
Improving the Solubility, Stability, and Bioavailability of Albendazole through Synthetic Salts

Abstract

1. Introduction
2. Results and Discussion
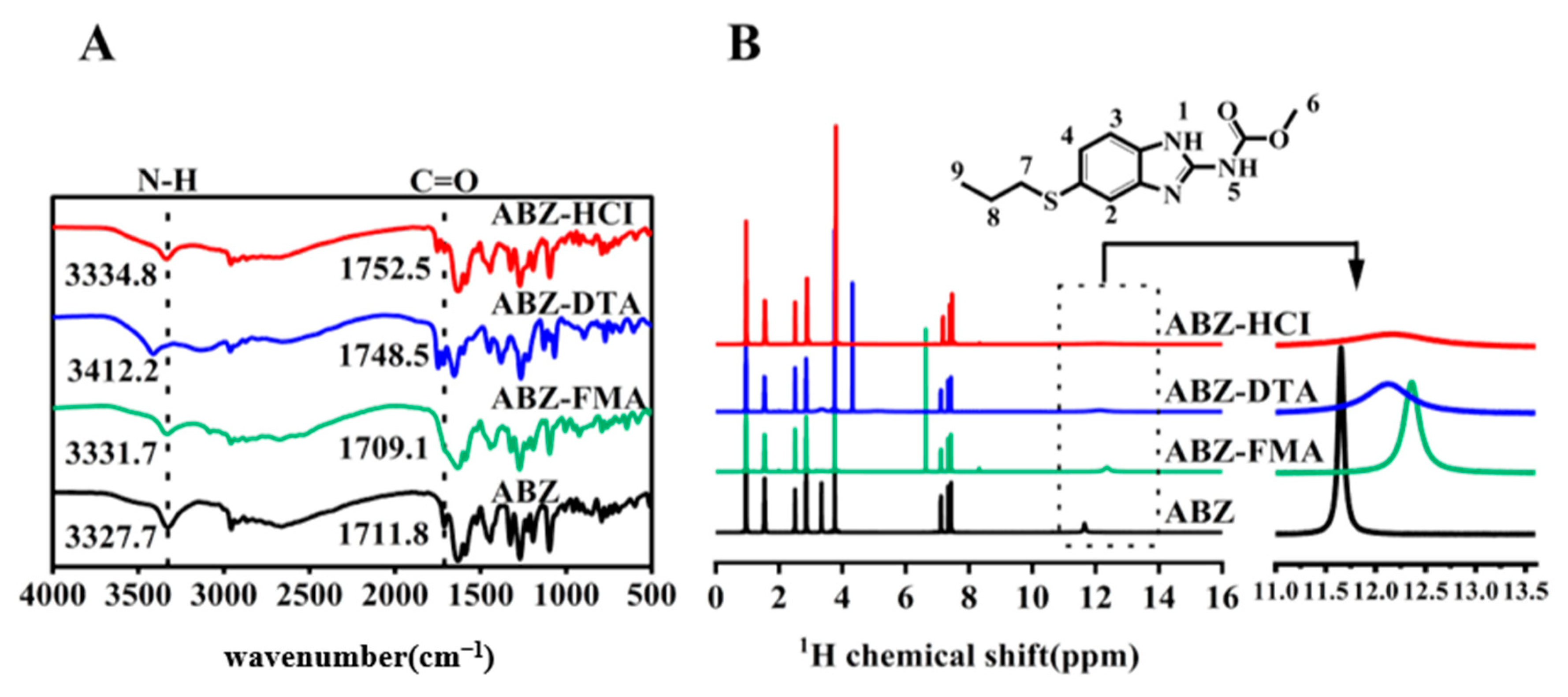
2.1. Characterization
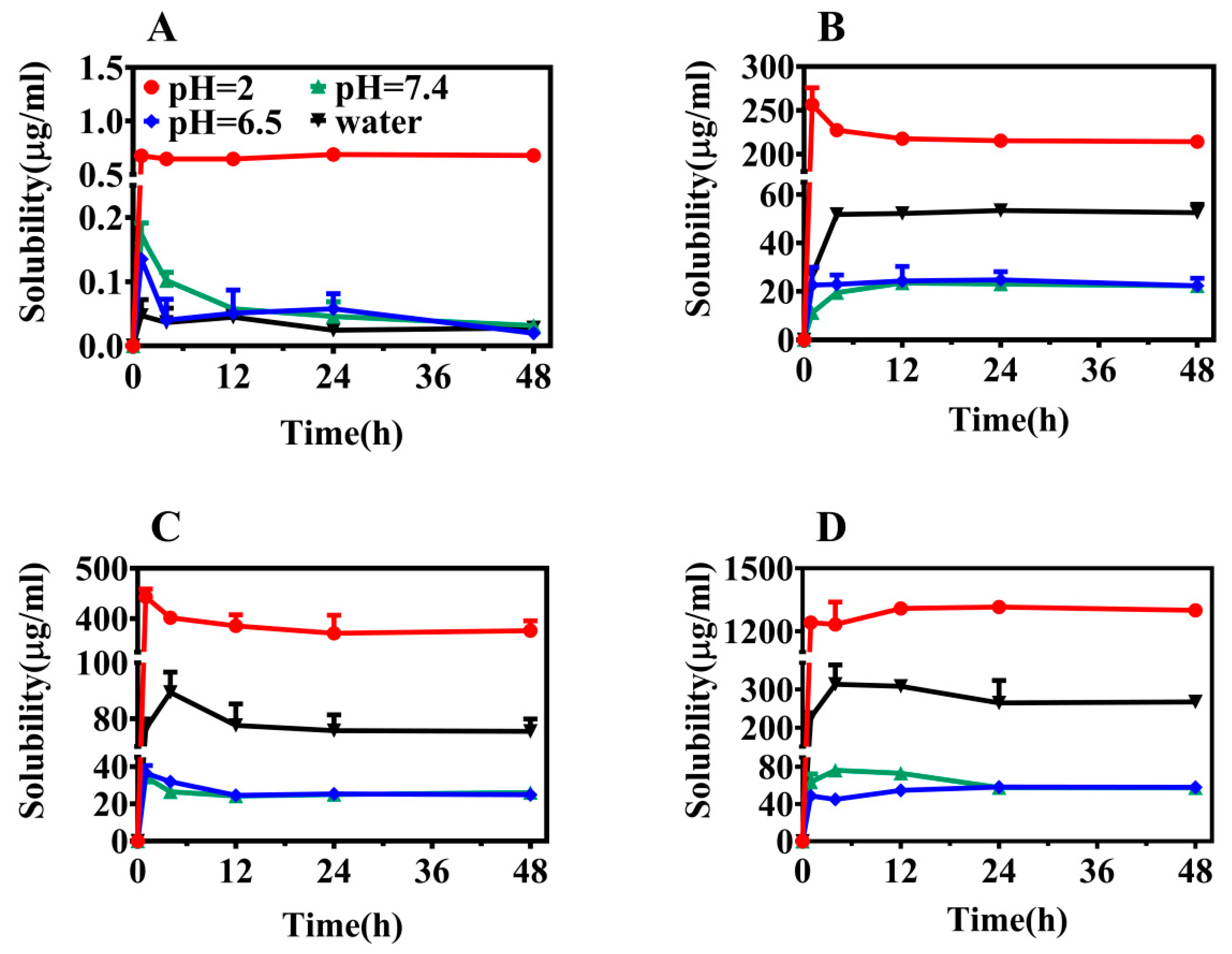
2.2. Solubility
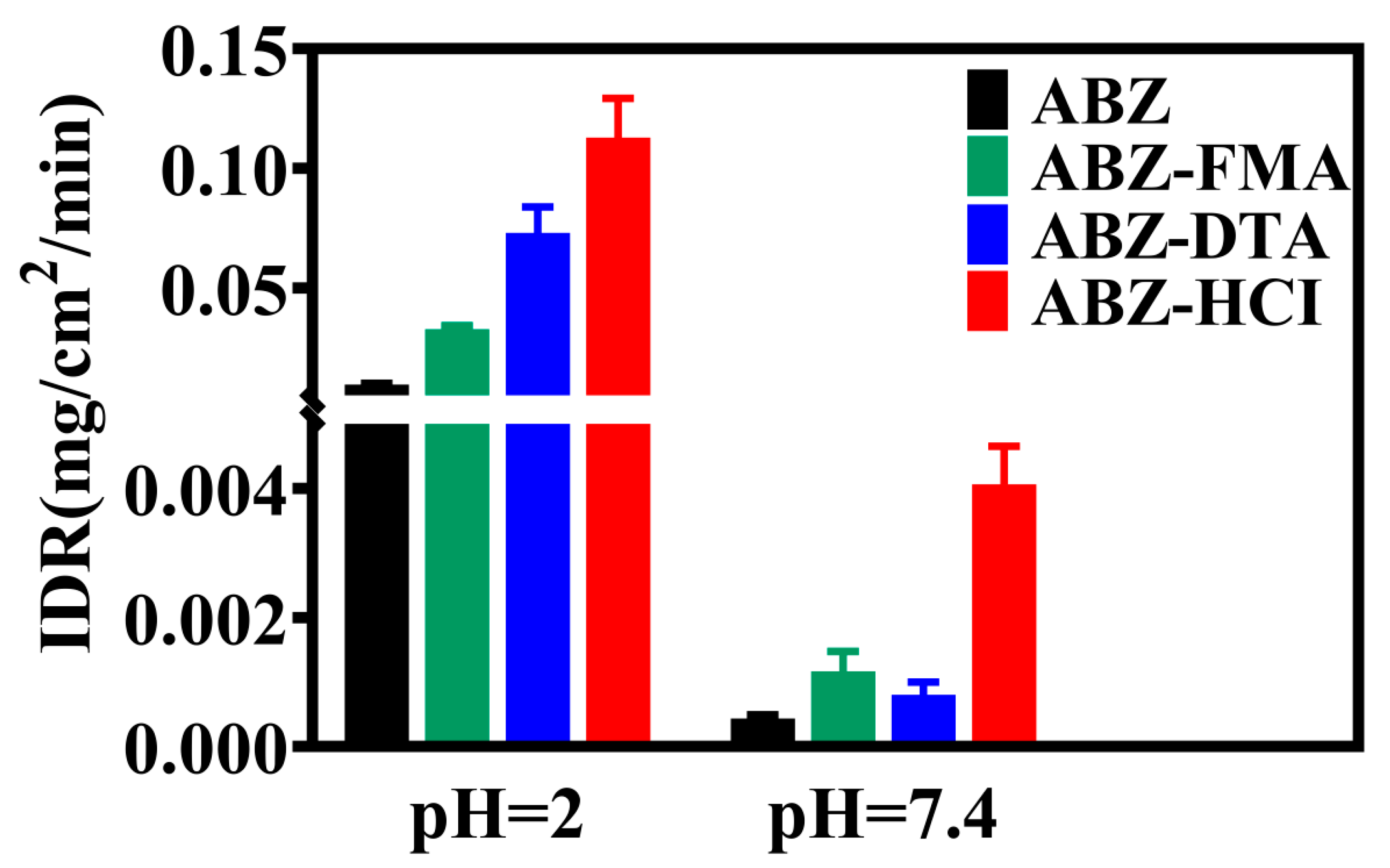
2.3. Intrinsic Dissolution Rate
2.4. Solid-State Stability
2.4.1. Influencing Factor Testing
2.4.2. Accelerated Stability
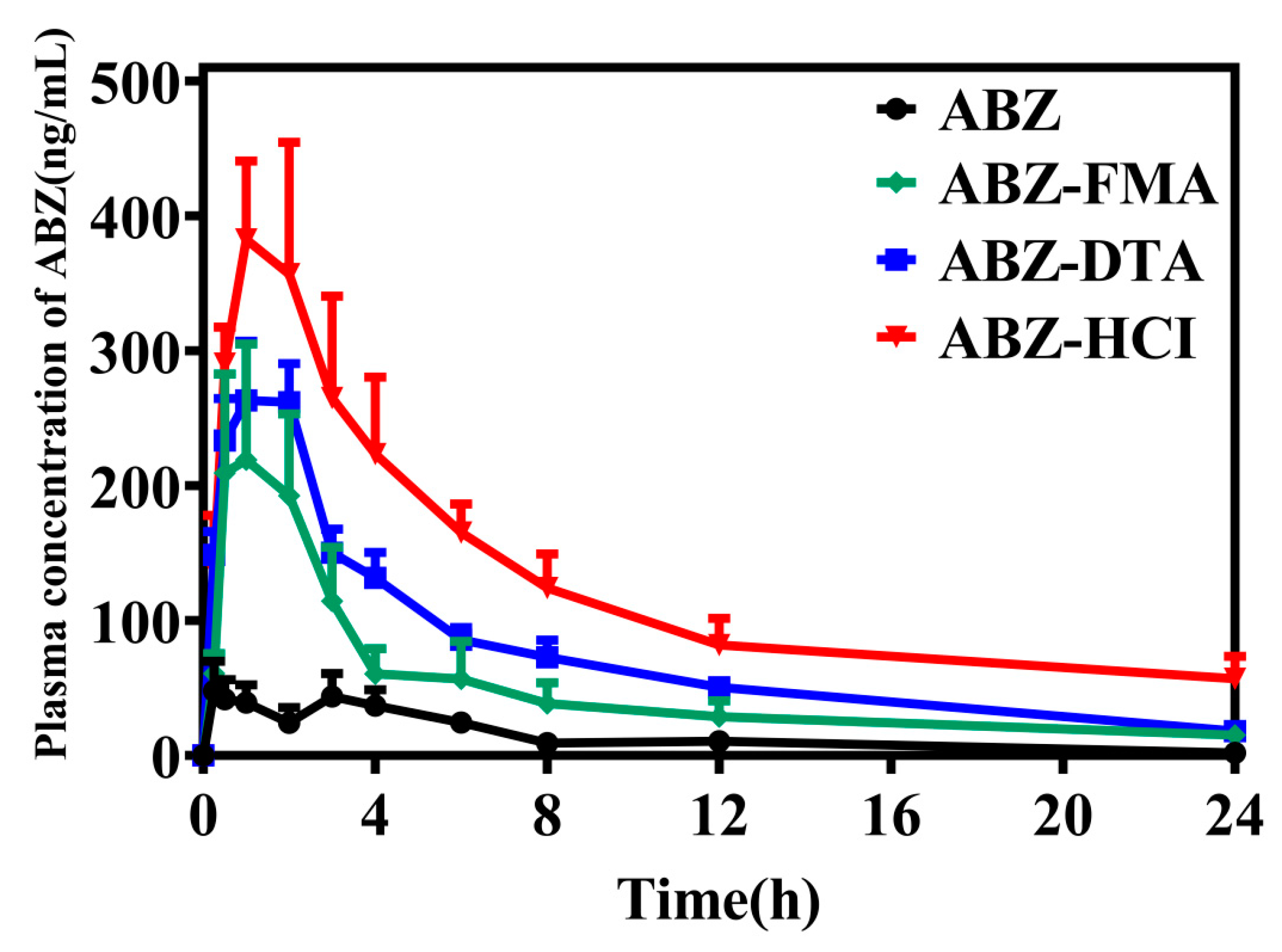
2.5. Pharmacokinetic Comparison in Wistar Rats
3. Materials and Methods
3.1. Materials and Reagents
3.2. Preparation of Albendazole Salts
3.3. Characterization of Albendazole Salts
3.3.1. Fourier Transform Infrared Spectroscopy (FT-IR)
3.3.2. Nuclear Magnetic Resonance (1H-NMR)
3.3.3. Powder X-ray Diffraction (PXRD)
3.3.4. Dynamic Vapor Sorption (DVS)
3.3.5. Thermogravimetric Analysis (TGA)
3.3.6. Differential Scanning Calorimetry (DSC)
3.3.7. Scanning Electron Microscopy (SEM)
3.4. Solubility
3.5. Intrinsic Dissolution Rate (IDR)
3.6. Solid-State Stability
3.6.1. Influencing Factor Testing
3.6.2. Accelerated Testing
3.7. Pharmacokinetic Comparison in Wistar Rats
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hu, C.; Liu, Z.; Liu, C.; Li, J.; Wang, Z.; Xu, L.; Chen, C.; Fan, H.; Qian, F. Enhanced Oral Bioavailability and Anti-Echinococcosis Efficacy of Albendazole Achieved by Optimizing the “Spring” and “Parachute”. Mol. Pharm. 2019, 16, 4978–4986. [Google Scholar] [CrossRef] [PubMed]
