
Calcium-Dependent Chemiluminescence Catalyzed by a Truncated c-MYC Promoter G-Triplex DNA



Abstract

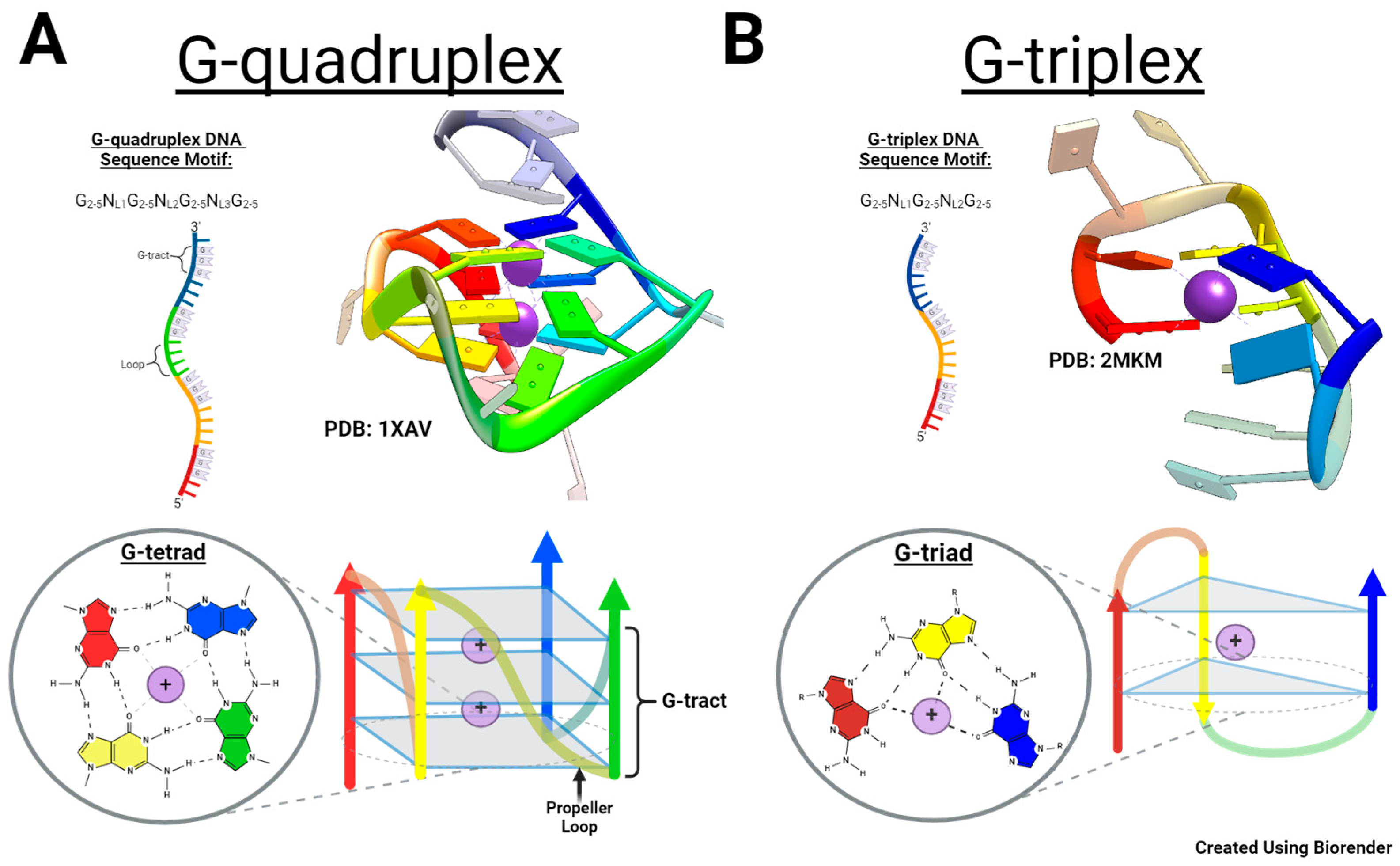
1. Introduction
2. Results and Discussion
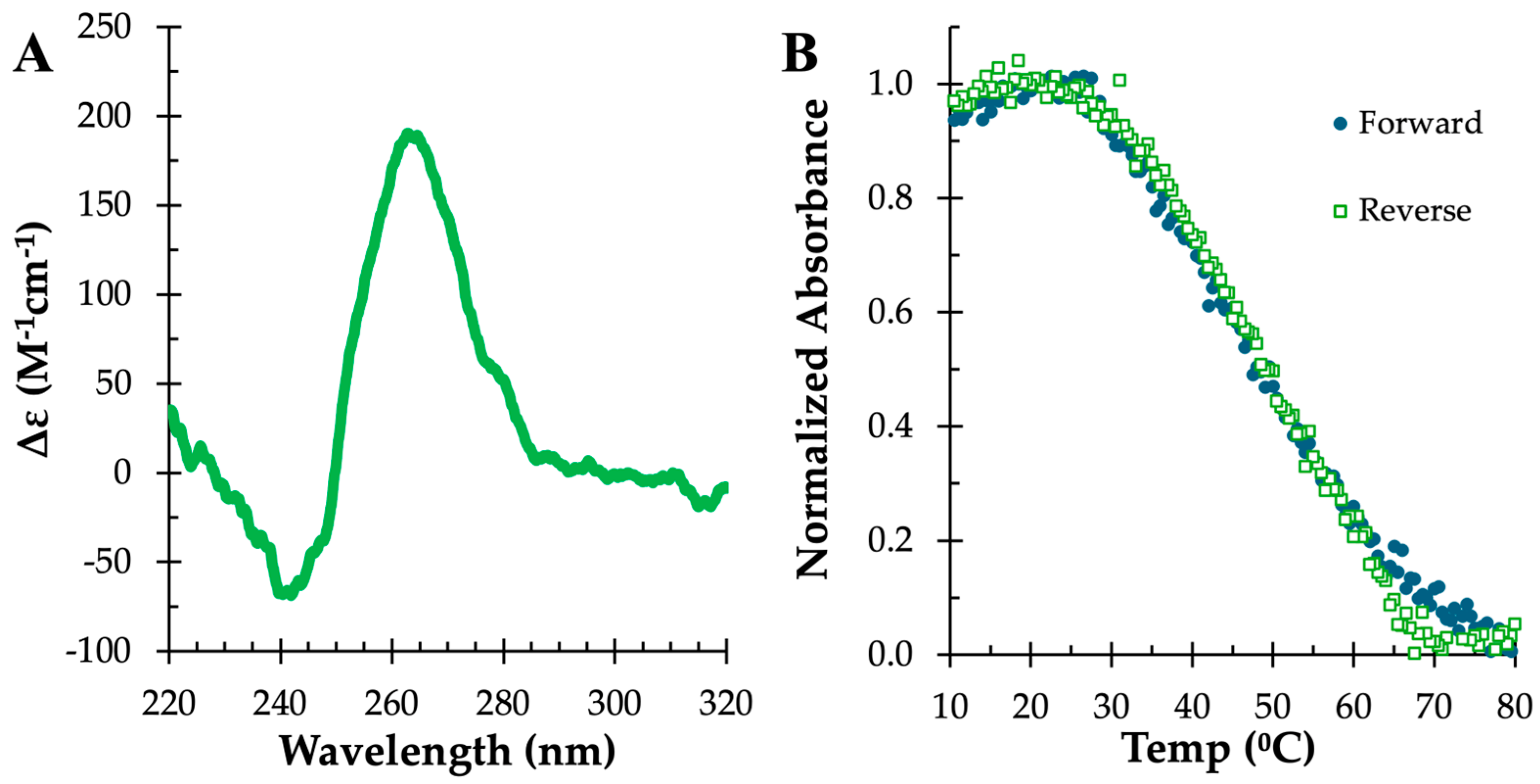
2.1. Characterization of c-MYC-G3 G-Triplex
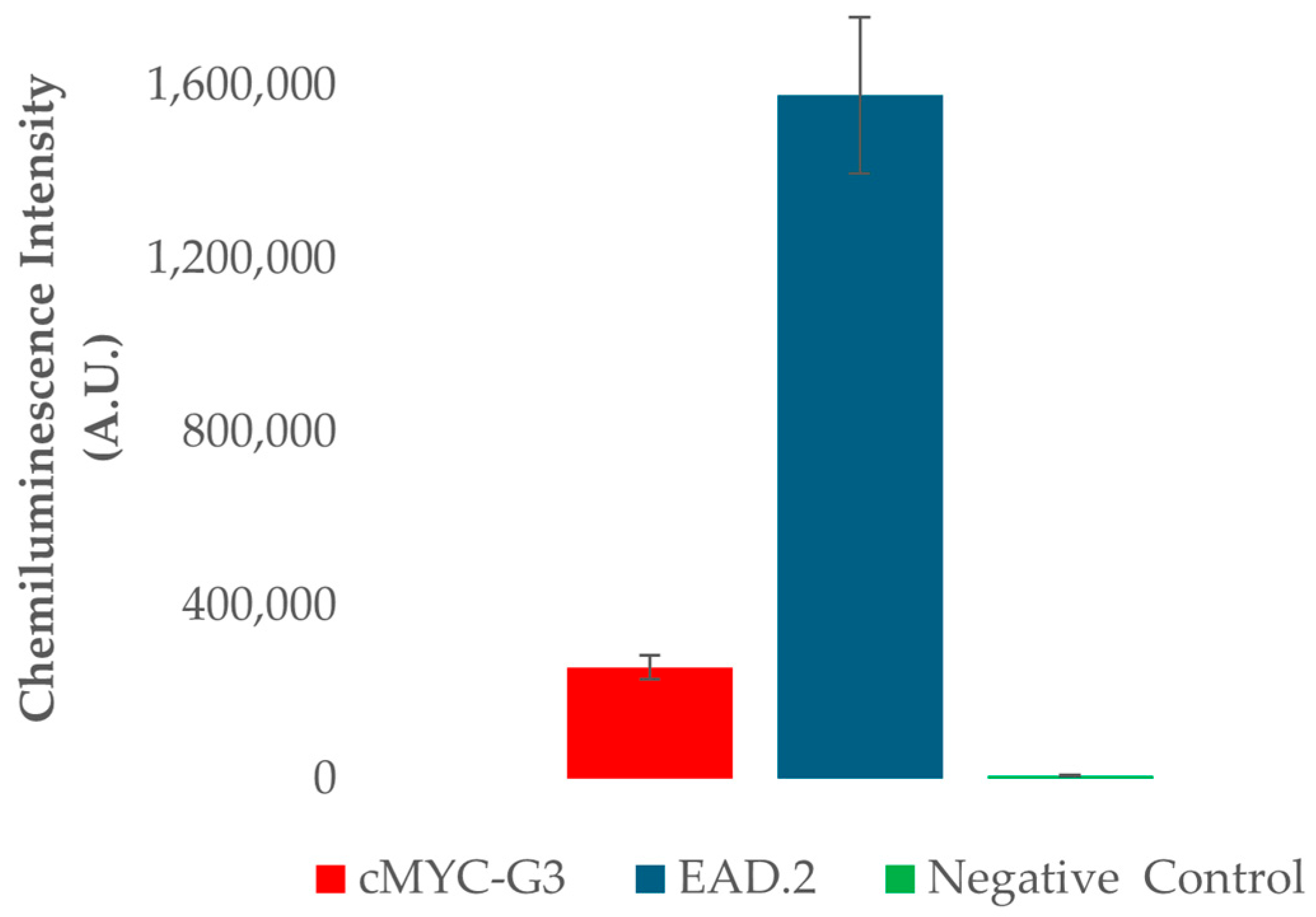
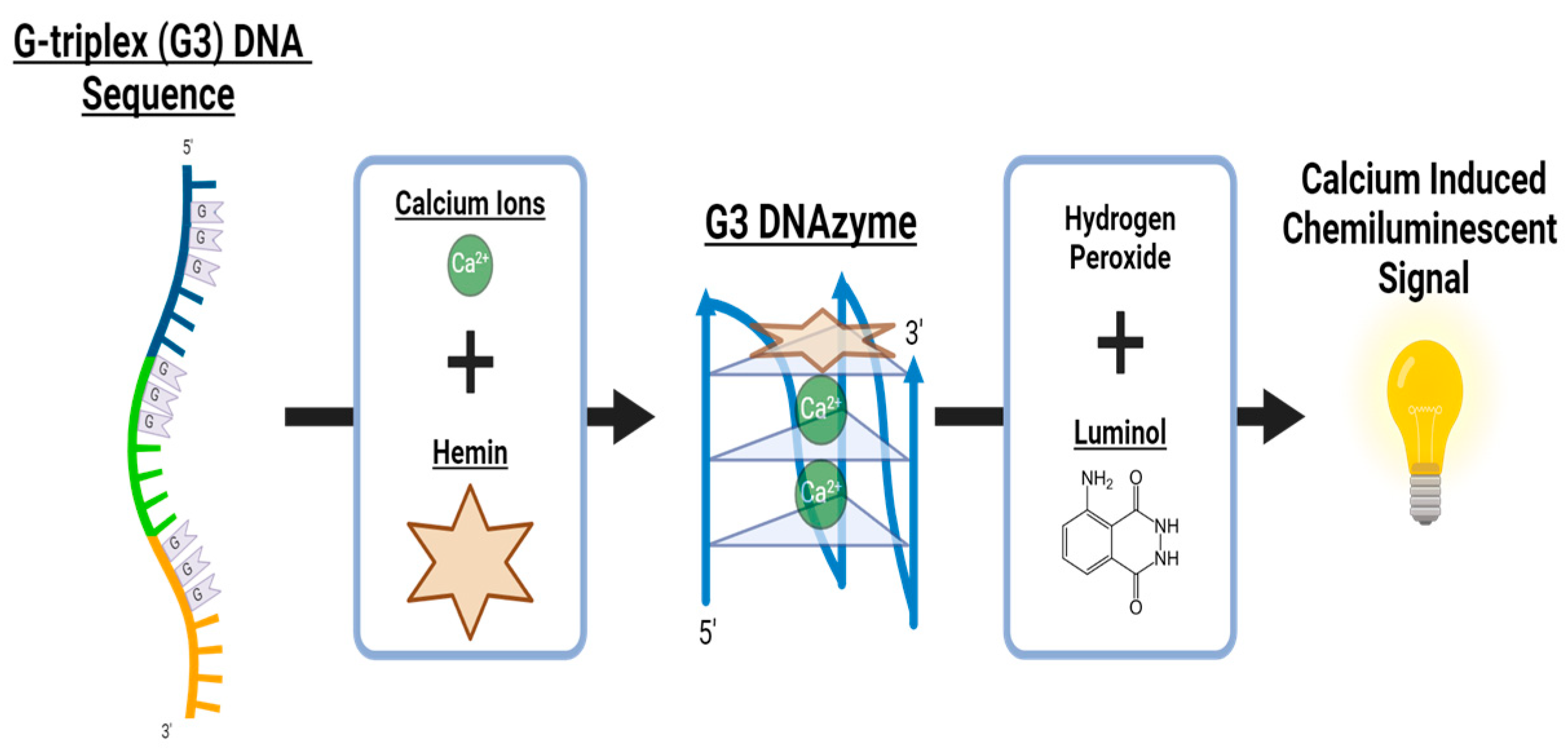
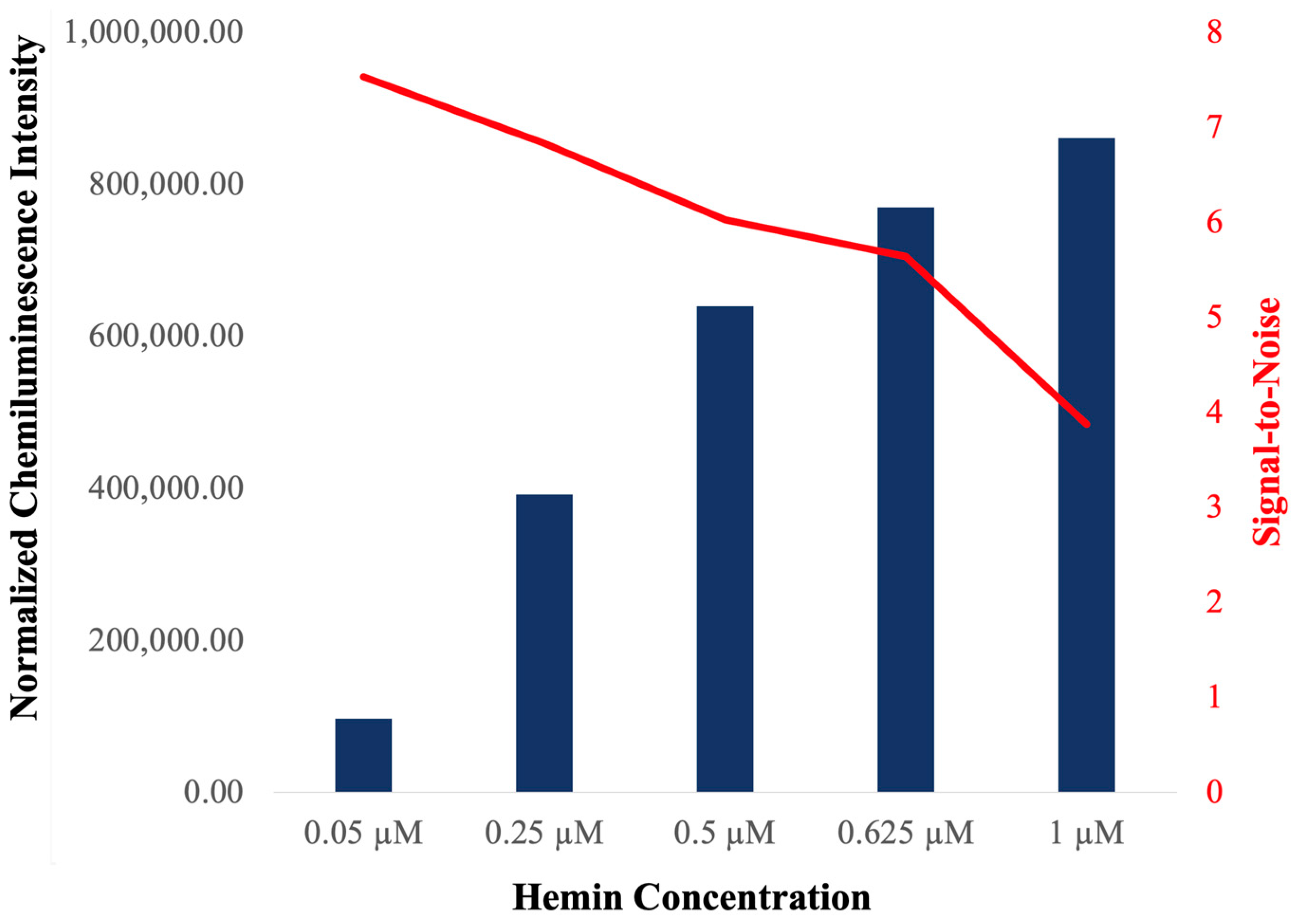
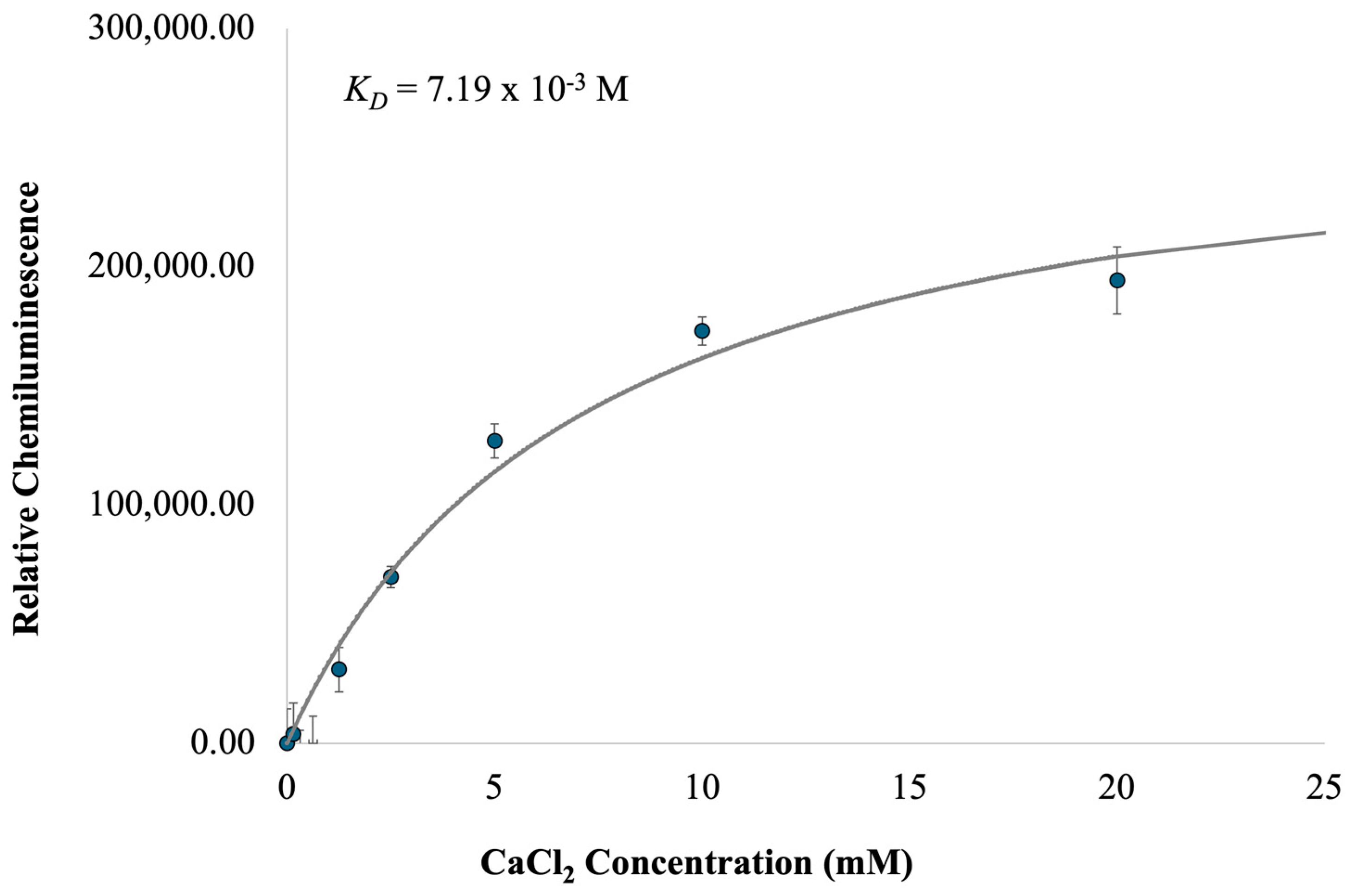
2.2. Characterization of c-MYC-G3 G-Triplex as a Peroxidase-Mimicking DNAzyme through Chemiluminescence
3. Materials and Methods
3.1. Experimental Reagents and Materials
3.2. Apparatus and Software
3.3. Circular Dichroism
3.4. UV Melting Temperature Analysis
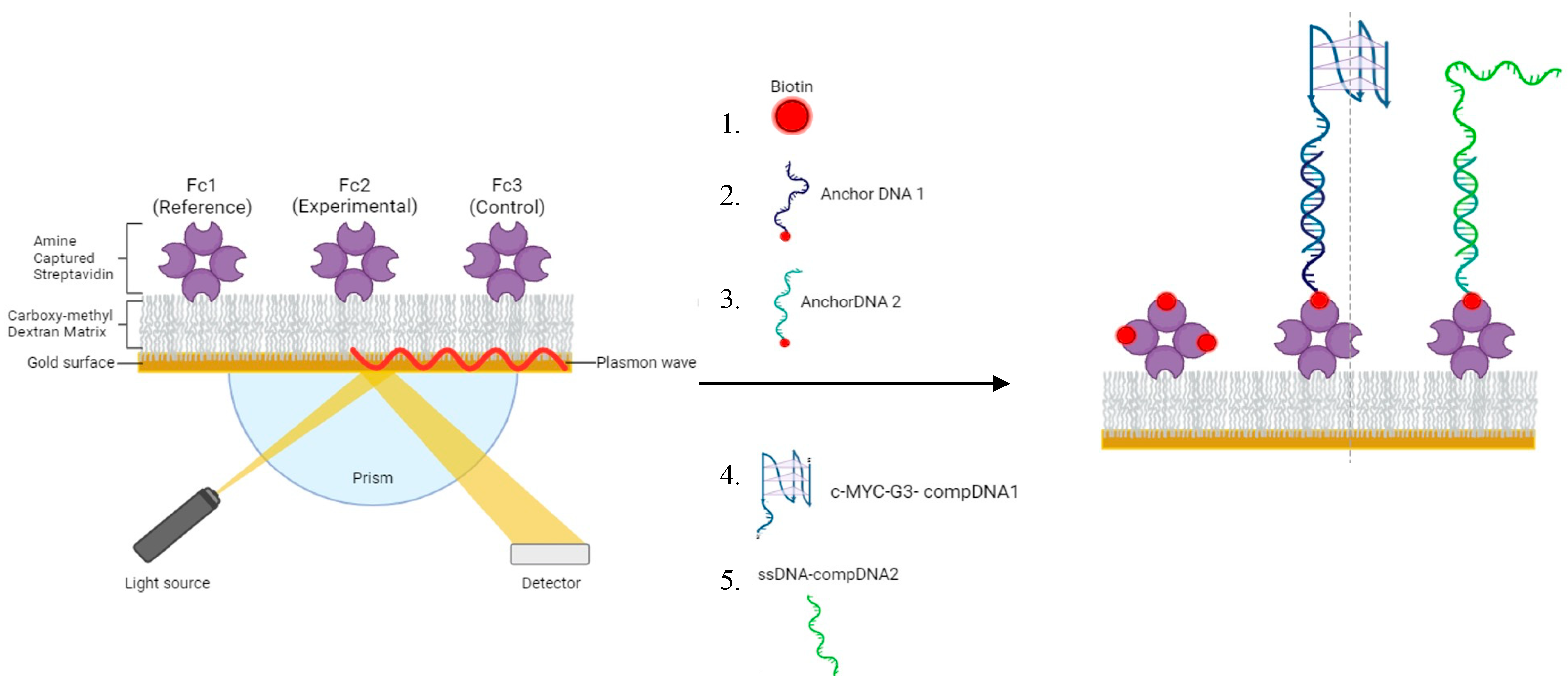
3.5. SPR Chip Setup
3.6. SPR Binding Experiments
3.7. Chemiluminescence Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, G.; Vasquez, K.M. Impact of Alternative DNA Structures on DNA Damage, DNA Repair, and Genetic Instability. DNA Repair 2014, 19, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Saini, N.; Zhang, Y.; Usdin, K.; Lobachev, K.S. When Secondary Comes First-The Importance of Non-Canonical DNA Structures. Biochimie 2013, 95, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Asamitsu, S.; Obata, S.; Yu, Z.; Bando, T.; Sugiyama, H. Recent Progress of Targeted G-Quadruplex-Preferred Ligands toward Cancer Therapy. Molecules 2019, 24, 429. [Google Scholar] [CrossRef]
- Bahls, B.; Aljnadi, I.M.; Emídio, R.; Mendes, E.; Paulo, A. G-Quadruplexes in c-Myc Promoter as Targets for Cancer Therapy. Biomedicines 2023, 11, 969. [Google Scholar] [CrossRef]
