
Magnesium(II) Porphyrazine with Thiophenylmethylene Groups-Synthesis, Electrochemical Characterization, UV–Visible Titration with Palladium Ions, and Density Functional Theory Calculations

 , ,
, ,  ,
,  and
and
Abstract
1. Introduction
2. Results and Discussion
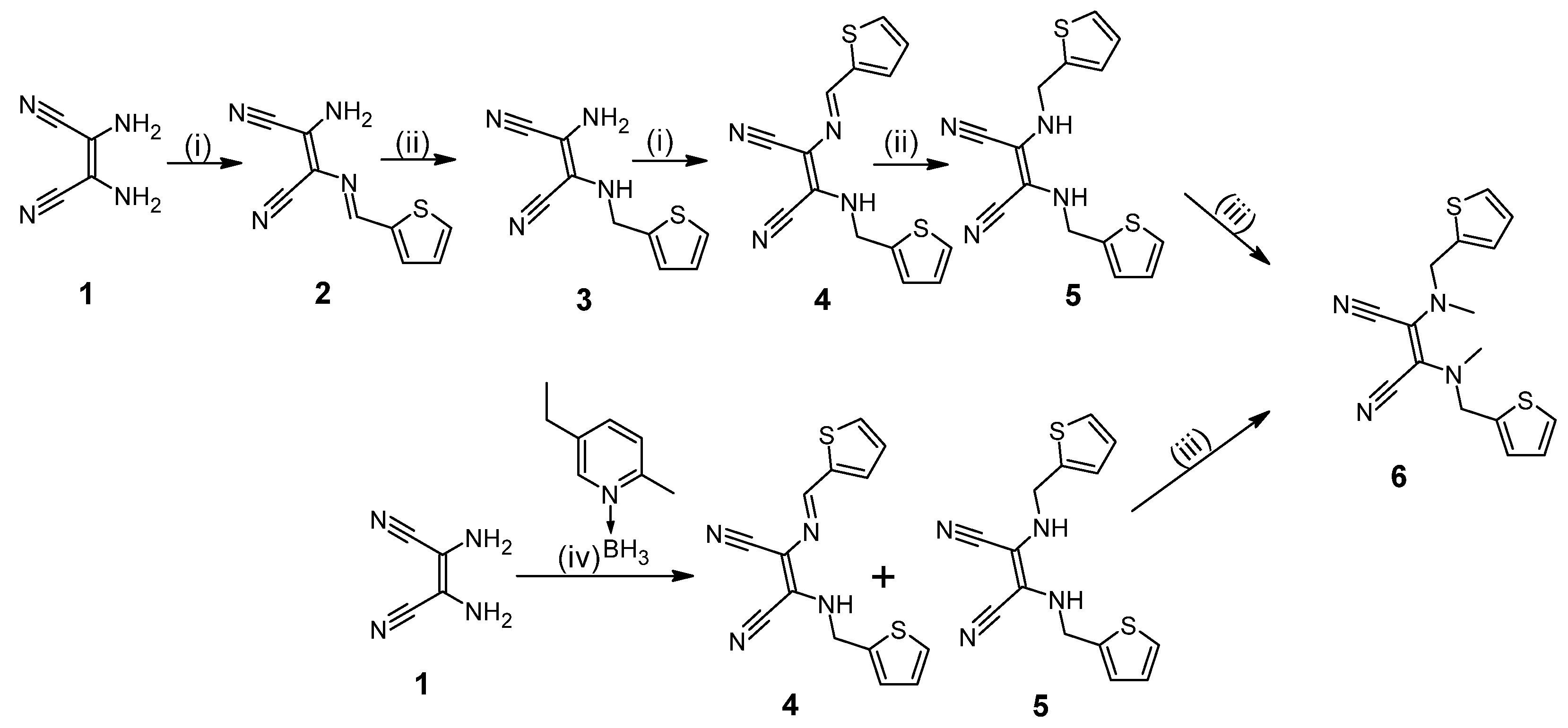
2.1. Synthesis and Physicochemical Characterization
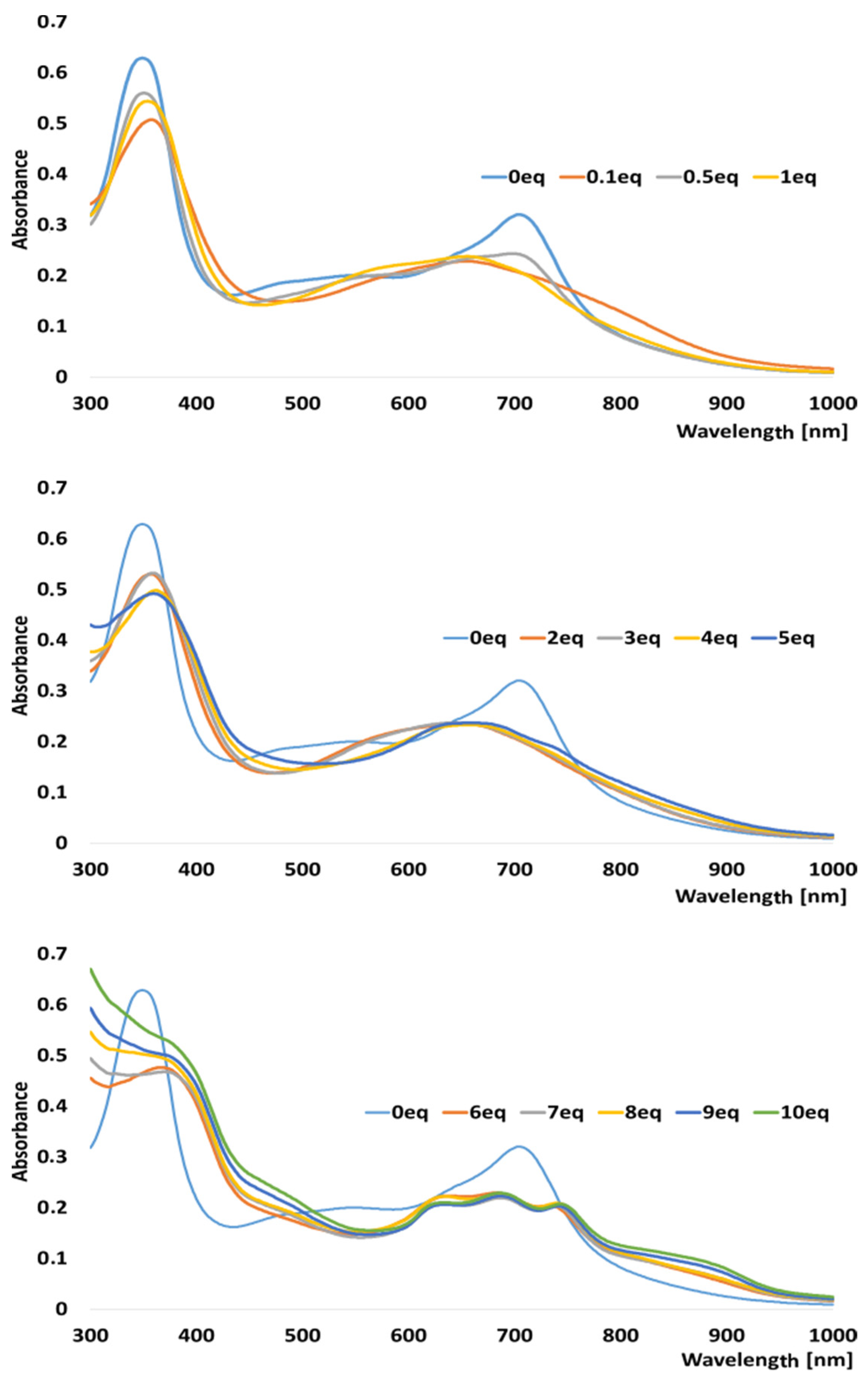
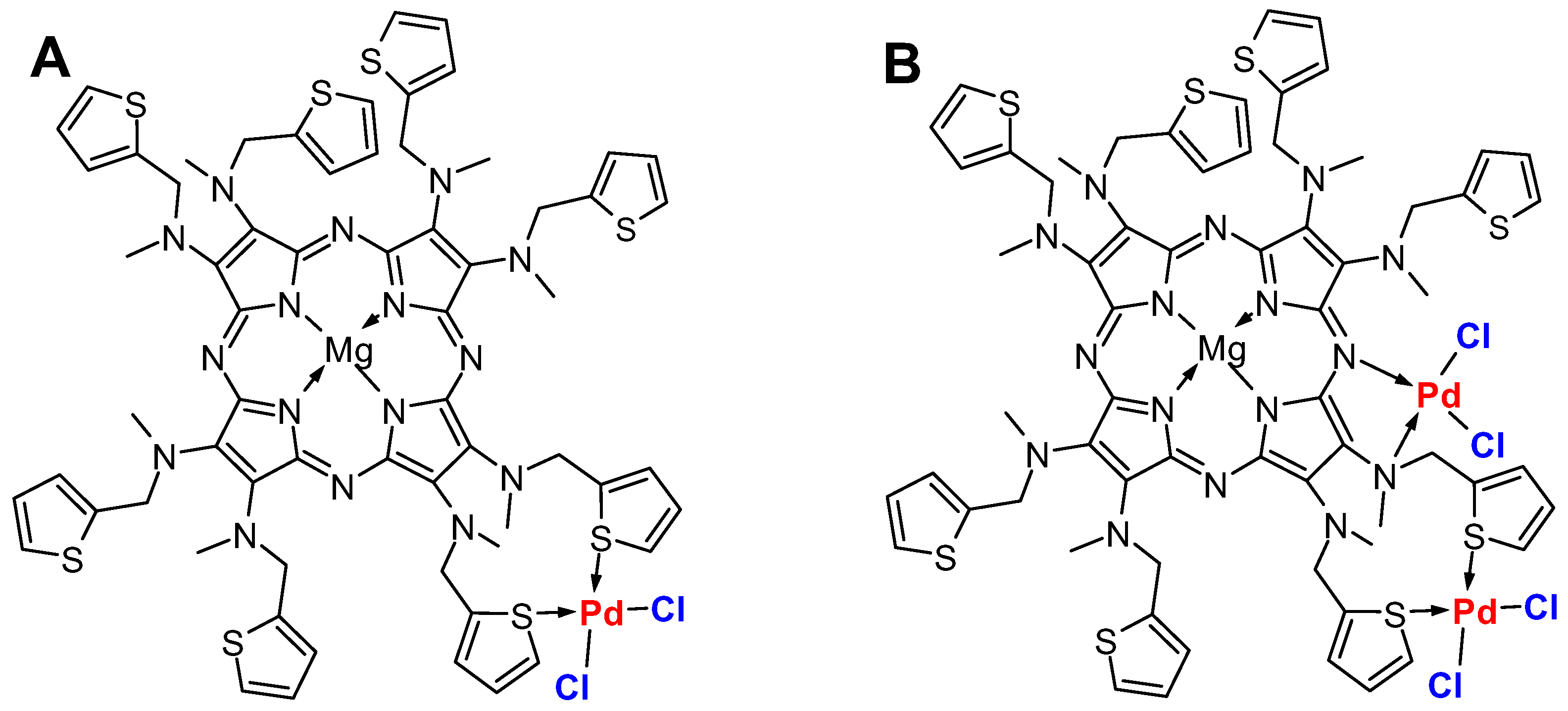
2.2. Titration of Porphyrazine 7 with Palladium Ions
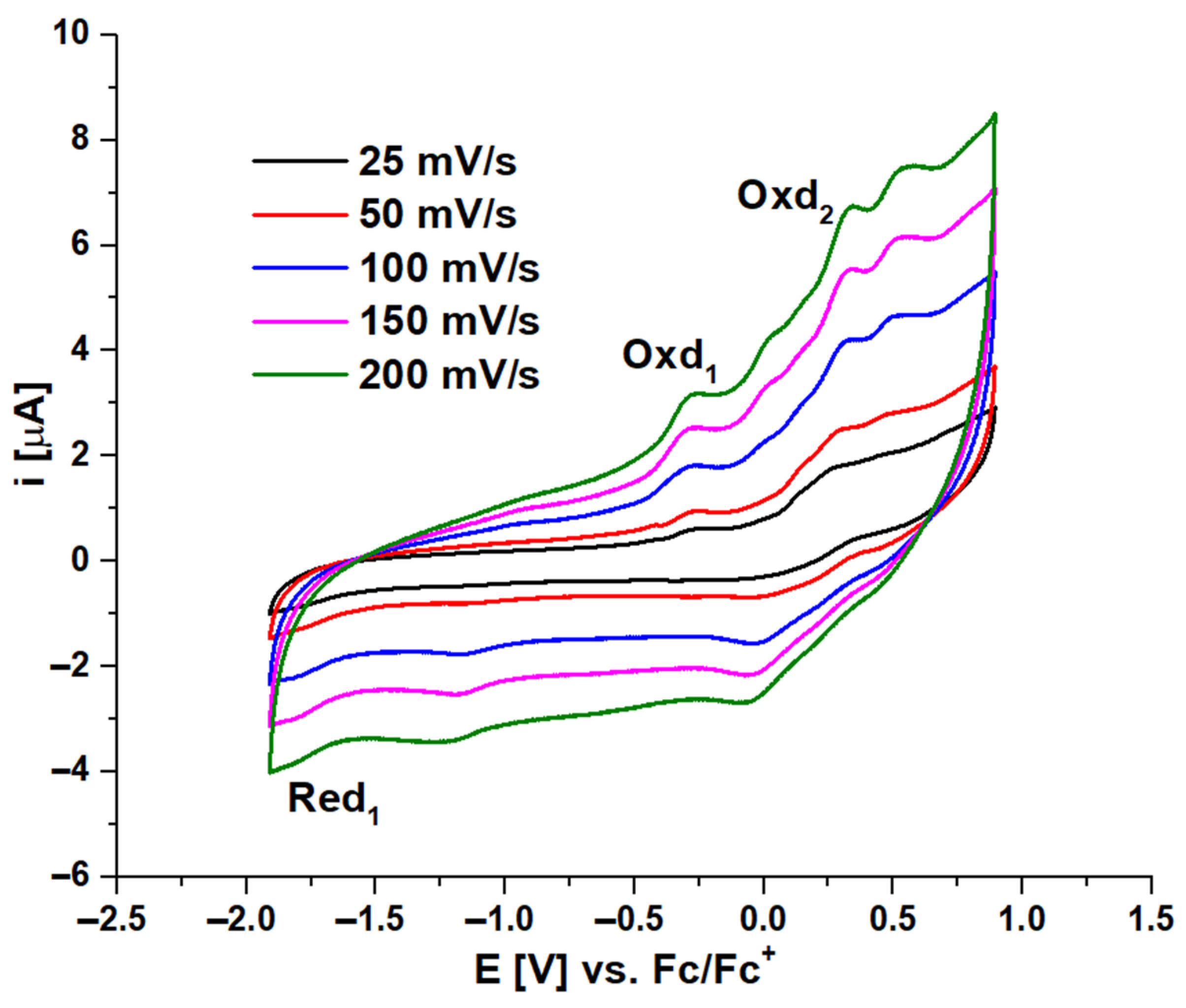
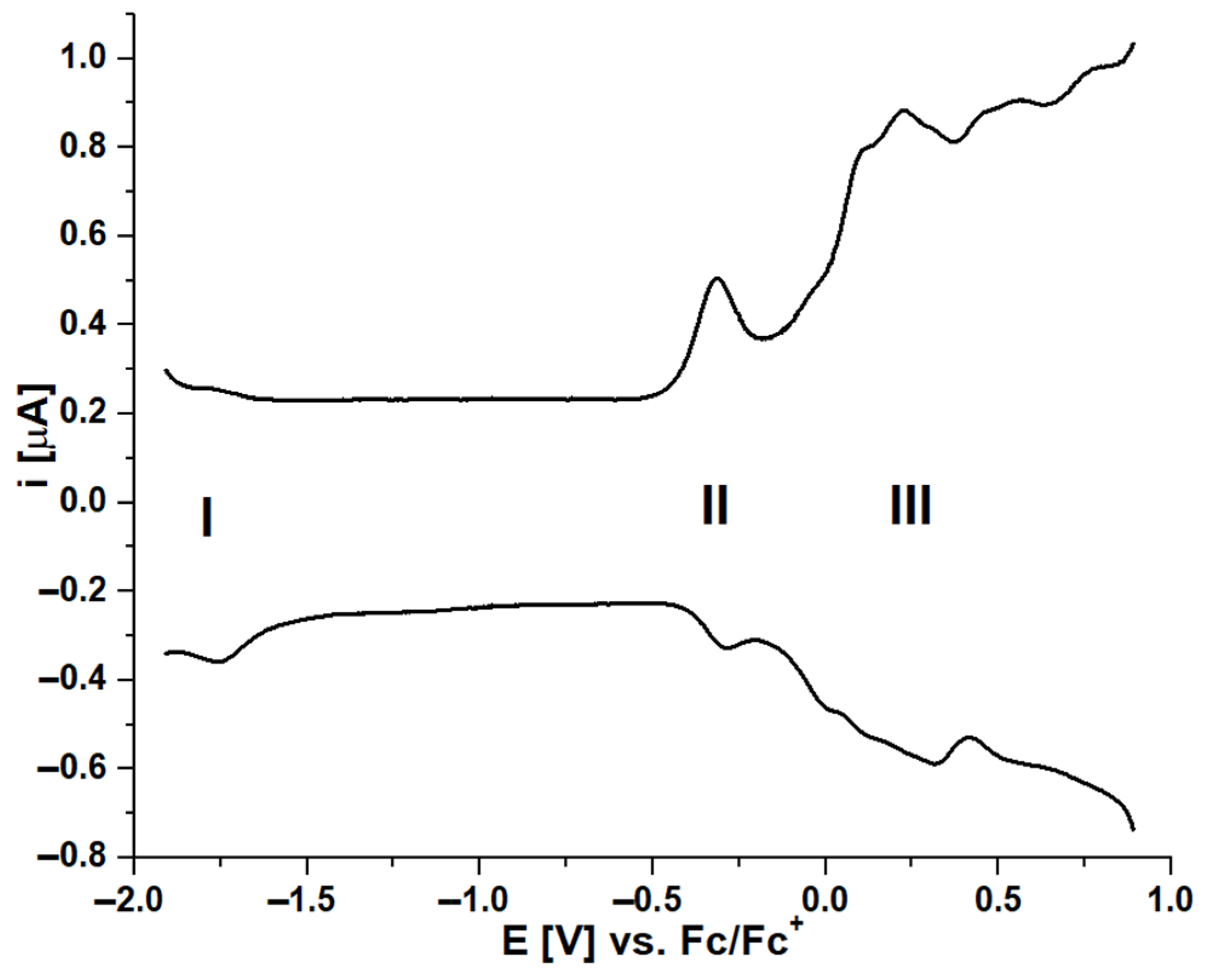
2.3. Electrochemical and Spectroelectrochemical Studies
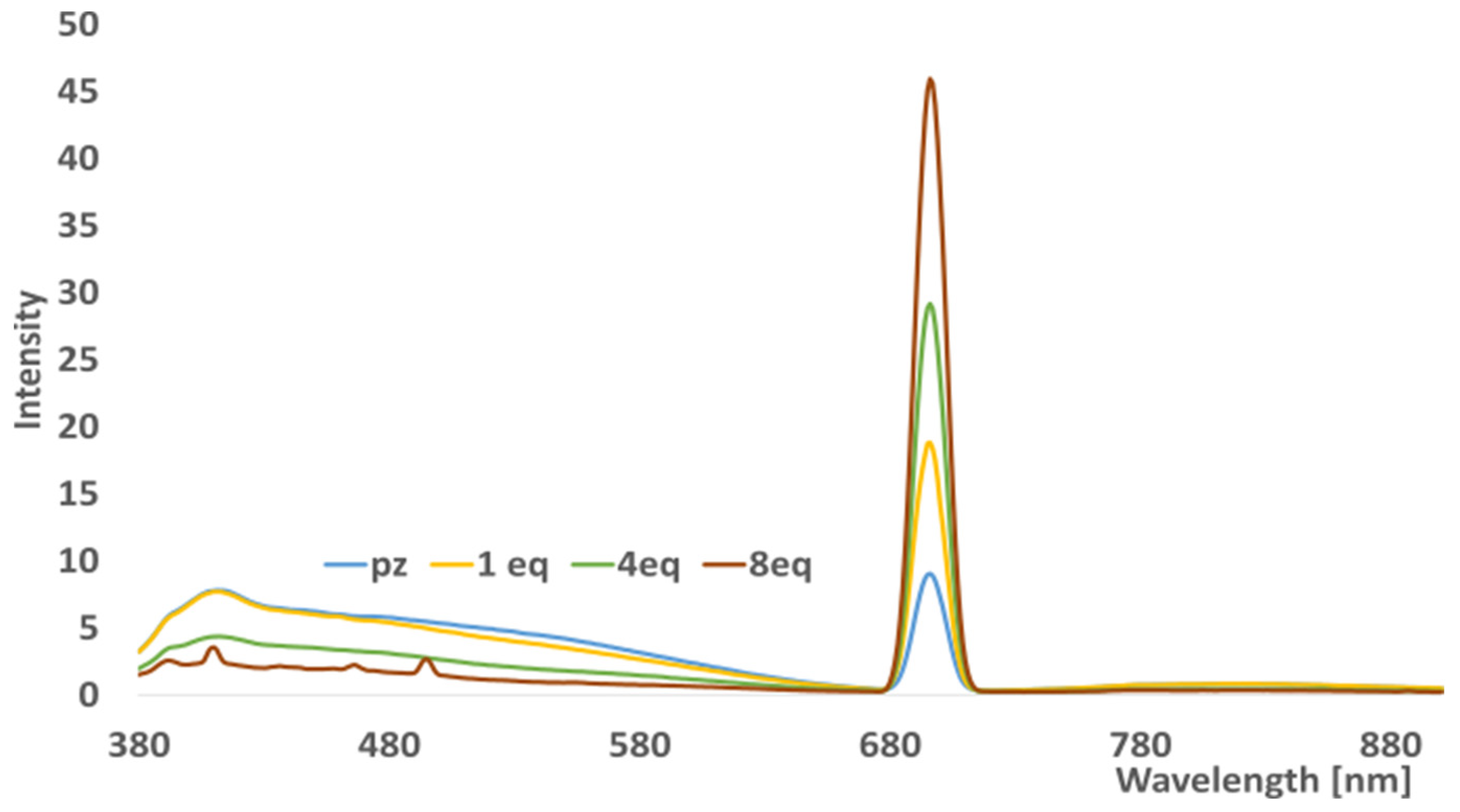
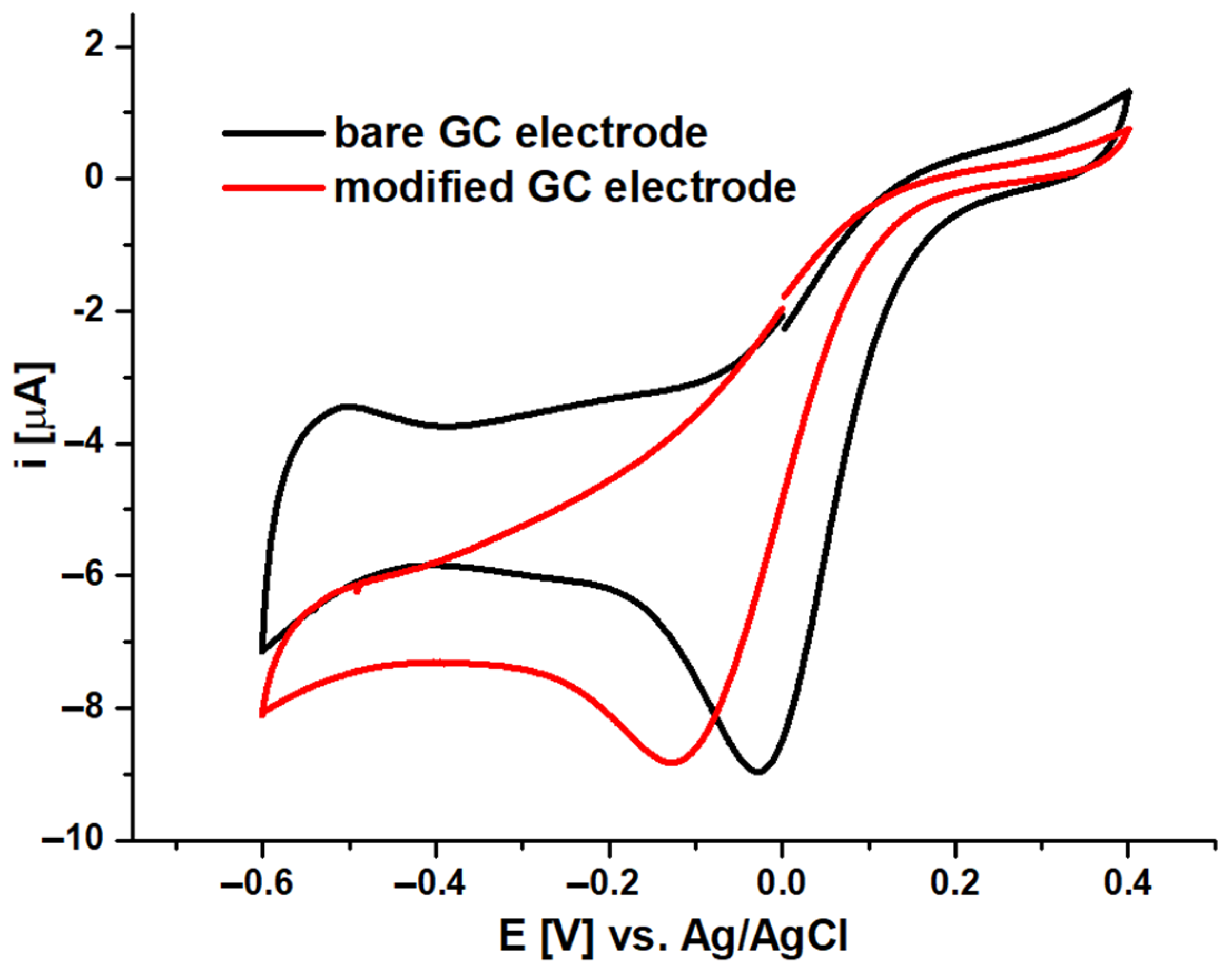
2.4. Electrocatalytic Studies
2.5. Computational Studies
3. Materials and Methods
3.1. General Experimental Methods
3.2. Synthesis and Characterization of New Compounds
- 2-Amino-3-[(2-thiophenylmethylene)amino]-2-butene-1,4-dinitrile (2)
- 2-Amino-3-[(2-thiophenylmethyl)amino]-2-butene-1,4-dinitrile (3)
- 2-[(2-thiophenylmethylene)amino]-3-[(2-thiophenylmethyl)amino]-2-butene-1,4-dinitrile (4)
- 2,3-Bis-[(2-thiophenylmethyl)amino]-2(Z)-butene-1,4-dinitrile (5)
- 2,3-Bis-[methyl(2-thiophenylmethyl)amino]-2(Z)-butene-1,4-dinitrile (6)
- [2,3,7,8,12,13,17,18-Octakis-[methyl(2-thiophenylmethyl)amino]-porphyrazinato]magnesium (II) (7)
3.3. General Procedure for UV–Vis Titrations of Porphyrazine 7
3.4. Electrochemical Measurements
3.5. Spectroelectrochemical Measurements
3.6. Modification of Working Electrode and Electrocatalytic Studies
3.7. Computational Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Michel, S.L.J.; Hoffman, B.M.; Baum, S.M.; Barrett, A.G.M. Peripherally Functionalized Porphyrazines: Novel Metallomacrocycles with Broad, Untapped Potential. Prog. Inorg. Chem. 2001, 50, 473–590. [Google Scholar]