- Aydin, Y.; Ulas, A.B.; Ince, I.; Kalin, A.; Can, F.K.; Gundogdu, B.; Kasali, K.; Kerget, B.; Ogul, Y.; Eroglu, A. Evaluation of albendazole efficiency and complications in patients with pulmonary hydatid cyst. Interact. Cardiovasc. Thorac. Surg. 2021, 34, 245–249. [Google Scholar] [CrossRef]
- Neumayr, A.; Schunk, M.; Theunissen, C.; Van Esbroeck, M.; Mechain, M.; Hatz, C.; Mørch, K.; Soriano Pérez, M.J.; Sydow, V.; Sothmann, P.; et al. Efficacy and tolerability of quinacrine monotherapy and albendazole plus chloroquine combination therapy in nitroimidazole-refractory giardiasis: A TropNet study. Clin. Infect. Dis. 2021, 73, 1517–1523. [Google Scholar] [CrossRef] [PubMed]
- Pene, P.; Mojon, M.; Garin, J.P.; Rossignol, J.F.; Coulaud, J.P. Albendazole: A New Broad Spectrum Anthelmintic. Am. J. Trop. Med. Hyg. 1982, 31, 263–266. [Google Scholar] [CrossRef]
- Haifeng, C.; Zhen, W.; Chunfang, X. Albendazole suppresses cell proliferation and migration and induces apoptosis in human pancreatic cancer cells. Anticancer Drugs. 2020, 31, 431–439. [Google Scholar]
- Zhang, X.; Zhao, J.; Gao, X.; Pei, D.; Gao, C. Anthelmintic drug albendazole arrests human gastric cancer cells at the mitotic phase and induces apoptosis. Exp. Ther. Med. 2017, 13, 595–603. [Google Scholar] [CrossRef]
- Yang, M.H.; Ha, I.J.; Um, J.Y.; Ahn, K.S. Albendazole Exhibits Anti-Neoplastic Actions against Gastric Cancer Cells by Affecting STAT3 and STAT5 Activation by Pleiotropic Mechanism(s). Biomedicines 2021, 9, 362. [Google Scholar] [CrossRef]
