M.; Miao, S.; Li, C. Highly selective conversion of carbon dioxide to aromatics over tandem catalysts. Joule 2019, 3, 570–583. [Google Scholar] [CrossRef]
- Cui, X.; Gao, P.; Li, S.; Yang, C.; Liu, Z.; Wang, H.; Zhong, L.; Sun, Y. Selective production of aromatics directly from carbon dioxide hydrogenation. ACS Catal. 2019, 9, 3866–3876. [Google Scholar] [CrossRef]
- Li, K.; Chen, J.G. CO2 hydrogenation to methanol over ZrO2-containing catalysts: Insights into ZrO2 induced synergy. ACS Catal. 2019, 9, 7840–7861. [Google Scholar] [CrossRef]
- Jiang, X.; Nie, X.; Guo, X.; Song, C.; Chen, J.G. Recent advances in carbon dioxide hydrogenation to methanol via heterogeneous catalysis. Chem. Rev. 2020, 120, 7984–8034. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, J.; Noritatsu, T. Clever nanomaterials fabrication techniques encounter sustainable C1 catalysis. Acc. Chem. Res. 2023, 56, 2341–2353. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zeng, C.; Noritatsu, T. Recent advancements and perspectives of the CO2 hydrogenation reaction. Green Carbon 2023, 1, 133–145. [Google Scholar] [CrossRef]
- Shyama, C.M.; Amitabha, D.; Diptendu, R.; Sandeep, D.; Akhil, S.N.; Long, C.; Biswarup, P. Developments of the heterogeneous and homogeneous CO2 hydrogenation to value-added C2+-based hydrocarbons and oxygenated products. Coord. Chem. Rev. 2022, 471, 214737. [Google Scholar]
- Ho, T.L.; Cecilia, M.; Daniel, C.F.; Joseph, A.S.; Javier, P.R. Status and prospects in higher alcohols synthesis from syngas. Chem. Soc. Rev. 2017, 46, 1358. [Google Scholar]
- Xu, D.; Wang, Y.; Ding, M.; Hong, X.; Liu, G.; Tsang, S.C.E. Advances in higher alcohol synthesis from CO2 hydrogenation. Chem 2021, 7, 849–881. [Google Scholar] [CrossRef]
- Torres Galvis, H.M.; de Jong, K.P. Catalysts for production of lower olefins from synthesis gas: A review. ACS Catal. 2013, 3, 2130–2149. [Google Scholar] [CrossRef]
- Hirsa, M.T.G.; Johannes, H.B.; Krijn, P.J. Supported iron nanoparticles as catalysts for sustainable production of lower olefins. Science 2012, 335, 835–838. [Google Scholar]
- Francisco, R.R. The role of carbon materials in heterogeneous catalysis. Pergamon 1998, 36, 159–175. [Google Scholar]
- Xiong, H.; Jewell, L.L.; Coville, N.J. Shaped carbons as supports for the catalytic conversion of syngas to clean fuels. ACS Catal. 2015, 5, 2640–2658. [Google Scholar] [CrossRef]
- Cheng, Y.; Lin, J.; Xu, K.; Wang, H.; Yao, X.; Pei, Y.; Yan, S.; Qiao, M.; Zong, B. Fischer–Tropsch synthesis to lower olefins over potassium-promoted reduced graphene oxide supported iron catalysts. ACS Catal. 2015, 6, 389–399. [Google Scholar] [CrossRef]
- Santos, V.P.; Wezendonk, T.A.; Jaén, J.J.D.; Dugulan, A.I.; Nasalevich, M.A.; Islam, H.-U.; Chojecki, A.; Sartipi, S.; Sun, X.; Hakeem, A.A.; et al. Metal organic framework-mediated synthesis of highly active and stable Fischer-Tropsch catalysts. Nat. Commun. 2015, 6, 6451. [Google Scholar] [CrossRef]
- Wezendonk, T.A.; Santos, V.P.; Nasalevich, M.A.; Warringa, Q.S.E.; Dugulan, A.I.; Chojecki, A.; Koeken, A.C.J.; Ruitenbeek, M.; Meima, G.; Islam, H.-U.; et al. Elucidating the nature of Fe species during pyrolysis of the Fe-BTC MOF into highly active and stable Fischer–Tropsch catalysts. ACS Catal. 2016, 6, 3236–3247. [Google Scholar] [CrossRef]