- Rodríguez-Morgade, M.S.; Stuzhin, P.A. The Chemistry of Porphyrazines: An Overview. J. Porphyr. Phthalocyanines 2004, 8, 1129–1165. [Google Scholar] [CrossRef]
- Khelevina, O.G.; Chizhovan, N.V.; Stuzhin, P.A. Modification of β-Positions in Porphyrazines by Substitution Reactions. J. Porphyr. Phthalocyanines 2000, 4, 555–563. [Google Scholar] [CrossRef]
- Fuchter, M.J.; Zhong, C.; Zong, H.; Hoffman, B.M.; Barrett, A.G.M.; Fuchter, M.J.; Zhong, C.; Zong, H.; Hoffman, B.M.; Barrett, A.G.M. Porphyrazines: Designer Macrocycles by Peripheral Substituent Change. Aust. J. Chem. 2008, 61, 235–255. [Google Scholar] [CrossRef]
- Kahn, O. Magnetism of Heterobimetallics: Toward Molecular-Based Magnets. In Advances in Inorganic Chemistry; Elsevier: Amsterdam, The Netherlands, 1995; Volume 43, pp. 179–259. ISBN 978-0-12-023643-5. [Google Scholar]
- Sammes, P.G.; Yahioglu, G. 1,10-Phenanthroline: A Versatile Ligand. Chem. Soc. Rev. 1994, 23, 327. [Google Scholar] [CrossRef]
- Montalban, A.G.; Sakellariou, E.G.; Riguet, E.; McCubbin, Q.J.; Barrett, A.G.M.; Hoffman, B.M. Phenanthroline-Appended Porphyrazines: Synthesis and Conversion into Solitaire Ru(II) Complexes. Inorganica Chim. Acta 2001, 317, 143–148. [Google Scholar] [CrossRef]
- Kabay, N.; Baygu, Y.; Alpoguz, H.K.; Kaya, A.; Gök, Y. Synthesis and Characterization of Porphyrazines as Novel Extractants for the Removal of Ag(I) and Hg(II) from Aqueous Solution. Dye. Pigment. 2013, 96, 372–376. [Google Scholar] [CrossRef]