- Pourgholami, M.H.; Akhter, J.; Wang, L.; Lu, Y.; Morris, D.L. Antitumor activity of albendazole against the human colorectal cancer cell line HT-29: In vitro and in a xenograft model of peritoneal carcinomatosis. Cancer Chemother. Pharmacol. 2005, 55, 425–432. [Google Scholar] [CrossRef]
- García, J.J.; Bolás, F.; Torrado, J.J. Bioavailability and efficacy characteristics of two different oral liquid formulations of albendazole. Int. J. Pharm. 2003, 250, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Geetha, B.; Ashwini, N. Novel pharmaceutical salts of albendazole. Crystengcomm 2018, 20, 6394–6405. [Google Scholar]
- Moro, P.; Schantz, P.M. Echinococcosis: A review. Int. J. Infect. Dis. 2008, 13, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Mansuri, S.; Kesharwani, P.; Tekade, R.K.; Jain, N.K. Lyophilized mucoadhesive-dendrimer enclosed matrix tablet for extended oral delivery of albendazole. Eur. J. Pharm. Biopharm. 2016, 102, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Kawakami, K.; Fukiage, M.; Oikawa, M.; Nishida, Y.; Matsuda, M.; Fujita, T. Relevance of Liquid-Liquid Phase Separation of Supersaturated Solution in Oral Absorption of Albendazole from Amorphous Solid Dispersions. Pharmaceutics 2021, 13, 220. [Google Scholar] [CrossRef] [PubMed]
- Pensel, P.; Paredes, A.; Albani, C.M.; Allemandi, D.; Bruni, S.S.; Palma, S.D.; Elissondo, M.C. Albendazole nanocrystals in experimental alveolar echinococcosis: Enhanced chemoprophylactic and clinical efficacy in infected mice. Vet. Parasitol. 