
Engineering Novel Amphiphilic Platinum(IV) Complexes to Co-Deliver Cisplatin and Doxorubicin

 , , ,
, , ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
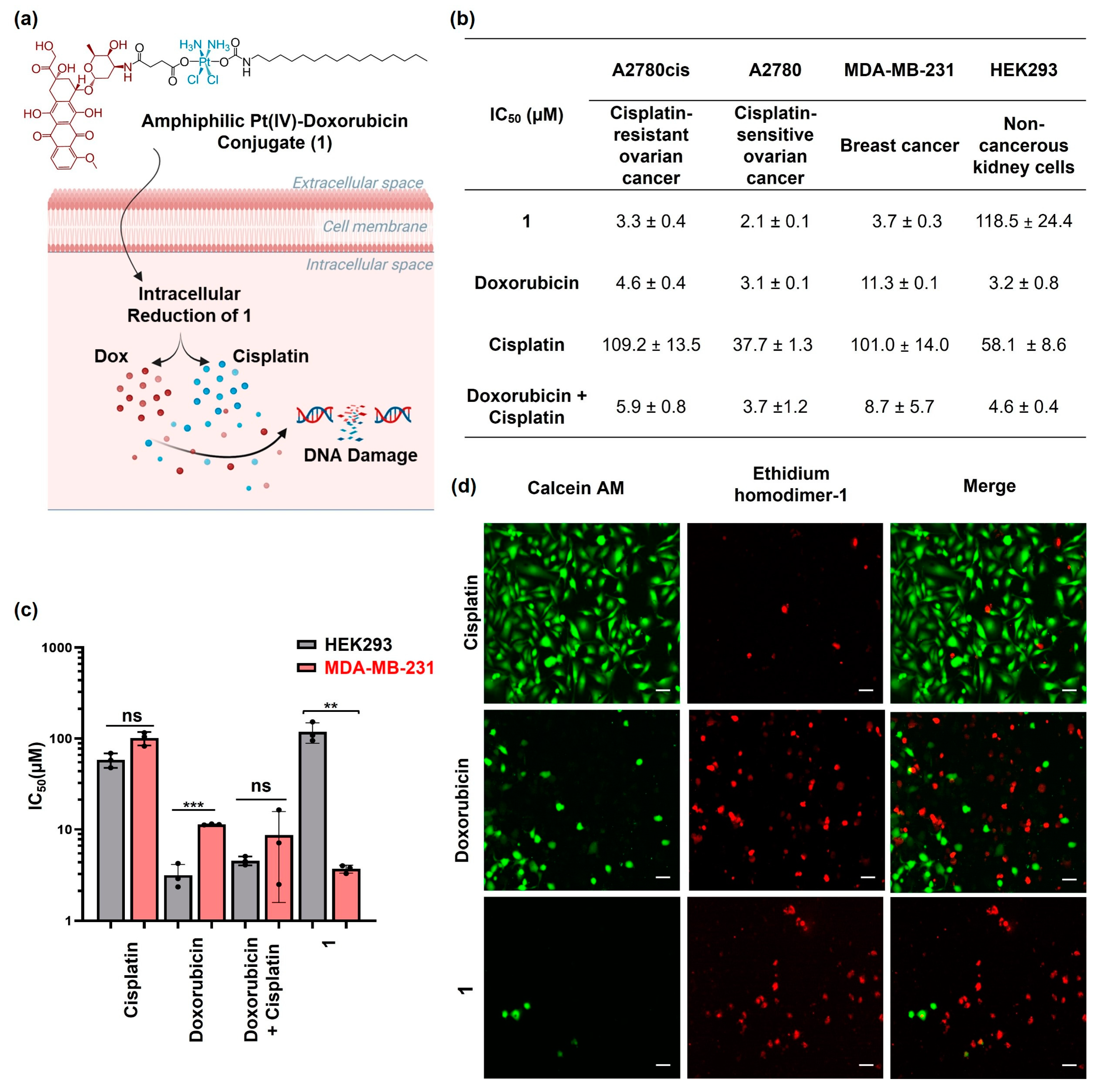
2.1. Synthesis and Characterization of the Amphiphilic Pt(IV)–Doxorubicin Conjugate (1)
2.2. Cytotoxicity Profiles of Amphiphilic Pt(IV)–Doxorubicin Conjugate (1)
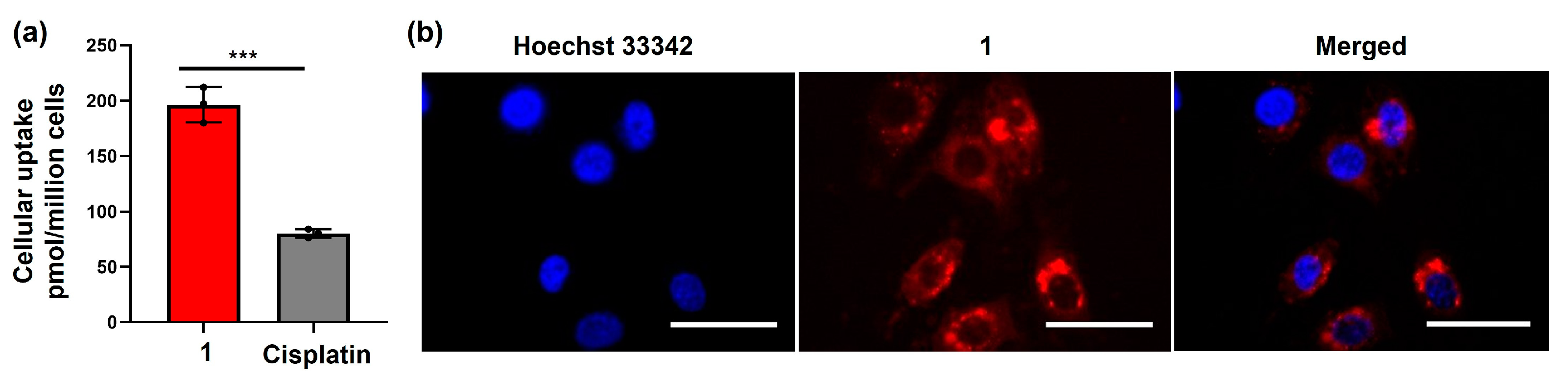
2.3. Cellular Accumulation of the Amphiphilic Pt(IV)–Doxorubicin Conjugate (1)
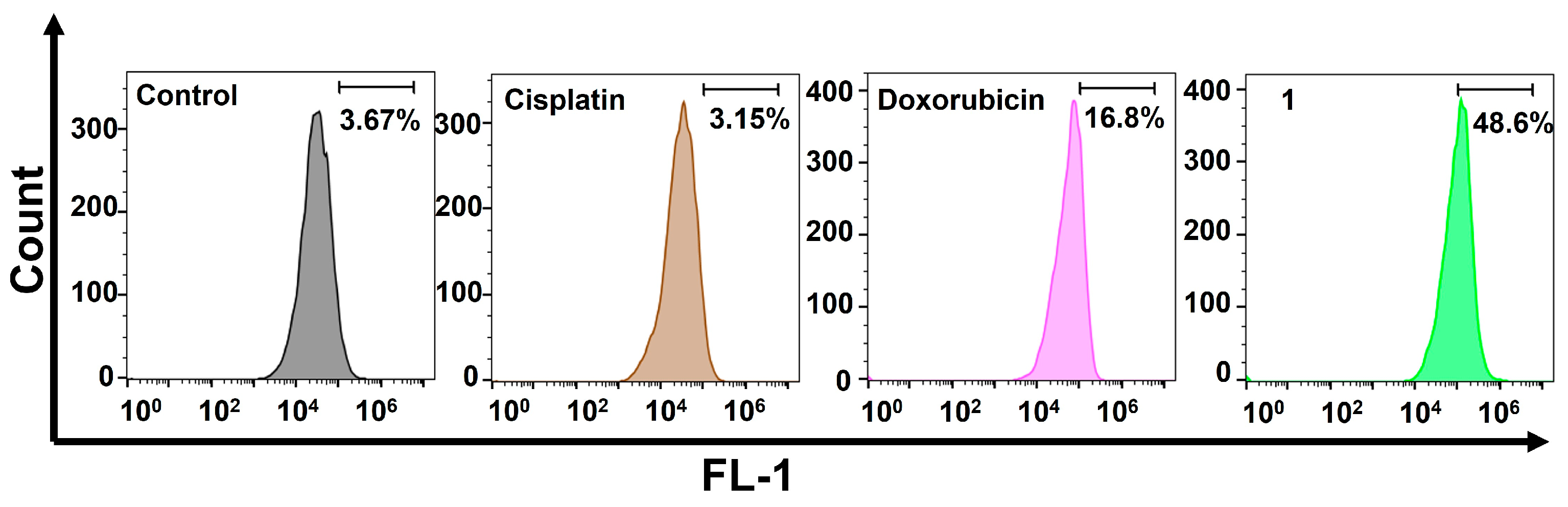
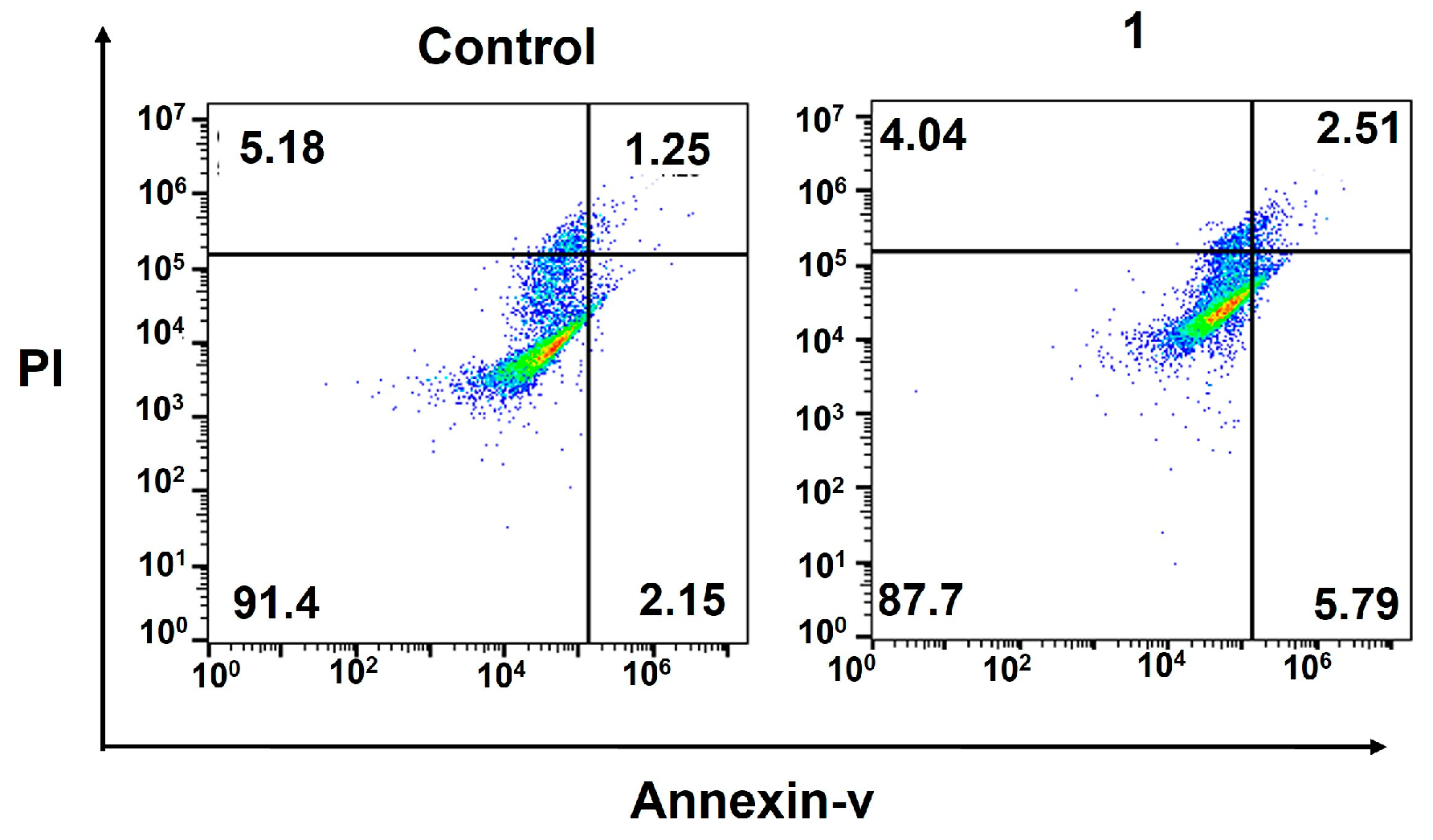
2.4. Cellular Responses of the Amphiphilic Pt(IV)–Doxorubicin Conjugate (1)
3. Conclusions
4. Materials and Methods
4.1. Cell Culture
4.2. MTT Assays
4.3. Live/Dead Cell Viability Assays
GFAAS Analysis of Cellular Pt Contents in MDA-MB-231 Cells
4.4. Fluorescence Imaging
4.5. Flow Cytometric Analysis of γH2AX
4.6. Flow Cytometric Analysis of Apoptosis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, D.; Lippard, S.J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 2005, 4, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, E.R.; Lippard, S.J. Structure, recognition, and processing of cisplatin-DNA adducts. Chem. Rev. 1999, 99, 2467–2498. [Google Scholar] [CrossRef] [PubMed]
- Todd, R.C.; Lippard, S.J. Inhibition of transcription by platinum antitumor compounds. Metallomics 2009, 1, 280–291. [Google Scholar] [CrossRef]
- Johnstone, T.C.; Suntharalingam, K.; Lippard, S.J. The Next Generation of Platinum Drugs: Targeted Pt(II) Agents, Nanoparticle Delivery, and Pt(IV) Prodrugs. Chem. Rev. 2016, 116, 3436–3486. [Google Scholar] [CrossRef]
