
Inhibition of DNA Topoisomerase Ι by Flavonoids and Polyacetylenes Isolated from Bidens pilosa L.


Abstract

1. Introduction
2. Results and Discussion
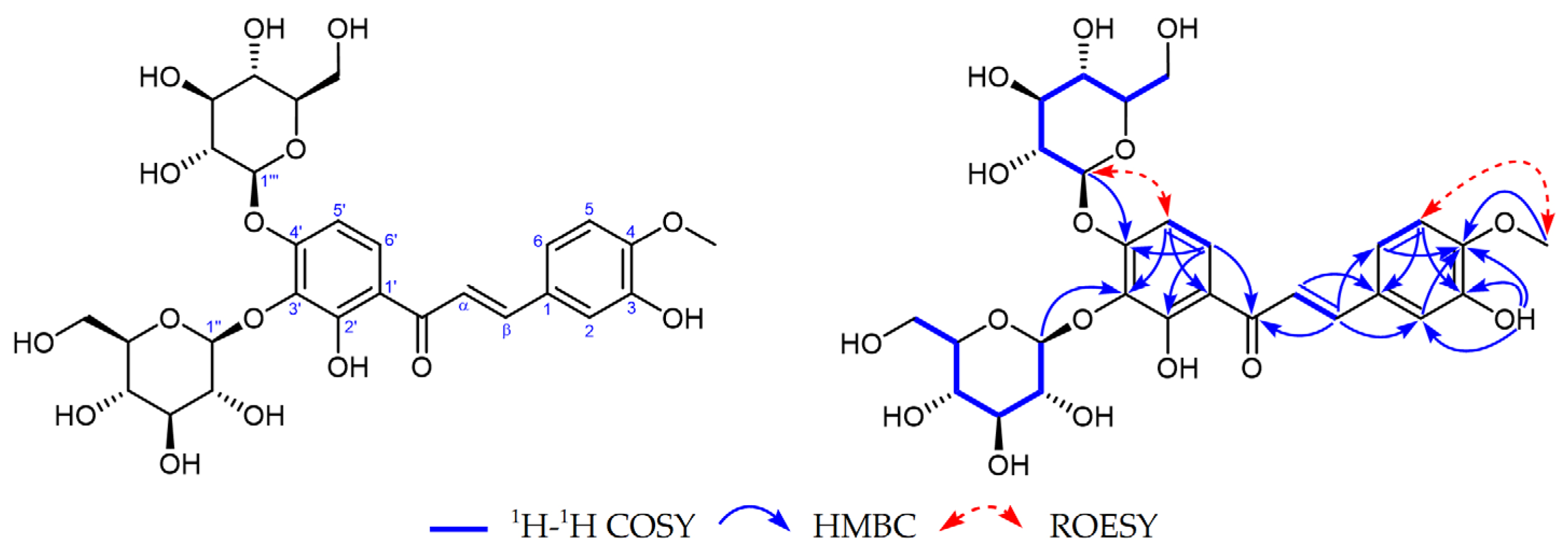
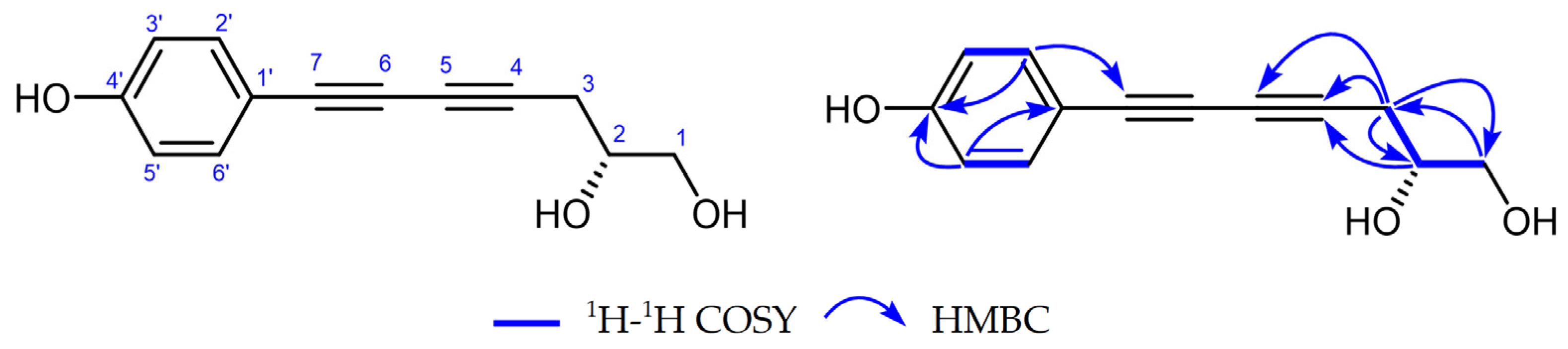
2.1. Identification of Two New Compounds
2.2. Structures of Compounds 1–29
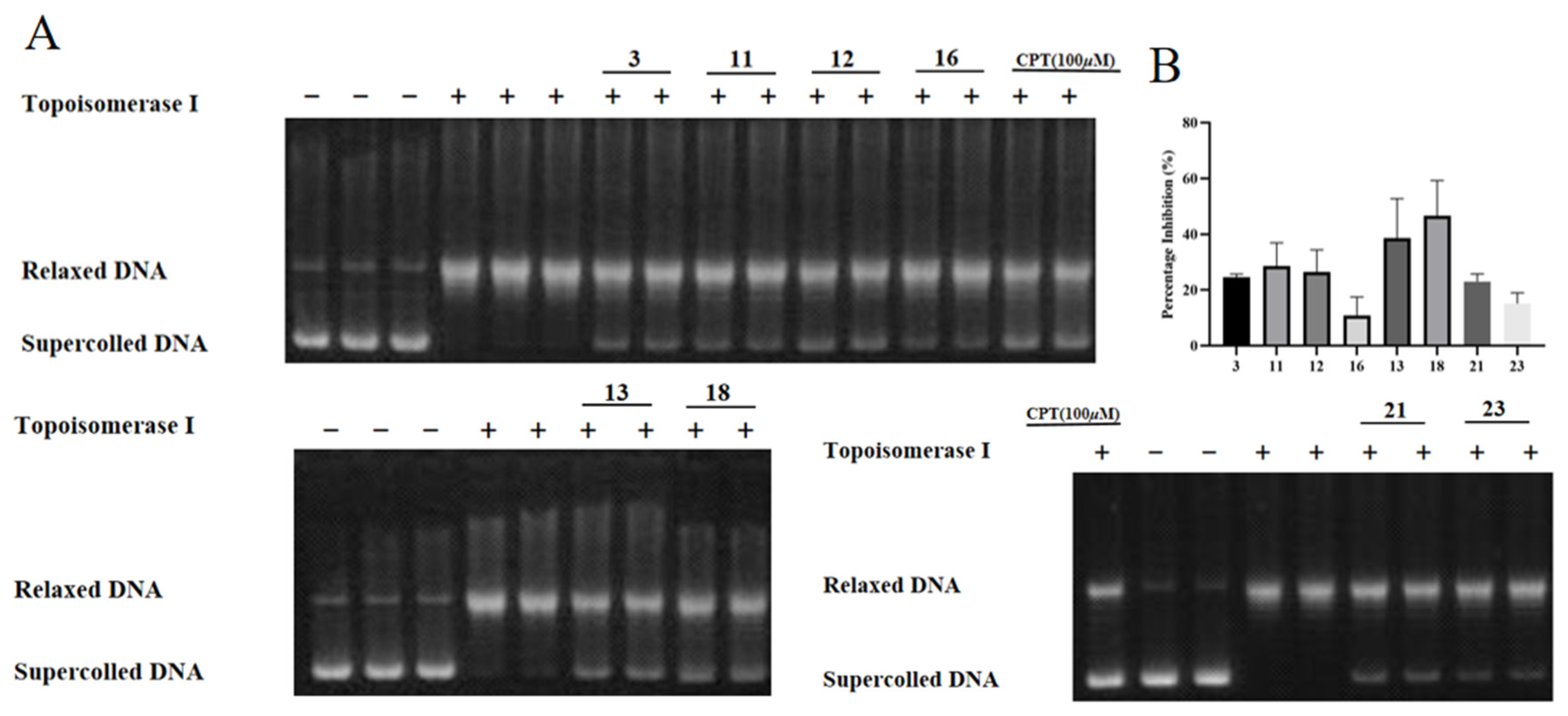
2.3. Compounds 1–29 Showed Cytotoxicities and Topo I Inhibitory Activities
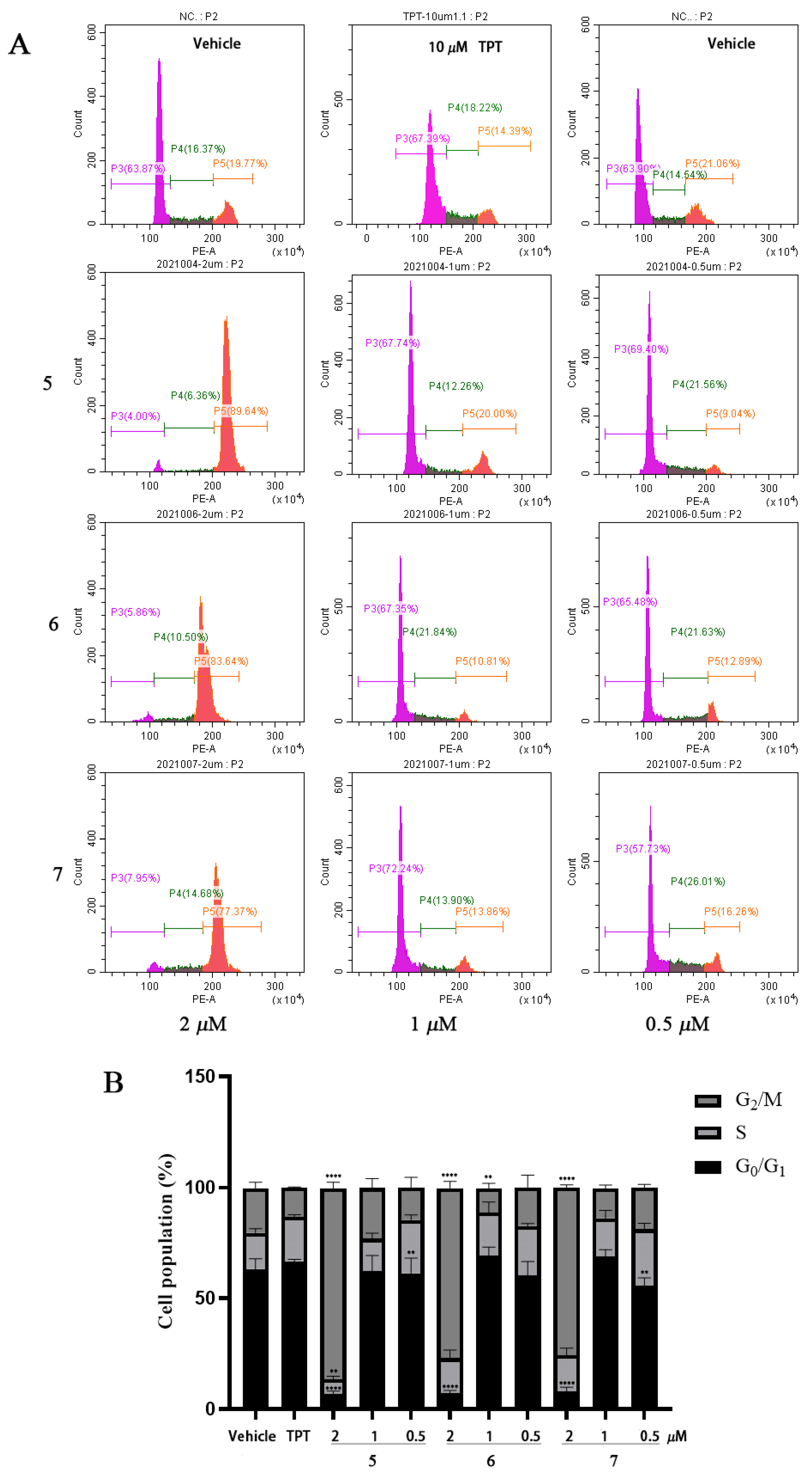
2.4. Compounds 5–7 Arrested Cell Cycle in Cancer Cells
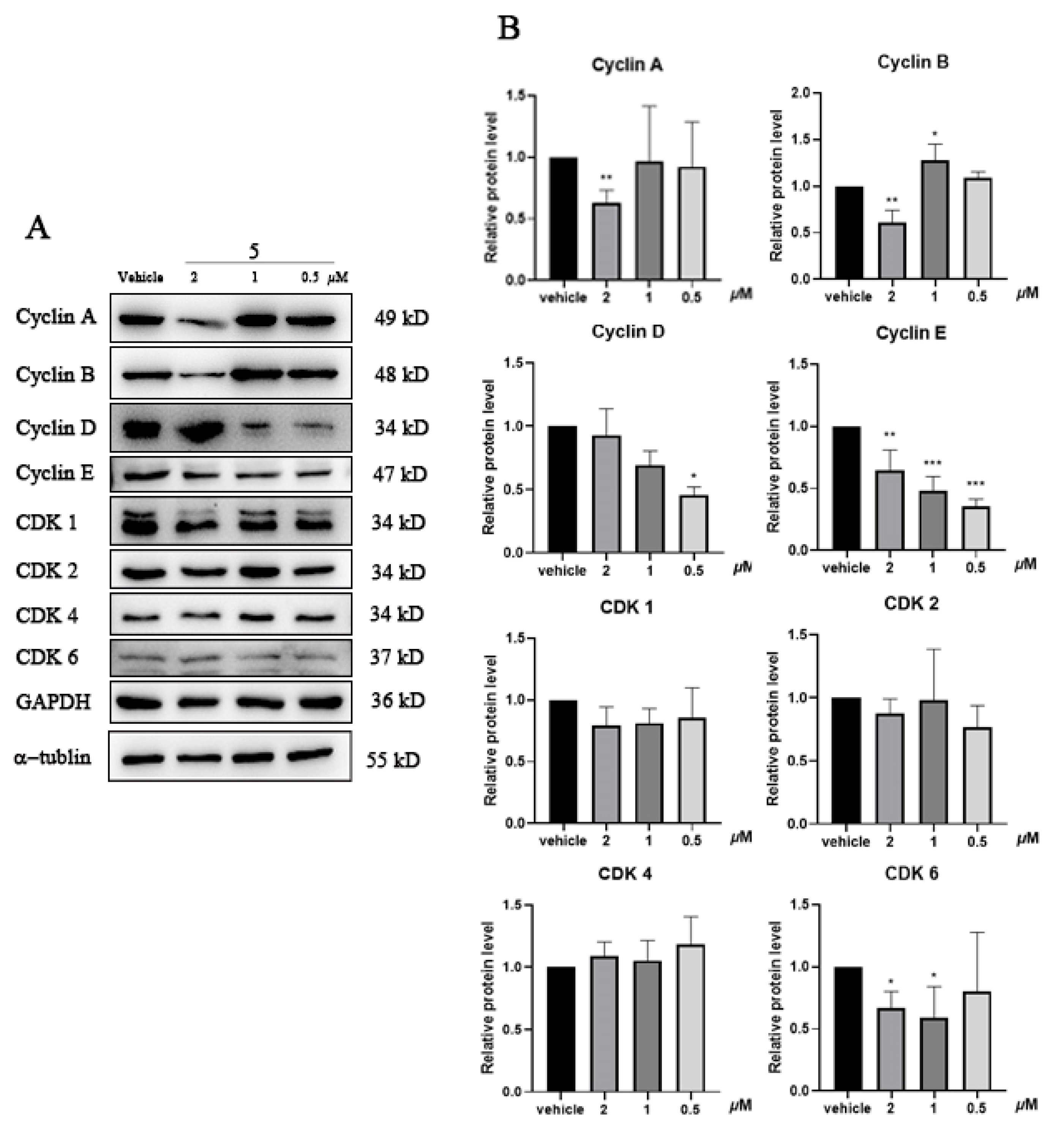
2.5. Compound 5 Regulated Cell Cycle-Related Protein Expression in Cancer Cells
3. Materials and Methods
3.1. Plant Material
3.2. General Experimental Procedures
3.3. Extraction and Isolation
3.4. Cell Culture
3.5. Cytotoxicity Assay
3.6. Topo I Inhibition Assay
3.7. DNA Content Analysis
3.8. Western Blot Analysis
3.9. Statistical Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Silva, F.L.; Fischer, D.C.; Tavares, J.F.; Silva, M.S.; de Athayde-Filho, P.F.; Barbosa-Filho, J.M. Compilation of secondary metabolites from Bidens pilosa L. Molecules 2011, 16, 1070–1102. [Google Scholar] [CrossRef] [PubMed]
- Pozharitskaya, O.N.; Shikov, A.N.; Makarova, M.N.; Kosman, V.M.; Faustova, N.M.; Tesakova, S.V.; Makarov, V.G.; Galambosi, B. Anti-inflammatory activity of a HPLC-fingerprinted aqueous infusion of aerial part of Bidens tripartita L. Phytomedicine 2010, 17, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Wu, Q.X.; Shi, Y.P. Polyacetylenes and flavonoids from the aerial parts of Bidens pilosa. Planta Med. 2010, 76, 893–896. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wan, Z.; Yi, J.; Wu, Y.; Peng, W.; Wu, J. Investigation of the extracts from Bidens pilosa Linn. var. radiata Sch. Bip. for antioxidant activities and cytotoxicity against human tumor cells. J. Nat. Med. 2013, 67, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Tobinaga, S.; Sharma, M.K.; Aalbersberg, W.G.; Watanabe, K.; Iguchi, K.; Narui, K.; Sasatsu, M.; Waki, S. Isolation and identification of a potent antimalarial and antibacterial polyacetylene from Bidens pilosa. Planta Med. 2009, 75, 624–628. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.P.; Chen, F.H.; Ling, L.; Dou, P.F.; Bo, H.; Zhong, M.M.; Xia, L.J. Protective effects of total flavonoids of Bidens pilosa L. (TFB) on animal liver injury and liver fibrosis. J. Ethnopharmacol. 2008, 116, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Kumari, P.; Misra, K.; Sisodia, B.S.; Faridi, U.; Srivastava, S.; Luqman, S.; Darokar, M.P.; Negi, A.S.; Gupta, M.M.; Singh, S.C.; et al. A promising anticancer and antimalarial component from the leaves of Bidens pilosa. Planta Med. 2009, 75, 59–61. [Google Scholar] [CrossRef] [PubMed]
- Chien, S.C.; Young, P.H.; Hsu, Y.J.; Chen, C.H.; Tien, Y.J.; Shiu, S.Y.; Li, T.H.; Yang, C.W.; Marimuthu, P.; Tsai, L.F.; et al. Anti-diabetic properties of three common Bidens pilosa variants in Taiwan. Phytochemistry 2009, 70, 1246–1254. [Google Scholar] [CrossRef]
- Delgado, J.L.; Hsieh, C.M.; Chan, N.L.; Hiasa, H. Topoisomerases as anticancer targets. Biochem. J. 2018, 475, 373–398. [Google Scholar] [CrossRef]
- Liang, X.; Wu, Q.; Luan, S.; Yin, Z.; He, C.; Yin, L.; Zou, Y.; Yuan, Z.; Li, L.; Song, X.; et al. A comprehensive review of topoisomerase inhibitors as anticancer agents in the past decade. Eur. J. Med. Chem. 2019, 171, 129–168. [Google Scholar] [CrossRef]