2018, 251, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Fateh, R.; Norouzi, R.; Mirzaei, E.; Nissapatron, V.; Nawaz, M.; Khalifeh-Gholi, M.; Hamta, A.; Sadati, S.J.A.; Siyadatpanah, A.; Bafghi, A.F. In vitro evaluation of albendazole nanocrystals against Echinococcus granulosus protoscolices. Ann. Parasitol. 2021, 67, 203–212. [Google Scholar] [PubMed]
- Castro Alpízar, J.A.; Vargas Monge, R.; Madrigal Redondo, G.; Pacheco Molina, J.A. Development of novel microstructured lipid carriers for dissolution rate enhancement of albendazole. Int. J. Appl. Pharm. 2020, 12, 173–178. [Google Scholar]
- Eriksen, J.B.; Christensen, S.B.; Bauer-Brandl, A.; Brandl, M. Dissolution/permeation of albendazole in the presence of cyclodextrin and bile salts: A mechanistic in-vitro study into factors governing oral bioavailability. J. Pharm. Sci. 2021, 111, 1667–1673. [Google Scholar] [CrossRef]
- Evrard, B.; Chiap, P.; DeTullio, P.; Ghalmi, F.; Piel, G.; Van Hees, T.; Crommen, J.; Losson, B.; Delattre, L. Oral bioavailability in sheep of albendazole from a suspension and from a solution containing hydroxypropyl-β-cyclodextrin. J. Control Release 2002, 85, 45–50. [Google Scholar] [CrossRef]
- Pacheco, P.A.; Rodrigues, L.N.C.; Ferreira, J.F.S.; Gomes, A.C.P.; Veríssimo, C.J.; Louvandini, H.; Costa, R.L.D.; Katiki, L.M. Inclusion complex and nanoclusters of cyclodextrin to increase the solubility and efficacy of albendazole. Parasitol. Res. 2018, 117, 705–712. [Google Scholar] [CrossRef]
- Meena, A.K.; Sharma, K.; Kandaswamy, M.; Rajagopal, S.; Mullangi, R. Formulation development of an albendazole self-emulsifying drug delivery system (SEDDS) with enhanced systemic exposure. Acta Pharm. 2012, 62, 563–580. [Google Scholar]