- Hou, R.; Jin, L.; Yin, B. Synthesis and Electron Donating Property of Novel Porphyrazines Containing Tetrathiacrown Ether-Linked Tetrathiafulvalene Moieties. Inorg. Chem. Commun. 2009, 12, 739–743. [Google Scholar] [CrossRef]
- Goslinski, T.; White, A.J.P. Synthesis, characterization and spectroscopic properties of novel periphery –functionalized unsymmetrical porphyrazines containing mixed dithienylpyrrolyl and dimethylamino groups. Polyhedron 2009, 28, 2579–2584. [Google Scholar] [CrossRef]
- Zagal, J.H.; Griveau, S.; Silva, J.F.; Nyokong, T.; Bedioui, F. Metallophthalocyanine-Based Molecular Materials as Catalysts for Electrochemical Reactions. Coord. Chem. Rev. 2010, 254, 2755–2791. [Google Scholar] [CrossRef]
- Pereira-Rodrigues, N.; Cofré, R.; Zagal, J.H.; Bedioui, F. Electrocatalytic Activity of Cobalt Phthalocyanine CoPc Adsorbed on a Graphite Electrode for the Oxidation of Reduced L-Glutathione (GSH) and the Reduction of Its Disulfide (GSSG) at Physiological pH. Bioelectrochemistry 2007, 70, 147–154. [Google Scholar] [CrossRef]
- Sun, J.; Sun, Y.; Deng, K.; Hou, H.; Wang, D. Oxidative Degradation of Organic Pollutants by Hydrogen Peroxide in the Presence of FePz(dtnCl2)4 under Visible Irradiation. Chem. Lett. 2007, 36, 586–587. [Google Scholar] [CrossRef]
- Deng, K.; Huang, F.; Wang, D.; Peng, Z.; Zhou, Y. A Novel Catalyst Iron(II) Tetra(1,4-Dithin)Porphyrazine for Oxygenating Degradation of Organic Pollutants in Aqueous Solutions. Chem. Lett. 2004, 33, 34–35. [Google Scholar] [CrossRef]
- Garrido Montalban, A.; Baum, S.M.; Barrett, A.G.M.; Hoffman, B.M. Studies on Seco-Porphyrazines: A Case Study on Serendipity. Dalton Trans. 2003, 2093–2102. [Google Scholar] [CrossRef]
- Wasserman, H.H.; Ives, J.L. Singlet Oxygen in Organic Synthesis. Tetrahedron 1981, 37, 1825–1852. [Google Scholar] [CrossRef]