- Gu, L.; Hickey, R.J.; Malkas, L.H. Therapeutic Targeting of DNA Replication Stress in Cancer. Genes 2023, 14, 1346. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Pandey, V.P.; Yadav, K.; Yadav, A.; Dwivedi, U.N. Natural Products as Anti-Cancerous Therapeutic Molecules Targeted towards Topoisomerases. Curr. Protein Pept. Sci. 2020, 21, 1103–1142. [Google Scholar] [CrossRef] [PubMed]
- Jain, C.K.; Majumder, H.K.; Roychoudhury, S. Natural Compounds as Anticancer Agents Targeting DNA Topoisomerases. Curr. Genom. 2017, 18, 75–92. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.Y.; Ma, Q.T.; Wu, Y.H.; Duan, W.L.; Zeng, G.Z.; Yin, J.L. Chemical constituents of the whole plant of Bidens pilosa L. J. Yunnan Minzu Univ. Nat. Sci. Ed. 2022, 31, 627–632. [Google Scholar]
- Redl, K.; Davis, B.; Bauer, R. Chalcone glycosides from Bidens campylotheca. Phytochemistry 1992, 32, 218–220. [Google Scholar] [CrossRef]
- Cui, S.; Zou, Y.; Wu, Y.; Gao, P. Synthesis and Absolute Configuration of the 7-Phenylhepta-4,6-diyne-1,2-diol Isolated from Bidens pilosa. Synthesis 2012, 44, 3108. [Google Scholar]
- Li, C.H.; Zhu, Y.J.; Yu, S.H.; Jiang, H.Q.; Zhou, H.L. A new polyacetylene and a new isonicotinic acid glucoside from Bidens parviflora Willd. Acta Pharm. Sin. 2020, 55, 489–494. [Google Scholar]
- Tian, X.; Zhou, S.X.; Wei, H.L.; Hu, N.; Dai, Z.; Liu, Z.G.; Han, Z.Z.; Tu, P.F. Flavonoids from the herb of Bidens pilosa L. J. Chin. Pharm. Sci. 2011, 20, 518–522. [Google Scholar] [CrossRef]
- Liu, J.; Chen, R.; Nie, Y.; Feng, L.; Li, H.D.; Liang, J.Y. A new carbamate with cytotoxic activity from the aerial parts of Siegesbeckia pubecens. Chin. J. Nat. Med. 2012, 10, 13–15. [Google Scholar] [CrossRef]