- Bayat Mokhtari, R.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef]
- Kar, A.; Agarwal, S.; Singh, A.; Bajaj, A.; Dasgupta, U. Insights into molecular mechanisms of chemotherapy resistance in cancer. Transl. Oncol. 2024, 42, 101901. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lin, W. Platinum-based combination nanomedicines for cancer therapy. Curr. Opin. Chem. Biol. 2023, 74, 102290. [Google Scholar] [CrossRef]
- Zhong, T.; Yu, J.; Pan, Y.; Zhang, N.; Qi, Y.; Huang, Y. Recent Advances of Platinum-Based Anticancer Complexes in Combinational Multimodal Therapy. Adv. Healthc. Mater. 2023, 12, e2300253. [Google Scholar] [CrossRef]
- Jogadi, W.; Zheng, Y.R. Supramolecular platinum complexes for cancer therapy. Curr. Opin. Chem. Biol. 2023, 73, 102276. [Google Scholar] [CrossRef]
- Zhang, P.; Zhou, Z.; Long, W.; Yan, Y.; Li, Y.; Fu, T.; Liu, Y.; Zhao, Z.; Tan, W.; Stang, P.J. Self-assembled Pt(II) metallacycles enable precise cancer combination chemotherapy. Proc. Natl. Acad. Sci. USA 2022, 119, e2202255119. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, Y.; Meng, Z.; Guo, L.; Yuan, X.; Chai, Y.; Sessler, J.L.; Meng, Q.; Li, C. Supramolecular combination chemotherapy: A pH-responsive co-encapsulation drug delivery system. Chem. Sci. 2020, 11, 6275–6282. [Google Scholar] [CrossRef] [PubMed]
- Samanta, S.; Quigley, J.; Vinciguerra, B.; Briken, V.; Isaacs, L. Cucurbit[7]uril Enables Multi-Stimuli-Responsive Release from the Self-Assembled Hydrophobic Phase of a Metal Organic Polyhedron. J. Am. Chem. Soc. 2017, 139, 9066–9074. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Zhu, Y.; Wang, Y.; Wei, D.; Wu, Y.; Zheng, L.; Bai, H.; Xiao, H.; Zhang, Z. pH/redox sensitive nanoparticles with platinum(iv) prodrugs and doxorubicin enhance chemotherapy in ovarian cancer. RSC Adv. 2019, 9, 20513–20517. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.; Askevold, B.; Mikula, H.; Kohler, R.; Pirovich, D.; Weissleder, R. Nano-palladium is a cellular catalyst for in vivo chemistry. Nat. Commun. 2017, 8, 15906. [Google Scholar] [CrossRef]
- Samanta, S.K.; Moncelet, D.; Briken, V.; Isaacs, L. Metal–Organic Polyhedron Capped with Cucurbit[8]uril Delivers Doxorubicin to Cancer Cells. J. Am. Chem. Soc. 2016, 138, 14488–14496. [Google Scholar] [CrossRef]
- Hess, U.; Shahabi, S.; Treccani, L.; Streckbein, P.; Heiss, C.; Rezwan, K. Co-delivery of cisplatin and doxorubicin from calcium phosphate beads/matrix scaffolds for osteosarcoma therapy. Mater. Sci. Eng. C Mater. Biol. Appl. 2017, 77, 427–435. [Google Scholar] [CrossRef]
- Sreekanth, V.; Medatwal, N.; Pal, S.; Kumar, S.; Sengupta, S.; Bajaj, A. Molecular Self-Assembly of Bile Acid-Phospholipids Controls the Delivery of Doxorubicin and Mice Survivability. Mol. Pharm. 2017, 14, 2649–2659. [Google Scholar] [CrossRef]
- Jin, Y.; Wang, Y.; Liu, X.; Zhou, J.; Wang, X.; Feng, H.; Liu, H. Synergistic Combination Chemotherapy of Lung Cancer: Cisplatin and Doxorubicin Conjugated Prodrug Loaded, Glutathione and pH Sensitive Nanocarriers. Drug Des. Dev. Ther. 2020, 14, 5205–5215. [Google Scholar] [CrossRef]
- Xue, X.; Wu, Y.; Xu, X.; Xu, B.; Chen, Z.; Li, T. pH and Reduction Dual-Responsive Bi-Drugs Conjugated Dextran Assemblies for Combination Chemotherapy and In Vitro Evaluation. Polymers 2021, 13, 1515. [Google Scholar] [CrossRef]