- Hosu, I.S.; Wang, Q.; Vasilescu, A.; Peteu, S.F.; Raditoiu, V.; Railian, S.; Zaitsev, V.; Turcheniuk, K.; Wang, Q.; Li, M.; et al. Cobalt Phthalocyanine Tetracarboxylic Acid Modified Reduced Graphene Oxide: A Sensitive Matrix for the Electrocatalytic Detection of Peroxynitrite and Hydrogen Peroxide. RSC Adv. 2015, 5, 1474–1484. [Google Scholar] [CrossRef]
- Liu, D.; Long, Y.-T. Superior Catalytic Activity of Electrochemically Reduced Graphene Oxide Supported Iron Phthalocyanines toward Oxygen Reduction Reaction. ACS Appl. Mater. Interfaces 2015, 7, 24063–24068. [Google Scholar] [CrossRef]
- Koczorowski, T.; Cerbin-Koczorowska, M.; Rębiś, T. Azaporphyrins Embedded on Carbon-Based Nanomaterials for Potential Use in Electrochemical Sensing—A Review. Nanomaterials 2021, 11, 2861. [Google Scholar] [CrossRef]
- Koczorowski, T.; Ber, J.; Sokolnicki, T.; Teubert, A.; Szczolko, W.; Goslinski, T. Electrochemical and Catalytic Assessment of Peripheral Bromoaryl-Substituted Manganese and Iron Porphyrazines. Dye. Pigment. 2020, 178, 108370. [Google Scholar] [CrossRef]
- Fuchter, M.J.; Beall, L.S.; Baum, S.M.; Montalban, A.G.; Sakellariou, E.G.; Mani, N.S.; Miller, T.; Vesper, B.J.; White, A.J.P.; Williams, D.J.; et al. Synthesis of Porphyrazine-Octaamine, Hexamine and Diamine Derivatives. Tetrahedron 2005, 61, 6115–6130. [Google Scholar] [CrossRef]
- Belviso, S.; Cammarota, F.; Rossano, R.; Lelj, F. Effect of Polyfluorination on Self-Assembling and Electronic Properties of Thioalkyl-Porphyrazines. J. Porphyr. Phthalocyanines 2016, 20, 223–233. [Google Scholar] [CrossRef]