- Guillory, J.K. Handbook of Pharmaceutical Salts: Properties Selection and Use Edited by P. H. Stahl and C. G. Wermuth. Verlag Helvetica Chimica Acta/Wiley-VCH: Zurich. 2002. 374 + XII pp. £85. ISBN 3-906-390-26-8. Org. Process. Res. Dev. 2003, 7, 1277. [Google Scholar]
- Bharate, S.S. Recent developments in pharmaceutical salts: FDA approvals from 2015 to 2019. Drug Discov. Today 2021, 26, 384–398. [Google Scholar] [CrossRef] [PubMed]
- Paulekuhn, G.S.; Dressman, J.B.; Saal, C. Salt screening and characterization for poorly soluble, weak basic compounds: Case study albendazole. Pharmazie 2013, 68, 555–564. [Google Scholar] [PubMed]
- Elder, D.P.; Holm, R.; de Diego, H.L. Use of pharmaceutical salts and cocrystals to address the issue of poor solubility. Int. J. Pharm. 2013, 453, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Cabeza, A.J. Acid-base crystalline complexes and the pKa rule. Crystengcomm 2012, 14, 6362–6365. [Google Scholar] [CrossRef]
- Williams, H.D.; Trevaskis, N.L.; Charman, S.A.; Shanker, R.M.; Charman, W.N.; Pouton, C.W.; Porter, C.J.H. Strategies to address low drug solubility in discovery and development. Pharmacol. Rev. 2013, 65, 315–499. [Google Scholar]
- Gunasekaran, S.; Uthra, D. Vibrational Spectra and Qualitative Analysis of Albendazole and Mebendazole. Asian J. Chem. Int. Q. Res. J. Chem. 2008, 20, 6310–6324. [Google Scholar]