- Silverman, S.K. Catalytic Dna: Scope, Applications, and Biochemistry of Deoxyribozymes. Trends Biochem. Sci. 2016, 41, 595–609. [Google Scholar] [CrossRef]
- He, H.Z.; Wang, M.; Chan, D.S.H.; Leung, C.H.; Lin, X.; Lin, J.M.; Ma, D.L. A Parallel G-Quadruplex-Selective Luminescent Probe for the Detection of Nanomolar Calcium(II) Ion. Methods 2013, 64, 212–217. [Google Scholar] [CrossRef]
- Li, T.; Dong, S.; Wang, E. Label-Free Colorimetric Detection of Aqueous Mercury Ion (Hg2+) Using Hg2+-Modulated G-quadruplex-Based DNAzymes. Anal. Chem. 2009, 81, 2144–2149. [Google Scholar] [CrossRef] [PubMed]
- Sen, D.; Poon, L.C.H. RNA and DNA Complexes with Hemin [Fe(III) Heme] are Efficient Peroxidases and Peroxygenases: How Do They Do It and What Does It Mean? Crit. Rev. Biochem. Mol. Biol. 2011, 46, 478–492. [Google Scholar] [CrossRef]
- Kosman, J.; Juskowiak, B. Peroxidase-Mimicking DNAzymes for Biosensing Applications: A review. Anal. Chim. Acta 2011, 707, 7–17. [Google Scholar] [CrossRef]
- Robert, D. Gray, Robert Buscaglia, and J.B.C. Populated Intermediates in The Thermal Unfolding of the Human Telomeric Quadruplex. J. Am. Chem. Soc. 2008, 134, 16834–168447. [Google Scholar]
- Limongelli, V.; De Tito, S.; Cerofolini, L.; Fragai, M.; Pagano, B.; Trotta, R.; Cosconati, S.; Marinelli, L.; Novellino, E.; Bertini, I.; et al. The G-triplex DNA. Angew. Chem. Int. Ed. 2013, 52, 2269–2273. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Che, B.; Dai, H. A New G-Triplex-Based Strategy for Sensitivity Enhancement of the Detection of Endonuclease Activity and Inhibition. RSC Adv. 2021, 11, 28008–28013. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Liu, Q.; Jin, Y.; Li, B. Sensitive Colorimetric Determination of MicroRNA Let-7a through Rolling Circle Amplification and a Peroxidase-Mimicking System Composed of Trimeric G-Triplex and Hemin DNAzyme. Mikrochim. Acta 2020, 187, 139. [Google Scholar] [CrossRef]
- Wang, S.; Fu, B.; Wang, J.; Long, Y.; Zhang, X.; Peng, S.; Guo, P.; Tian, T.; Zhou, X. Novel Amplex Red Oxidases Based on Noncanonical DNA Structures: Property Studies And Applications In MicroRNA Detection. Anal. Chem. 2014, 86, 2925–2930. [Google Scholar] [CrossRef]
- Li, R.; Liu, Q.; Jin, Y.; Li, B. G-Triplex/Hemin DNAzyme: An Ideal Signal Generator for Isothermal Exponential Amplification Reaction-Based Biosensing Platform. Anal. Chim. Acta 2019, 1079, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.M.; Yang, B.; Shi, L.; Tang, Q.R.; Wang, J.; Liu, W.; Li, B.X.; Jin, Y. Label-Free and Portable Detection of HIV-DNA by a Handheld Luminometer. Anal. Chim. Acta 2024, 1304, 342553. [Google Scholar] [CrossRef] [PubMed]
- Pang, L.D.; Wang, L.E.; Liang, Y.Q.; Wang, Z.H.; Zhang, W.; Zhao, Q.Y.; Yang, X.Y.; Jiang, Y.J. G-Triplex/Hemin DNAzyme Mediated Colorimetric Aptasensor for Escherichia Coli O157:H7 Detection Based on Exonuclease III-Assisted Amplification and Aptamers-Functionalized Magnetic Beads. Talanta 2024, 269, 125457. [Google Scholar] [CrossRef]