- Zheng, Y.-R.; Suntharalingam, K.; Johnstone, T.C.; Yoo, H.; Lin, W.; Brooks, J.G.; Lippard, S.J. Pt(IV) Prodrugs Designed to Bind Non-Covalently to Human Serum Albumin for Drug Delivery. J. Am. Chem. Soc. 2014, 136, 8790–8798. [Google Scholar] [CrossRef] [PubMed]
- Jayawardhana, A.M.D.S.; Stilgenbauer, M.; Datta, P.; Qiu, Z.; Mckenzie, S.; Wang, H.; Bowers, D.; Kurokawa, M.; Zheng, Y.R. Fatty acid-like Pt(IV) prodrugs overcome cisplatin resistance in ovarian cancer by harnessing CD36. Chem. Commun. 2020, 56, 10706–10709. [Google Scholar] [CrossRef]
- Jayawardhana, A.M.D.S.; Zheng, Y.R. Interactions between mitochondria-damaging platinum(IV) prodrugs and cytochrome c. Dalton Trans. 2022, 51, 2012–2018. [Google Scholar] [CrossRef] [PubMed]
- Awuah, S.G.; Zheng, Y.R.; Bruno, P.M.; Hemann, M.T.; Lippard, S.J. A Pt(IV) Pro-drug Preferentially Targets Indoleamine-2,3-dioxygenase, Providing Enhanced Ovarian Cancer Immuno-Chemotherapy. J. Am. Chem. Soc. 2015, 137, 14854–14857. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.; Yu, Y.; Zhang, X.; Wang, Y.; Chen, H.; Zhao, Y.; Wang, F.; Rong, G.; Wang, W.; Kang, X.; et al. Breaking the Intracellular Redox Balance with Diselenium Nanoparticles for Maximizing Chemotherapy Efficacy on Patient-Derived Xenograft Models. ACS Nano 2020, 14, 16984–16996. [Google Scholar] [CrossRef] [PubMed]
- Jayawardhana, A.M.D.S.; Bhandari, S.; Kaspi-Kaneti, A.W.; Kshetri, M.; Qiu, Z.; Cheline, M.; Shen, H.; Dunietz, B.D.; Zheng, Y.R. Visible light-activatable platinum(IV) prodrugs harnessing CD36 for ovarian cancer therapy. Dalton Trans. 2023, 52, 10942–10950. [Google Scholar] [CrossRef]