- Hassani, M.; Leda, A.; Porolnik, W.; Falkowski, M.; Rębiś, T.; Piskorz, J.; Popenda, L.; Wicinski, M.; Mlynarczyk, D.T.; Düzgüneş, N.; et al. Synthesis, Electrochemical and Photochemical Properties of Sulfanyl Porphyrazine with Ferrocenyl Substituents. Molecules 2023, 28, 5215. [Google Scholar] [CrossRef]
- Bucher, C.; Devillers, C.; Moutet, J.; Royal, G.; Saintaman, E. Ferrocene-Appended Porphyrins: Syntheses and Properties. Coord. Chem. Rev. 2009, 253, 21–36. [Google Scholar] [CrossRef]
- González-Cabello, A.; Vázquez, P.; Torres, T. A New Phthalocyanine–Ferrocene Conjugated Dyad. J. Organomet. Chem. 2001, 637–639, 751–756. [Google Scholar] [CrossRef]
- Goslinski, T.; Tykarska, E.; Szczolko, W.; Osmalek, T.; Smigielska, A.; Walorczyk, S.; Zong, H.; Gdaniec, M.; Hoffman, B.M.; Mielcarek, J.; et al. Synthesis and Characterization of Periphery-Functionalized Porphyrazines Containing Mixed Pyrrolyl and Pyridylmethylamino Groups. J. Porphyr. Phthalocyanines 2009, 13, 223–234. [Google Scholar] [CrossRef]
- Burkhardt, E.R.; Coleridge, B.M. Reductive Amination with 5-Ethyl-2-Methylpyridine Borane. Tetrahedron Lett. 2008, 49, 5152–5155. [Google Scholar] [CrossRef]
- Begland, R.W.; Hartter, D.R.; Jones, F.N.; Sam, D.J.; Sheppard, W.A.; Webster, O.W.; Weigert, F.J. Hydrogen Cyanide Chemistry. VIII. New Chemistry of Diaminomaleonitrile. Heterocyclic Synthesis. J. Org. Chem. 1974, 39, 2341–2350. [Google Scholar] [CrossRef]
- Piskorz, J.; Skupin, P.; Lijewski, S.; Korpusinski, M.; Sciepura, M.; Konopka, K.; Sobiak, S.; Goslinski, T.; Mielcarek, J. Synthesis, Physical–Chemical Properties and in Vitro Photodynamic Activity against Oral Cancer Cells of Novel Porphyrazines Possessing Fluoroalkylthio and Dietherthio Substituents. J. Fluor. Chem. 2012, 135, 265–271. [Google Scholar] [CrossRef]