- Wang, S.; Fu, B.; Peng, S.; Zhang, X.; Tian, T.; Zhou, X. The G-Triplex DNA Could Function as a New Variety of DNA Peroxidase. Chem. Comm. 2013, 49, 7920–7922. [Google Scholar] [CrossRef]
- Zhao, L.L.; Cao, T.; Zhou, Q.Y.; Zhang, X.H.; Zhou, Y.L.; Yang, L.; Zhang, X.X. The Exploration of a New Stable G-Triplex DNA and Its Novel Function in Electrochemical Biosensing. Anal. Chem. 2019, 91, 10731–10737. [Google Scholar] [CrossRef]
- Zhou, H.; Wu, Z.F.; Han, Q.J.; Zhong, H.M.; Peng, J.B.; Li, X.; Fan, X.L. Stable and Label-Free Fluorescent Probe Based on G-Triplex DNA and Thioflavin T. Anal. Chem. 2018, 90, 3220–3226. [Google Scholar] [CrossRef] [PubMed]
- Wen, C.J.; Wen, C.J.; Gong, J.Y.; Gong, J.Y.; Zheng, K.W.; He, Y.D.; Zhang, J.Y.; Hao, Y.H.; Tan, Z.; Tan, Z. Targeting Nucleic Acids with a G-Triplex-to-G-Quadruplex Transformation and Stabilization Using a Peptide-PNA G-Tract Conjugate. Chem. Commun. 2020, 56, 6567–6570. [Google Scholar] [CrossRef] [PubMed]
- Cerofolini, L.; Amato, J.; Giachetti, A.; Limongelli, V.; Novellino, E.; Parrinello, M.; Fragai, M.; Randazzo, A.; Luchinat, C. G-Triplex Structure and Formation Propensity. Nucleic Acids Res. 2014, 42, 13393–13404. [Google Scholar] [CrossRef] [PubMed]
- Grün, J.T.; Blümler, A.; Burkhart, I.; Wirmer-Bartoschek, J.; Heckel, A.; Schwalbe, H. Unraveling the Kinetics of Spare-Tire DNA G-Quadruplex Folding. J. Am. Chem. Soc. 2021, 143, 6185–6193. [Google Scholar] [CrossRef]
- Zhao, L.-L.; Gu, Y.-X.; Dong, J.-J.; Li, X.-T.; Pan, H.-Y.; Xue, C.-Y.; Liu, Y.; Zhou, Y.-L.; Zhang, X.-X. New G-Triplex DNA Dramatically Activates the Fluorescence of Thioflavin T and Acts as a Turn-On Fluorescent Sensor for Uracil-DNA Glycosylase Activity Detection. Anal. Chem. 2024, 96, 8458–8466. [Google Scholar] [CrossRef]