- Glasl, S.; Mucaji, P.; Werner, I.; Presser, A.; Jurenitsch, J. Sesquiterpenes and flavonoid aglycones from a Hungarian taxon of the Achillea millefolium group. Z. Naturforsch C J. Biosci. 2002, 57, 976–982. [Google Scholar] [CrossRef]
- Yuan, K.; Xu, R.; Chen, C. A new flavonol glycoside from Tridax procumbens Linn. J. Chem. Res. 2009, 2009, 165–166. [Google Scholar] [CrossRef]
- Lee, H.J.; Sim, M.O.; Woo, K.W.; Jeong, D.E.; Jung, H.K.; An, B.; Cho, H.W. Antioxidant and Antimelanogenic Activities of Compounds Isolated from the Aerial Parts of Achillea alpina L. Chem. Biodivers. 2019, 16, e1900033. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.J.; Zhu, J.X.; Zhu, Y.; Yan, L.; Jin, H.Z.; Zhang, W.D. Flavonoids from the Aerial Parts of Inula japonica. Chin. J. Nat. Med. 2010, 8, 257–259. [Google Scholar] [CrossRef]
- Xu, R.; Zhang, J.; Yuan, K. Two new flavones from Tridax procumbens Linn. Molecules 2010, 15, 6357–6364. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.W.; Hang, M.Z.; Zhao, W.Q.; Jin, Y.S.; Chen, H.S. Studies on Chemical Constituents of Bidens bipinnata. Pharm. J. Chin. People’s Lib. Army 2009, 25, 283–286. [Google Scholar]
- Kim, G.R.; Kim, E.N.; Jeong, G.S. Simultaneous quantitative analysis of flavonoids isolated from the leaves of Diospyros kaki. Korean J. Pharmacogn. 2020, 51, 139–145. [Google Scholar]
- Shafiq, N.; Riaz, N.; Saleem, M. Isolation, characterization of flavonoids from Seriphidium oliverianum and their antioxidant and anti-urease activities. J. Chem. Soc. Pak. 2014, 36, 517–523. [Google Scholar]
- Laraoui, H.; Long, C.; Haba, H.; Benkhaled, M. New methylated flavonol glucosides from Fumana montana Pomel. Nat. Prod. Res. 2013, 27, 1770–1775. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Y.; Chen, G.R.; Deng, Z.Y.; Zhao, J.; Ge, J.F.; Li, N.; Chen, F.H. Chemical constituents from Bidens bipinnata. China J. Chin. Mater. Med. 2014, 39, 1838–1844. [Google Scholar]