- Chadha, R.; Singh, P.; Khullar, S.; Mandal, S.K. Ciprofloxacin Hippurate Salt: Crystallization Tactics, Structural Aspects, and Biopharmaceutical Performance. Cryst. Growth Des. 2016, 16, 4960–4967. [Google Scholar] [CrossRef]
- Saladi, V.N.; Kammari, B.R.; Mandad, P.R.; Krishna, G.R.; Sajja, E.; Thirumali, R.S.; Marutapilli, A.; Mathad, V.T. Novel Pharmaceutical Cocrystal of Apalutamide, a Nonsteroidal Antiandrogen Drug: Synthesis, Crystal Structure, Dissolution, Stress, and Excipient Compatibility. Cryst. Growth Des. 2022, 22, 1130–1142. [Google Scholar] [CrossRef]
- Ray, S.; Roy, N.; Barman, B.K.; Karmakar, P.; Bomzan, P.; Rajbanshi, B.; Dakua, V.K.; Dutta, A.; Kumar, A.; Roy, M.N. Synthesis and Characterization of an Inclusion Complex of dl-Aminoglutethimide with β-Cyclodextrin and Its Innovative Application in a Biological System: Computational and Experimental Investigations. Acs Omega 2022, 7, 11208–11216. [Google Scholar] [CrossRef]
- Al-Muhtaseb, A.H.; McMinn WA, M.; Magee TR, A. Water sorption isotherms of starch powders. J. Food Eng. 2004, 61, 297–307. [Google Scholar] [CrossRef]
- de Miguel, Y.; Rohr, T.; Sherrington, D.C. Structure, Morphology, Physical Formats and Characterization of Polymer Supports. In Polymeric Materials in Organic Synthesis and Catalysis; John Wiley & Sons: Hoboken, NJ, USA, 2003; pp. 1–52. [Google Scholar]
- Ozawa, T. Thermal analysis—Review and prospect. Thermochim. Acta 2000, 355, 35–42. [Google Scholar] [CrossRef]