- Goslinski, T.; Dutkiewicz, Z.; Kryjewski, M.; Tykarska, E.; Sobotta, L.; Szczolko, W.; Gdaniec, M.; Mielcarek, J. Experimental and Computational Study on the Reactivity of 2,3-Bis[(3-Pyridylmethyl)Amino]-2(Z)-Butene-1,4-Dinitrile, a Key Intermediate for the Synthesis of Tribenzoporphyrazine Bearing Peripheral Methyl(3-Pyridylmethyl)Amino Substituents. Monatsh. Chem. 2011, 142, 599–608. [Google Scholar] [CrossRef]
- Tuncer, S.; Koca, A.; Gül, A.; Avcıata, U. Synthesis, Characterization, Electrochemistry and Spectroelectrochemistry of Novel Soluble Porphyrazines Bearing Unsaturated Functional Groups. Dye. Pigment. 2012, 92, 610–618. [Google Scholar] [CrossRef]
- Koczorowski, T.; Szczolko, W.; Teubert, A.; Goslinski, T. Sulfanyl Porphyrazines with Morpholinylethyl Periphery—Synthesis, Electrochemistry, and Photocatalytic Studies after Deposition on Titanium(IV) Oxide P25 Nanoparticles. Molecules 2021, 26, 2280. [Google Scholar] [CrossRef]
- BIOVIA. Dassault Systèmes, Discovery Studio, Release 3.5; Dassault Systèmes: San Diego, CA, USA, 2012. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R. Gaussian 16, Revision A.03 Gaussian; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Parr, R.G.; Weitao, Y. Density-Functional Theory of Atoms and Molecules; Oxford University Press: Oxford, UK, 2015; ISBN 978-0-19-535773-8. [Google Scholar]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Petersson, G.A.; Bennett, A.; Tensfeldt, T.G.; Al-Laham, M.A.; Shirley, W.A.; Mantzaris, J. A Complete Basis Set Model Chemistry. I. The Total Energies of Closed-Shell Atoms and Hydrides of the First-Row Elements. J. Chem. Phys. 1988, 89, 2193–2218. [Google Scholar] [CrossRef]