- Ma, W.; Ding, Y.; Lin, L. Fischer-Tropsch synthesis over activated-carbon-supported cobalt catalysts: Effect of Co loading and promoters on catalyst performance. Ind. Eng. Chem. Res. 2004, 43, 2391–2398. [Google Scholar] [CrossRef]
- Wang, T.; Ding, Y.; Xiong, J.; Yan, L.; Zhu, H.; Lu, Y.; Lin, L. Effect of vanadium promotion on activated carbon-supported cobalt catalysts in Fischer–Tropsch synthesis. Catal. Lett. 2006, 107, 47–52. [Google Scholar] [CrossRef]
- Du, H.; Zhu, H.; Liu, T.; Zhao, Z.; Chen, X.; Dong, W.; Lu, W.; Luo, W.; Ding, Y. Higher alcohols synthesis via CO hydrogenation on Fe-promoted Co/AC catalysts. Catal. Today 2017, 281, 549–558. [Google Scholar] [CrossRef]
- Oschatz, M.; Krans, N.; Xie, J.; de Jong, K.P. Systematic variation of the sodium/sulfur promoter content on carbon-supported iron catalysts for the Fischer–Tropsch to olefins reaction. J. Energy Chem. 2016, 25, 985–993. [Google Scholar] [CrossRef]
- Chen, Y.; Ma, L.; Zhang, R.; Ye, R.; Liu, W.; Wei, J.; Ordomsky, V.V.; Liu, J. Carbon-supported Fe catalysts with well-defined active sites for highly selective alcohol production from Fischer-Tropsch synthesis. Appl. Catal. B-Environ. 2022, 312, 121393. [Google Scholar] [CrossRef]
- Rehman, A.; Heo, Y.J.; Nazir, G.; Park, S.J. Solvent-free, one-pot synthesis of nitrogen-tailored alkali-activated microporous carbons with an efficient CO2 adsorption. Carbon 2021, 172, 71–82. [Google Scholar] [CrossRef]
- Zeng, Y.; Zou, R.; Zhao, Y. Covalent organic frameworks for CO2 capture. Adv. Mater. 2016, 28, 2855–2873. [Google Scholar] [CrossRef]
- Li, X.; Zhao, Y.; Yang, Y.; Gao, S. A universal strategy for carbon-based ORR–active electrocatalyst: One porogen, two pore–creating mechanisms, three pore types. Nano Energy 2019, 62, 628–637. [Google Scholar] [CrossRef]
- Jozwiak, W.K.; Kaczmarek, E.; Maniecki, T.P.; Ignaczak, W.; Maniukiewicz, W. Reduction behavior of iron oxides in hydrogen and carbon monoxide atmospheres. Appl. Catal. A-Gen. 2007, 326, 17–27. [Google Scholar] [CrossRef]
- Zeng, Z.; Li, Z.; Guan, T.; Guo, S.; Hu, Z.; Wang, J.; Rykov, A.; Lv, J.; Huang, S.; Wang, Y.; et al. CoFe alloy carbide catalysts for higher alcohols synthesis from syngas: Evolution of active sites and Na promoting effect. J. Catal. 2022, 405, 430–444. [Google Scholar] [CrossRef]
- Orege, J.I.; Wei, J.; Han, Y.; Yang, M.; Sun, X.; Zhang, J.; Amoo, C.C.; Ge, Q.; Sun, J. Highly stable Sr and Na co-decorated Fe catalyst for high-valued olefin synthesis from CO2 hydrogenation. Appl. Catal. B-Environ. 2022, 316, 121640. [Google Scholar] [CrossRef]
- Gu, Z.; Li, K.; Qing, S.; Zhu, X.; Wei, Y.; Li, Y.; Wang, H. Enhanced reducibility and redox stability of Fe2O3 in the presence of CeO2 nanoparticles. RSC Adv. 2014, 4, 47191. [Google Scholar] [CrossRef]
- Huang, J.; Dai, W.L.; Fan, K. Remarkable support crystal phase effect in Au/FeOx catalyzed oxidation of 1,4-butanediol to γ-butyrolactone. J. Catal. 2009, 266, 228–235. [Google Scholar] [CrossRef]