- Li, Y.L.; Li, J.; Wang, N.L.; Yao, X.S. Flavonoids and a new polyacetylene from Bidens parviflora Willd. Molecules 2008, 13, 1931–1941. [Google Scholar] [CrossRef]
- Cai, F.J.; Li, C.H.; Sun, X.H.; Wang, L.; Tian, J.L.; Zhao, W.; Kong, D.G.; Liu, Q.; Zhou, H.L. A new dihydroflavone and a new polyacetylene glucoside from Bidens parviflora. J. Asian Nat. Prod. Res. 2022, 24, 963–970. [Google Scholar] [CrossRef]
- Deng, K.Z.; Liu, C.; Chang, M.Y.; Zong, Q.; Xiong, Y. Isolation and Identification of Phenols Compounds in Aurantii Fructus Immaturus. China Pharm. 2020, 31, 1040–1043. [Google Scholar]
- Li, P.; Wang, Y.L.; Tang, L.; Guo, Z.Y.; Long, C.L. Studies on flavonoids from Bidens pilosa. Chin. Tradit. Herb. Drugs 2023, 44, 2498–2501. [Google Scholar]
- Okuyama, T.; Li, S.; Kuang, H.X.; Okada, Y. A New Aurone Glucoside and a New Chalcone Glucoside from Bidens bipinnata Linne. Heterocycles 2003, 61, 557–561. [Google Scholar] [CrossRef]
- Chang, M.H.; Wang, G.J.; Kuo, Y.H.; Lee, C.K. The Low Polar Constituents from Bidens pilosa L. var. Minor (Blume) Sherff. J. Chin. Chem. Soc. 2013, 47, 1131–1136. [Google Scholar] [CrossRef]
- Kimura, Y.; Hiraoka, K.; Kawano, T.; Fujioka, S.; Shimada, A. Nematicidal Activities of Acetylene Compounds from Coreopsis lanceolata L. Z. Naturforschung C 2015, 63, 1121–1124. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.D.; Chen, Z.K.; Xie, F.D.; Hu, J.J.; Liang, H.J.; Li, Q.R.; Yuan, J. Studies on Isolation of Polyacetylenes from Bidens pilosa var.radiata and their biological activities. J. Trop. Subtrop. Bot. 2021, 29, 216–220. [Google Scholar]