- Cammi, R.; Mennucci, B.; Tomasi, J. Fast Evaluation of Geometries and Properties of Excited Molecules in Solution: A Tamm-Dancoff Model with Application to 4-Dimethylaminobenzonitrile. J. Phys. Chem. A 2000, 104, 5631–5637. [Google Scholar] [CrossRef]
- Davidson, N.R. Statistical Mechanics; Dover, Ed.; Dover Publications: Mineola, NY, USA, 2003; ISBN 978-0-486-43264-9. [Google Scholar]
- Sychrovský, V.; Gräfenstein, J.; Cremer, D. Nuclear Magnetic Resonance Spin–Spin Coupling Constants from Coupled Perturbed Density Functional Theory. J. Chem. Phys. 2000, 113, 3530–3547. [Google Scholar] [CrossRef]
- Sychrovský, V.; Šponer, J.; Trantírek, L.; Schneider, B. Indirect NMR Spin−Spin Coupling Constants 3 J (P,C) and 2 J (P,H) across the P−O···H−C Link Can Be Used for Structure Determination of Nucleic Acids. J. Am. Chem. Soc. 2006, 128, 6823–6828. [Google Scholar] [CrossRef] [PubMed]
- Trani, F.; Scalmani, G.; Zheng, G.; Carnimeo, I.; Frisch, M.J.; Barone, V. Time-Dependent Density Functional Tight Binding: New Formulation and Benchmark of Excited States. J. Chem. Theory Comput. 2011, 7, 3304–3313. [Google Scholar] [CrossRef]
- Wolinski, K.; Hinton, J.F.; Pulay, P. Efficient Implementation of the Gauge-Independent Atomic Orbital Method for NMR Chemical Shift Calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locant | Exp. | Conformers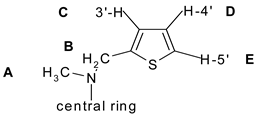 | E1 | E2 | E3 | E4 | E5 | E6 | E | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| I | II | III | IV | V | VI | |||||||||
| A | 3.61 | 3.23 | 3.58 | 3.65 | 3.67 | 3.54 | 3.55 | 11 | 1 | 1 | 2 | 2 | 2 | 3 |
| B | 5.68 | 4.26 | 4.51 | 4.51 | 4.52 | 4.54 | 4.54 | 25 | 21 | 21 | 20 | 20 | 20 | 21 |