- Feng, W.W.; Wilkins, O.; Bang, S.; Ung, M.; Li, J.; An, J.; Del Genio, C.; Canfield, K.; DiRenzo, J.; Wells, W.; et al. CD36-Mediated Metabolic Rewiring of Breast Cancer Cells Promotes Resistance to HER2-Targeted Therapies. Cell Rep. 2019, 29, 3405–3420.e5. [Google Scholar] [CrossRef]
- Miller, M.; Zheng, Y.; Suresh, G.; Pfirschke, C.; Zope, H.; Engblom, C.; Kohler, R.; Iwamoto, Y.; Yang, K.; Askevold, B.; et al. Tumour-associated macrophages act as a slow-release reservoir of nano-therapeutic Pt(IV) pro-drug. Nat. Commun. 2015, 6, 8692. [Google Scholar] [CrossRef]
- Zhou, F.; Feng, B.; Yu, H.; Wang, D.; Wang, T.; Ma, Y.; Wang, S.; Li, Y. Tumor Microenvironment-Activatable Prodrug Vesicles for Nanoenabled Cancer Chemoimmunotherapy Combining Immunogenic Cell Death Induction and CD47 Blockade. Adv. Mater. 2019, 31, e1805888. [Google Scholar] [CrossRef]
- Kang, X.; Wang, Y.; Chen, Z.; Wu, Y.; Chen, H.; Yang, X.; Yu, C. Imidazole modified Pt(iv) prodrug-loaded multi-stage pH responsive nanoparticles to overcome cisplatin resistance. Chem. Commun. 2020, 56, 11271–11274. [Google Scholar] [CrossRef]
- Ma, J.; Wang, Q.; Huang, Z.; Yang, X.; Nie, Q.; Hao, W.; Wang, P.G.; Wang, X. Glycosylated Platinum(IV) Complexes as Substrates for Glucose Transporters (GLUTs) and Organic Cation Transporters (OCTs) Exhibited Cancer Targeting and Human Serum Albumin Binding Properties for Drug Delivery. J. Med. Chem. 2017, 60, 5736–5748. [Google Scholar] [CrossRef] [PubMed]