- Zhao, A.H.; Zhao, Q.S.; Peng, L.Y.; Zhang, J.X.; Lin, Z.W.; Sun, H.D. A New Chalcone Glycoside from Bidens pilosa. Acta Bot. Yunnanica 2004, 26, 121–126. [Google Scholar]
- Rücker, G.; Kehrbaum, S.; Sakulas, H.; Lawong, B.; Goeltenboth, F. Acetylenic glucosides from Microglossa pyrifolia. Planta Med. 1992, 58, 266–269. [Google Scholar] [CrossRef]
- Chiang, Y.M.; Chang, C.L.; Chang, S.L.; Yang, W.C.; Shyur, L.F. Cytopiloyne, a novel polyacetylenic glucoside from Bidens pilosa, functions as a T helper cell modulator. J. Ethnopharmacol. 2007, 110, 532–538. [Google Scholar] [CrossRef]
- Xi, F.M.; Li, C.T.; Han, J.; Yu, S.S.; Wu, Z.J.; Chen, W.S. Thiophenes, polyacetylenes and terpenes from the aerial parts of Eclipata prostrate. Bioorganic Med. Chem. 2014, 22, 6515–6522. [Google Scholar] [CrossRef]
- Lee, S.H.; Kim, J.G.; Le TP, L.; Han, J.S.; Cho, Y.B.; Lee, M.K.; Lee, D.; Hwang, B.Y. Polyacetylenes from the roots of Cirsium japonicum var. ussuriense. Phytochemistry 2022, 202, 113319. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Liu, Z.; He, Z.; Yi, J.; Qiao, X.; Tan, C.; Xing, Y.; Zeng, Y.; Yang, D.; Yin, J.; et al. Cantharidin analogue alleviates dextran sulfate sodium-induced colitis in mice by inhibiting the activation of NF-κB signaling. Eur. J. Med. Chem. 2023, 260, 115731. [Google Scholar] [CrossRef] [PubMed]
- Scotti, L.; Bezerra Mendonca, F.J.; Ribeiro, F.F.; Tavares, J.F.; da Silva, M.S.; Barbosa Filho, J.M.; Scotti, M.T. Natural Product Inhibitors of Topoisomerases: Review and Docking Study. Curr. Protein Pept. Sci. 2018, 19, 275–291. [Google Scholar] [CrossRef]