- Yao, X.; Kim, S.; Gui, Y.; Chen, Z.; Yu, J.; Jones, K.J.; Yu, L. Amorphous Drug-Polymer Salt with High Stability under Tropical Conditions and Fast Dissolution: The Challenging Case of Lumefantrine-PAA. J. Pharm. Sci. 2021, 110, 3670–3677. [Google Scholar] [CrossRef] [PubMed]
- Gelmboldt, V.O.; Anisimov, V.Y.; Shyshkin, I.O.; Fonari, M.S.; Kravtsov, V.C. Synthesis, crystal structures, properties and caries prevention efficiency of 2-, 3-, 4-carboxymethylpyridinium hexafluorosilicates. J. Fluor. Chem. 2018, 205, 15–21. [Google Scholar] [CrossRef]
- Berry, D.J.; Steed, J.W. Pharmaceutical cocrystals, salts and multicomponent systems; intermolecular interactions and property based design. Adv. Drug Deliv. Rev. 2017, 117, 3–24. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.X.; Carlin, A.S.; Amidon, G.L.; Hussain, A.S. Feasibility studies of utilizing disk intrinsic dissolution rate to classify drugs. Int. J. Pharm. 2004, 270, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.X.; Amidon, G.L.; Polli, J.E.; Zhao, H.; Mehta, M.U.; Conner, D.P.; Shah, V.P.; Lesko, L.J.; Chen, M.; Lee, V.H.L.; et al. Biopharmaceutics classification system: The scientific basis for biowaiver extensions. Pharm. Res. 2002, 19, 921–925. [Google Scholar] [CrossRef]
- Serajuddin, A.T.M. Salt formation to improve drug solubility. Adv. Drug Deliv. Rev. 2007, 59, 603–616. [Google Scholar] [CrossRef]
- Badawy, S.I.F.; Hussain, M.A. Microenvironmental pH Modulation in Solid Dosage Forms. J. Pharm. Sci. 2007, 96, 948–959. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | ABZ | ABZ-FMA | ABZ-DTA | ABZ-HCI |
|---|---|---|---|---|