- Abu Ammar, A.; Raveendran, R.; Gibson, D.; Nassar, T.; Benita, S. A Lipophilic Pt(IV) Oxaliplatin Derivative Enhances Antitumor Activity. J. Med. Chem. 2016, 59, 9035–9046. [Google Scholar] [CrossRef]
- Kshetri, M.; Jogadi, W.; Alqarni, S.; Datta, P.; Cheline, M.; Sharma, A.; Betters, T.; Broyles, D.; Zheng, Y.R. Exploring the Impact of Head Group Modifications on the Anticancer Activities of Fatty-Acid-like Platinum(IV) Prodrugs: A Structure-Activity Relationship Study. Int. J. Mol. Sci. 2023, 24, 13301. [Google Scholar] [CrossRef] [PubMed]
- Sreekanth, V.; Bajaj, A. Recent Advances in Engineering of Lipid Drug Conjugates for Cancer Therapy. ACS Biomater. Sci. Eng. 2019, 5, 4148–4166. [Google Scholar] [CrossRef]
- Morstein, J.; Capecchi, A.; Hinnah, K.; Park, B.; Petit-Jacques, J.; Van Lehn, R.C.; Reymond, J.L.; Trauner, D. Medium-Chain Lipid Conjugation Facilitates Cell-Permeability and Bioactivity. J. Am. Chem. Soc. 2022, 144, 18532–18544. [Google Scholar] [CrossRef] [PubMed]
- Couvreur, P.; Lepetre-Mouelhi, S.; Garbayo, E.; Blanco-Prieto, M.J. Self-assembled lipid–prodrug nanoparticles. Nat. Rev. Bioeng. 2023, 1, 749–768. [Google Scholar] [CrossRef]
- Huang, L.; Yang, J.; Wang, T.; Gao, J.; Xu, D. Engineering of small-molecule lipidic prodrugs as novel nanomedicines for enhanced drug delivery. J. Nanobiotechnol. 2022, 20, 49. [Google Scholar] [CrossRef]
- Coppens, E.; Desmaële, D.; Mougin, J.; Tusseau-Nenez, S.; Couvreur, P.; Mura, S. Gemcitabine Lipid Prodrugs: The Key Role of the Lipid Moiety on the Self-Assembly into Nanoparticles. Bioconjugate Chem. 2021, 32, 782–793. [Google Scholar] [CrossRef]