- Yoon, G.; Kang, B.Y.; Cheon, S.H. Topoisomerase I inhibition and cytotoxicity of licochalcones A and E from Glycyrrhiza inflata. Arch. Pharm. Res. 2007, 30, 313–316. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, A.R.; Sharma, S.; Mandal, S.; Goswami, A.; Mukhopadhyay, S.; Majumder, H.K. Luteolin, an emerging anti-cancer flavonoid, poisons eukaryotic DNA topoisomerase I. Biochem. J. 2002, 366, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Constantinou, A.; Mehta, R.; Runyan, C.; Rao, K.; Vaughan, A.; Moon, R. Flavonoids as DNA topoisomerase antagonists and poisons: Structure-activity relationships. J. Nat. Prod. 1995, 58, 217–225. [Google Scholar] [CrossRef]
- Müller, L.; Schütte, L.R.F.; Bücksteeg, D.; Alfke, J.; Uebel, T.; Esselen, M. Topoisomerase poisoning by the flavonoid nevadensin triggers DNA damage and apoptosis in human colon carcinoma HT29 cells. Arch. Toxicol. 2021, 95, 3787–3802. [Google Scholar] [CrossRef]
- Warren, N.J.H.; Eastman, A. Comparison of the different mechanisms of cytotoxicity induced by checkpoint kinase I inhibitors when used as single agents or in combination with DNA damage. Oncogene 2020, 39, 1389–1401. [Google Scholar] [CrossRef]
- Singh, B.N.; Mudgil, Y.; John, R.; Achary, V.M.; Tripathy, M.K.; Sopory, S.K.; Reddy, M.K.; Kaul, T. Cell cycle stage-specific differential expression of topoisomerase I in tobacco BY-2 cells and its ectopic overexpression and knockdown unravels its crucial role in plant morphogenesis and development. Plant Sci. 2015, 240, 182–192. [Google Scholar] [CrossRef]
- Johnson, D.G.; Walker, C.L. Cyclins and cell cycle checkpoints. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 295–312. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 13C NMR (δ ppm) | 1H NMR (δ ppm) | No. | 13C NMR (δ ppm) | 1H NMR (δ ppm) |
|---|---|---|---|---|---|
| C=O | 192.1 | 1″ | 103.8 | 4.85 (1H, d, J = 7.6) | |
| α | 119.3 | 7.77 (1H, d, J = 15.4) | 2″ | 74.1 | 3.29 (1H, overlapped) |