- Yang, D.; Hurley, L. Structure of the Biologically Relevant G-Quadruplex in the c-MYC Promoter. Nucleosides Nucleotides Nucleic Acids 2006, 25, 951–968. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, D.; Arachchilage, G.M.; Basu, S. Metal Cations in G-Quadruplex Folding and Stability. Front. Chem. 2016, 4, 38. [Google Scholar] [CrossRef]
- Lee, M.P.H.; Parkinson, G.N.; Hazel, P.; Neidle, S. Observation of Coexistence of Sodium and Calcium Ions in a DNA G-Quadruplex Ion Channel. J. Am. Chem. Soc. 2007, 129, 10106–10107. [Google Scholar] [CrossRef]
- Jiang, H.X.; Cui, Y.; Zhao, T.; Fu, H.W.; Koirala, D.; Punnoose, J.A.; Kong, D.M.; Mao, H. Divalent Cations and Molecular Crowding Buffers Stabilize G-Triplex at Physiologically Relevant Temperatures. Sci. Rep. 2015, 5, 9255. [Google Scholar] [CrossRef]
- Nanjunda, R.; Munde, M.; Liu, Y.; Wilson, W.D. Real-Time Monitoring of Nucleic Acid Interactions with Biosensor-Surface Plasmon Resonance. In Methods for Studying Nucleic Acid/Drug Interactions, 1st ed.; Wanini, M., Tor, Y., Eds.; CRC Press: Boca Raton, FL, USA, 2016; pp. 91–122. [Google Scholar]
- Halder, K.; Chowdhury, S. Kinetic Resolution of Bimolecular Hybridization versus Intramolecular Folding in Nucleic Acids by Surface Plasmon Resonance: Application to G-Quadruplex/Duplex Competition in Human c-Myc Promoter. Nucleic Acids Res. 2005, 33, 4466–4474. [Google Scholar] [CrossRef]