- Irby, D.; Du, C.; Li, F. Lipid–Drug Conjugate for Enhancing Drug Delivery. Mol. Pharm. 2017, 14, 1325–1338. [Google Scholar] [CrossRef]
- Stilgenbauer, M.; Jayawardhana, A.M.D.S.; Datta, P.; Yue, Z.; Gray, M.; Nielsen, F.; Bowers, D.J.; Xiao, H.; Zheng, Y.R. A spermine-conjugated lipophilic Pt(iv) prodrug designed to eliminate cancer stem cells in ovarian cancer. Chem. Commun. 2019, 55, 6106–6109. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jogadi, W.; Kshetri, M.B.; Alqarni, S.; Sharma, A.; Cheline, M.; Amin, M.A.; Sheets, C.; Nsoure-Engohang, A.; Zheng, Y.-R. Engineering Novel Amphiphilic Platinum(IV) Complexes to Co-Deliver Cisplatin and Doxorubicin. Molecules 2024, 29, 4095. https://doi.org/10.3390/molecules29174095
Jogadi W, Kshetri MB, Alqarni S, Sharma A, Cheline M, Amin MA, Sheets C, Nsoure-Engohang A, Zheng Y-R. Engineering Novel Amphiphilic Platinum(IV) Complexes to Co-Deliver Cisplatin and Doxorubicin. Molecules. 2024; 29(17):4095. https://doi.org/10.3390/molecules29174095
Chicago/Turabian StyleJogadi, Wjdan, Man B. Kshetri, Suha Alqarni, Arpit Sharma, May Cheline, Md Al Amin, Cynthia Sheets, Angele Nsoure-Engohang, and Yao-Rong Zheng. 2024. "Engineering Novel Amphiphilic Platinum(IV) Complexes to Co-Deliver Cisplatin and Doxorubicin" Molecules 29, no. 17: 4095. https://doi.org/10.3390/molecules29174095
APA StyleJogadi, W., Kshetri, M. B., Alqarni, S., Sharma, A., Cheline, M., Amin, M. A., Sheets, C., Nsoure-Engohang, A., & Zheng, Y.-R. (2024). Engineering Novel Amphiphilic Platinum(IV) Complexes to Co-Deliver Cisplatin and Doxorubicin. Molecules, 29(17), 4095. https://doi.org/10.3390/molecules29174095