| C | 7.05 | 6.62 | 7.15 | 7.44 | 7.21 | 5.89 | 5.91 | 6 | 1 | 5 | 2 | 16 | 16 | 8 |
| D | 6.90 | 6.50 | 6.93 | 7.13 | 7.00 | 6.44 | 6.44 | 6 | 0.4 | 3 | 1 | 7 | 7 | 4 |
| E | 7.30 | 7.03 | 7.16 | 7.24 | 7.03 | 7.12 | 7.10 | 4 | 2 | 0.8 | 4 | 2 | 3 | 3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szczolko, W.; Chornovolenko, K.; Kujawski, J.; Dutkiewicz, Z.; Koczorowski, T. Magnesium(II) Porphyrazine with Thiophenylmethylene Groups-Synthesis, Electrochemical Characterization, UV–Visible Titration with Palladium Ions, and Density Functional Theory Calculations. Molecules 2024, 29, 3610. https://doi.org/10.3390/molecules29153610
Szczolko W, Chornovolenko K, Kujawski J, Dutkiewicz Z, Koczorowski T. Magnesium(II) Porphyrazine with Thiophenylmethylene Groups-Synthesis, Electrochemical Characterization, UV–Visible Titration with Palladium Ions, and Density Functional Theory Calculations. Molecules. 2024; 29(15):3610. https://doi.org/10.3390/molecules29153610
Chicago/Turabian StyleSzczolko, Wojciech, Kyrylo Chornovolenko, Jacek Kujawski, Zbigniew Dutkiewicz, and Tomasz Koczorowski. 2024. "Magnesium(II) Porphyrazine with Thiophenylmethylene Groups-Synthesis, Electrochemical Characterization, UV–Visible Titration with Palladium Ions, and Density Functional Theory Calculations" Molecules 29, no. 15: 3610. https://doi.org/10.3390/molecules29153610
APA StyleSzczolko, W., Chornovolenko, K., Kujawski, J., Dutkiewicz, Z., & Koczorowski, T. (2024). Magnesium(II) Porphyrazine with Thiophenylmethylene Groups-Synthesis, Electrochemical Characterization, UV–Visible Titration with Palladium Ions, and Density Functional Theory Calculations. Molecules, 29(15), 3610. https://doi.org/10.3390/molecules29153610