- White, E.W.; Tanious, F.; Ismail, M.A.; Reszka, A.P.; Neidle, S.; Boykin, D.W.; Wilson, W.D. Structure-Specific Recognition of Quadruplex DNA by Organic Cations: Influence of Shape, Substituents and Charge. Biophys. Chem. 2007, 126, 140–153. [Google Scholar] [CrossRef]
- Plyler, J.; Jasheway, K.; Karr, J.; Brennan, J.S.; Kerwin, S.M.; David, W.M. Real-time Investigation of SV40 Large T-antigen Helicase Activity Using Surface Plasmon Resonance. Cell Biochem. Biophys. 2009, 53, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Bonnat, L.; Dautriche, M.; Saidi, T.; Revol-Cavalier, J.; Dejeu, J.; Defrancq, E.; Lavergne, T. Scaffold Stabilization of a G-Triplex and Study of Its Interactions with G-Quadruplex Targeting Ligands. Org. Biomol. Chem. 2019, 17, 8726–8736. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.X.; Zhao, Y.; Hao, Y.H.; Tan, Z. Tetraplex Formation of Surface-Immobilized Human Telomere Sequence Probed by Surface Plasmon Resonance Using Single-Stranded DNA Binding Protein. J. Mol. Recog. 2005, 18, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Prado, E.; Bonnat, L.; Bonnet, H.; Lavergne, T.; Van Der Heyden, A.; Pratviel, G.; Dejeu, J.; Defrancq, E. Influence of the SPR Experimental Conditions on the G-Quadruplex DNA Recognition by Porphyrin Derivatives. Langmuir 2018, 34, 13057–13064. [Google Scholar] [CrossRef] [PubMed]