- James, T.N.; Paul, G.T.; Vaishnavi, S.; Donald, R.B.; James, E.A.; Klaus, P.; Wang, G.; John, C.L.; Dean, W.M.; Penn, R.L.; et al. Characterization and properties of metallic iron nanoparticles: Spectroscopy, electrochemistry, and kinetics. Environ. Sci. Technol. 2005, 39, 1221–1230. [Google Scholar]
- Liu, B.; Geng, S.; Zheng, J.; Jia, X.; Jiang, F.; Liu, X. Unravelling the new roles of Na and Mn promoter in CO2 hydrogenation over Fe3O4-based catalysts for enhanced selectivity to light α-olefins. ChemCatChem 2018, 10, 4718–4732. [Google Scholar] [CrossRef]
- Peter, M.M.; Ruhksana, Q.; Helen, C.L.; Michael, L.T. Towards a chemical understanding of the Fischer–Tropsch reaction: Alkene formation. Appl. Catal. A-Gen. 1999, 186, 363–374. [Google Scholar]
- Yang, Q.; Vita, A.K.; Kondratenko, S.A.; Petrov, D.E.; Dmitry, E.D.; Erisa, S.; Henrik, L.; Aleks, A.; Ralph, K.; Andrey, S.S.; et al. Identifying performance descriptors in CO2 hydrogenation over iron-based catalysts promoted with alkali metals. Angew. Chem. Int. Ed. 2022, 61, e202116517. [Google Scholar] [CrossRef]
- Han, X.X.; Zhao, Q.; Gong, H.; Wei, C.; Lv, J.; Wang, Y.; Wang, M.; Huang, S.; Ma, X.B. Interface-induced phase evolution and spatial distribution of Fe-based catalysts for Fischer−Tropsch synthesis. ACS Catal. 2023, 13, 6525–6535. [Google Scholar] [CrossRef]
- Oschatz, M.; van Deelen, T.W.; Weber, J.L.; Lamme, W.S.; Wang, G.; Goderis, B.; Verkinderen, O.; Dugulan, A.I.; de Jong, K.P. Effects of calcination and activation conditions on ordered mesoporous carbon supported iron catalysts for production of lower olefins from synthesis gas. Catal. Sci. Technol. 2016, 6, 8464–8473. [Google Scholar] [CrossRef]
- Zhang, F.; Liu, T.; Hou, G.; Kou, T.; Yue, L.; Guan, R.; Li, Y. Hierarchically porous carbon foams for electric double layer capacitors. Nano Res. 2016, 9, 2875–2888. [Google Scholar] [CrossRef]
- Kim, B.-J.; Cho, K.-S.; Park, S.-J. Copper oxide-decorated porous carbons for carbon dioxide adsorption behaviors. J. Colloid Interface Sci. 2010, 342, 575–578. [Google Scholar] [CrossRef] [PubMed]
- Jang, D.-I.; Park, S.-J. Influence of nickel oxide on carbon dioxide adsorption behaviors of activated carbons. Fuel 2012, 102, 439–444. [Google Scholar] [CrossRef]
- Sun, N.; Tang, Z.; Wei, W.; Snape, C.E.; Sun, Y. Solid adsorbents for low-temperature CO2 capture with low-energy penalties leading to more effective integrated solutions for power generation and industrial processes. Front. Energy Res. 2015, 3, 9. [Google Scholar] [CrossRef]
- Goharibajestani, Z.; Yürüm, A.; Yürüm, Y. Effect of transition metal oxide nanoparticles on gas adsorption properties of graphene nanocomposites. Appl. Surf. Sci. 2019, 475, 1070–1076. [Google Scholar] [CrossRef]
- Zhang, D.; Luo, J.; Wang, J.; Xiao, X.; Liu, Y.; Qi, W.; Su, D.S.; Chu, W. Ru/FeOx catalyst performance design: Highly dispersed Ru species for selective carbon dioxide hydrogenation. Chin. J. Catal. 2018, 39, 157–166. [Google Scholar] [CrossRef]