| AUC(0–24) (ng/mL/h) | 352.44 ± 146.70 | 1205.56 ± 855.61 | 1835.37 ± 379.84 | 3089.28 ± 1228.16 |
| AUC(0–∞) (ng/mL/h) | 374.31 ± 135.98 | 1975.42 ± 1856.96 | 1955.94 ± 431.79 | 5371.96 ± 4618.85 |
| Cmax (ng/mL) | 69.61 ± 51.71 | 260.58 ± 179.73 | 322.68 ± 68.67 | 478.96 ± 138.62 |
| Tmax (hours) | 2.54 ± 1.57 | 1.67 ± 1.17 | 1.25 ± 0.61 | 1.21 ± 0.68 |
| T1/2 (hours) | 5.98 ± 3.27 | 14.07 ± 14.37 | 6.11 ± 1.21 | 16.85 ± 23.17 |
| CLZ/F (L/h/kg) | 73,576.20 ± 23,270.84 | 25,882.64 ± 19,061.91 | 13,340.60 ± 3054.71 | 7454.90 ± 4568.17 |
| MRT(0–24) (hours) | 7.07 ± 2.27 | 6.41 ± 0.66 | 6.67 ± 0.61 | 7.66 ± 2.08 |
| MRT(0–∞) (hours) | 9.19 ± 5.10 | 17.53 ± 15.90 | 8.92 ± 1.51 | 24.65 ± 33.76 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, H.; Zhong, X.; Liu, Y. Improving the Solubility, Stability, and Bioavailability of Albendazole through Synthetic Salts. Molecules 2024, 29, 3571. https://doi.org/10.3390/molecules29153571
Yan H, Zhong X, Liu Y. Improving the Solubility, Stability, and Bioavailability of Albendazole through Synthetic Salts. Molecules. 2024; 29(15):3571. https://doi.org/10.3390/molecules29153571
Chicago/Turabian StyleYan, Haiying, Xueping Zhong, and Yao Liu. 2024. "Improving the Solubility, Stability, and Bioavailability of Albendazole through Synthetic Salts" Molecules 29, no. 15: 3571. https://doi.org/10.3390/molecules29153571
APA StyleYan, H., Zhong, X., & Liu, Y. (2024). Improving the Solubility, Stability, and Bioavailability of Albendazole through Synthetic Salts. Molecules, 29(15), 3571. https://doi.org/10.3390/molecules29153571