- Špringer, T.; Šípová, H.; Vaisocherová, H.; Štepánek, J.; Homola, J. Shielding Effect of Monovalent and Divalent Cations on Solid-Phase DNA Hybridization: Surface Plasmon Resonance Biosensor Study. Nucleic Acids Res. 2010, 38, 7343–7351. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Yin, Z.; Xiao, R.; Huang, B.; Cui, Y.; Wang, H.; Xiang, Y.; Wang, L.; Lei, L.; Ye, J.; et al. G-Quadruplexes Sense Natural Porphyrin Metabolites for Regulation of Gene Transcription and Chromatin Landscapes. Genome Biol. 2022, 23, 259. [Google Scholar] [CrossRef] [PubMed]
- Gribas, A.V.; Zhao, S.; Sakharov, I.Y. Improved Method for Chemiluminescent Determination of Peroxidase-Mimicking DNAzyme Activity. Anal. Biochem. 2014, 466, 19–23. [Google Scholar] [CrossRef]
- Mergny, J.L.; Lacroix, L. UV Melting of G-Quadruplexes. Curr. Protoc. Nucleic Acid Chem. 2009, 17, 1–15. [Google Scholar] [CrossRef]
- Paul, A.; Musetti, C.; Nanjunda, R.; Wilson, W.D. Biosensor-Surface Plasmon Resonance: Label-Free Method for Investigation of Small Molecule-Quadruplex Nucleic Acid Interactions. Methods Mol. Biol. 2019, 2035, 63–85. [Google Scholar]
- Karlsson, R.; Katsamba, P.S.; Nordin, H.; Pol, E.; Myszka, D.G. Analyzing a Kinetic Titration Series Using Affinity Biosensors. Anal. Biochem. 2006, 349, 136–147. [Google Scholar] [CrossRef]
- Palau, W.; Di Primo, C. Single-Cycle Kinetic Analysis of Ternary DNA Complexes by Surface Plasmon Resonance on a Decaying Surface. Biochimie 2012, 94, 1891–1899. [Google Scholar] [CrossRef] [PubMed]
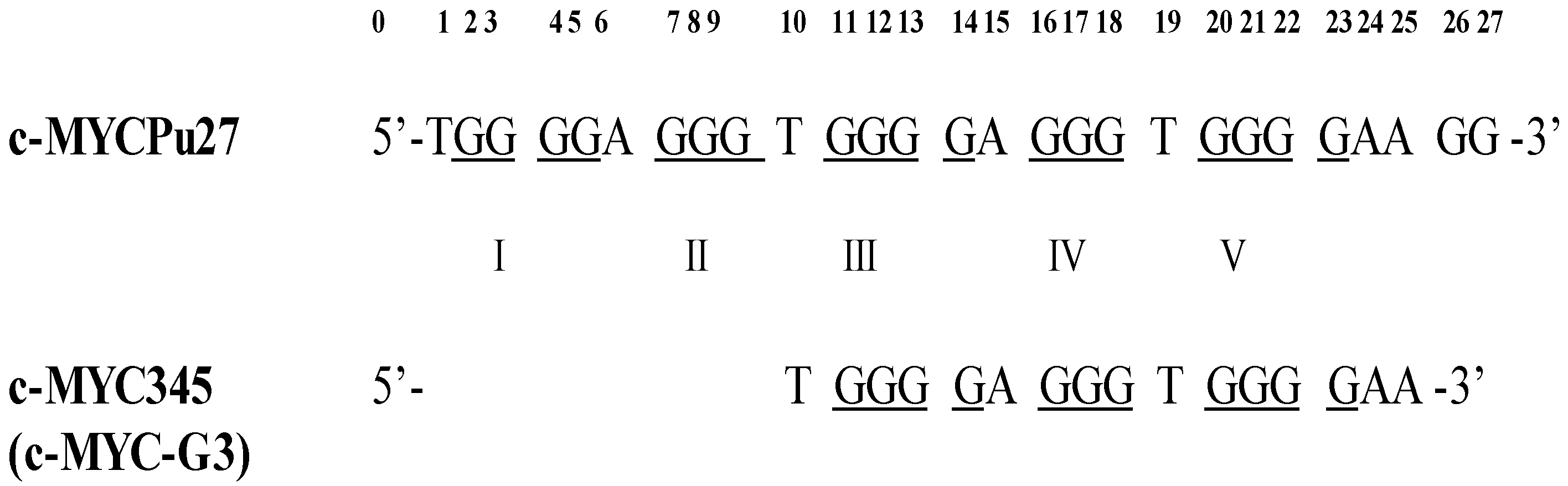



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| c-MYC-G3 Concentration | Tm a |
|---|---|
| 5 µM | 38.4 ± 1.1 °C |
| 20 µM | 37.7 ± 0.4 °C |
| 30 µM | 37.4 ± 0.5 °C |