- Wu, C.; Shen, J.; An, X.; Wu, Z.; Qian, S.; Zhang, S.; Wang, Z.; Song, B.; Cheng, Y.; Sham, T.K.; et al. Phosphorization-induced “Fence Effect” on the active hydrogen species migration enables tunable CO2 hydrogenation selectivity. ACS Catal. 2024, 14, 8592–8601. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, W.; He, R.; Li, M.; Zhang, J.; Cao, F.; Liu, J.; Lin, S.; Gao, X.; Yang, G.; et al. Carbon-based electron buffer layer on ZnOx-Fe5C2-Fe3O4 boosts ethanol synthesis from CO2 hydrogenation. Angew. Chem. Int. Ed. 2023, 135, e202311786. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | SBET (m2 g−1) | SBETmicro (m2 g−1) | SBETmeso (m2 g−1) | Vmicro (cm3 g−1) | Vmeso (cm3 g−1) | dsize (nm) |
|---|---|---|---|---|---|---|
| MC700 | 969.04 | 926.45 | 42.59 | 0.34 | 0.06 | 1.67 |
| MC800 | 1254.20 | 1178.86 | 75.34 | 0.44 | 0.09 | 1.70 |
| MC900 | 1732.19 | 1492.01 | 240.18 | 0.59 | 0.20 | 1.83 |
| MC1000 | 1645.13 | 734.38 | 910.74 | 0.25 | 0.68 | 2.54 |
| Catalyst | CO2 Conv. (%) | CO Sel. (%) | Hydrocarbons | MeOH | C2+OH | C2+OH STY mg gcat−1 h−1 | Yield | |||
|---|---|---|---|---|---|---|---|---|---|---|
| CH4 | C2–4 = | C5+ = | Paraffin | |||||||
| NaFe/MC700 | 6.1 | 87.0 | 90.9 | 1.1 | 0.0 | 8.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| NaFe/MC800 | 17.5 | 78.2 | 31.2 | 7.6 | 1.5 | 33.6 | 6.6 | 19.5 | 15.2 | 0.7 |
| NaFe/MC900 | 22.3 | 62.1 | 23.8 | 17.3 | 5.7 | 29.2 | 2.8 | 21.3 | 36.2 | 1.8 |
| NaFe/MC1000 | 22.8 | 70.8 | 22.5 | 28.2 | 3.9 | 20.0 | 2.7 | 22.6 | 30.3 | 1.5 |
| Fe/MC1000 | 18.4 | 87.05 | 36.8 | 30.3 | 1.3 | 31.2 | 0.1 | 4.5 | 0.8 | 0.8 |
| Na/MC1000 | 1.6 | - | - | - | - | - | - | - | - | - |
| Catalyst | SBET (m2 g−1) | SBETmicro (m2 g−1) | SBETmeso (m2 g−1) | Vmicro (cm3 g−1) | Vmeso (cm3 g−1) | dsize (nm) | Fe Content a (%) |
|---|---|---|---|---|---|---|---|
| NaFe/MC700 | 710.77 | 649.34 | 61.43 | 0.25 | 0.09 | 1.93 | 22.1 |
| NaFe/MC800 | 916.97 | 851.07 | 65.90 | 0.33 | 0.10 | 1.80 | 19.9 |
| NaFe/MC900 | 1224.74 | 1024.59 | 200.15 | 0.40 | 0.19 | 1.92 | 20.5 |
| NaFe/MC1000 | 862.76 | 505.27 | 357.49 | 0.19 | 0.36 | 2.55 | 21.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Jiang, L.; Lin, S.; Dong, P.; Fu, X.; Wang, Y.; Liu, Q.; Wu, M. Carbon-Supported Fe-Based Catalyst for Thermal-Catalytic CO2 Hydrogenation into C2+ Alcohols: The Effect of Carbon Support Porosity on Catalytic Performance. Molecules 2024, 29, 4628. https://doi.org/10.3390/molecules29194628
Chen Y, Jiang L, Lin S, Dong P, Fu X, Wang Y, Liu Q, Wu M. Carbon-Supported Fe-Based Catalyst for Thermal-Catalytic CO2 Hydrogenation into C2+ Alcohols: The Effect of Carbon Support Porosity on Catalytic Performance. Molecules. 2024; 29(19):4628. https://doi.org/10.3390/molecules29194628
Chicago/Turabian StyleChen, Yongjie, Lei Jiang, Simin Lin, Pei Dong, Xiaoli Fu, Yang Wang, Qiang Liu, and Mingbo Wu. 2024. "Carbon-Supported Fe-Based Catalyst for Thermal-Catalytic CO2 Hydrogenation into C2+ Alcohols: The Effect of Carbon Support Porosity on Catalytic Performance" Molecules 29, no. 19: 4628. https://doi.org/10.3390/molecules29194628
APA StyleChen, Y., Jiang, L., Lin, S., Dong, P., Fu, X., Wang, Y., Liu, Q., & Wu, M. (2024). Carbon-Supported Fe-Based Catalyst for Thermal-Catalytic CO2 Hydrogenation into C2+ Alcohols: The Effect of Carbon Support Porosity on Catalytic Performance. Molecules, 29(19), 4628. https://doi.org/10.3390/molecules29194628