| 50 µM | 37.2 ± 1.0 °C |
| Conditions | 0 mM CaCl2 | 3.75 mM CaCl2 | 7.5 mM CaCl2 |
|---|---|---|---|
| Tm (°C) | 38.4 ± 1.1 | 43.4 ± 1.2 | 51.0 ± 0.7 |
| DNA Name | Sequence (5′ to 3′) |
|---|---|
| EAD.2 | CTG GGA GGG AGG GAG GGA |
| c-MYC-G3 | TGG GGA GGG TGG GGA A |
| anchorDNA1 | BioTEG TTTTTTTT GAG CAG CAA TAC ACG A |
| anchorDNA2 | BioTEG TTTTTTTT ACA GGA TCT GCA TCT C |
| c-MYC-G3- compDNA1 | TGG GGA GGG TGG GGA A TTTTTTTT TCG TGT ATT GCT C |
| ssDNA-compDNA2 | TGT GTA GTG CGG GTT TTTTTTTT GAG ATG CAG ATC CTG T |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Das, M.K.; Williams, E.P.; Myhre, M.W.; David, W.M.; Kerwin, S.M. Calcium-Dependent Chemiluminescence Catalyzed by a Truncated c-MYC Promoter G-Triplex DNA. Molecules 2024, 29, 4457. https://doi.org/10.3390/molecules29184457
Das MK, Williams EP, Myhre MW, David WM, Kerwin SM. Calcium-Dependent Chemiluminescence Catalyzed by a Truncated c-MYC Promoter G-Triplex DNA. Molecules. 2024; 29(18):4457. https://doi.org/10.3390/molecules29184457
Chicago/Turabian StyleDas, Malay Kumar, Elizabeth P. Williams, Mitchell W. Myhre, Wendi M. David, and Sean M. Kerwin. 2024. "Calcium-Dependent Chemiluminescence Catalyzed by a Truncated c-MYC Promoter G-Triplex DNA" Molecules 29, no. 18: 4457. https://doi.org/10.3390/molecules29184457
APA StyleDas, M. K., Williams, E. P., Myhre, M. W., David, W. M., & Kerwin, S. M. (2024). Calcium-Dependent Chemiluminescence Catalyzed by a Truncated c-MYC Promoter G-Triplex DNA. Molecules, 29(18), 4457. https://doi.org/10.3390/molecules29184457