| β | 144.9 | 7.70 (1H, d, J = 15.4) | 3″ | 76.3 | 3.22 (1H, overlapped) |
| 1 | 127.5 | - | 4″ | 69.7 | 3.13 (1H, overlapped) |
| 2 | 115.1 | 7.34 (1H, d, J = 1.9) | 5″ | 77.2 | 3.10 (1H, overlapped) |
| 3 | 146.7 | - | 6″ | 60.8 | 3.44 (1H, overlapped) 3.62 (1H, m) |
| 4 | 150.6 | - | 1‴ | 100.7 | 4.98 (1H, d, J = 7.4) |
| 5 | 112.0 | 7.01 (1H, d, J = 8.4) | 2‴ | 73.4 | 3.35 (1H, overlapped) |
| 6 | 122.6 | 7.32 (1H, dd, J = 8.4, 1.9) | 3‴ | 76.0 | 3.30 (1H, overlapped) |
| 1′ | 116.8 | - | 4‴ | 69.8 | 3.17 (1H, overlapped) |
| 2′ | 156.5 | - | 5‴ | 77.4 | 3.40 (1H, overlapped) |
| 3′ | 133.6 | - | 6‴ | 60.7 | 3.47 (1H, overlapped) 3.71 (1H, m) |
| 4′ | 155.6 | - | 4-OCH3 | 55.8 | 3.84 (3H, s) |
| 5′ | 106.9 | 6.84 (1H, d, J = 9.3) | 3-OH | 9.25 (1H, s) | |
| 6′ | 127.1 | 8.02 (1H, d, J = 9.3) | 2′-OH | 12.90 (1H, br s) |
| No. | 13C NMR (δ ppm) | 1H NMR (δ ppm) | No. | 13C NMR (δ ppm) | 1H NMR (δ ppm) |
|---|---|---|---|---|---|
| 1 | 66.0 | 3.54 (1H, dd, J = 11.2, 5.8) 3.59 (1H, dd, J = 11.2, 4.9) | 1′ | 113.5 | - |
| 2 | 71.7 | 3.77 (1H, dddd, J = 6.2, 6.0, 5.8, 4.9) | 2′, 6′ | 135.1 | 7.28 (2H, d, J = 8.6) |
| 3 | 25.2 | 2.50 (1H, dd, J = 17.3, 6.2) 2.60 (1H, dd, J = 17.3, 6.0) | 3′, 5′ | 116.6 | 6.72 (2H, d, J = 8.6) |
| 4 | 80.8 | - | 4′ | 160.0 | - |
| 5 | 67.6 | - | |||
| 6 | 73.2 | - | |||
| 7 | 76.2 |
| Comp. | IC50 (μM) | ||||||
|---|---|---|---|---|---|---|---|
| Cytotoxicity | Topo I | ||||||
| A549 | HCT116 | MDA-MB-231 | HepG2 | DLD-1 | HL-7702 | ||
| 1 | >200 | >200 | >200 | >200 | >200 | >200 | 393.50 ± 38.99 |
| 2 | >200 | >200 | >200 | >200 | >200 | >200 | 328.98 ± 58.77 |
| 3 | 135.56 ± 25.04 | 177.96 ± 6.11 | 166.71 ± 5.63 | >200 | 117.49 ± 11.60 | >200 | >400 |
| 4 | 8.16 ± 4.04 | 4.52 ± 0.04 | >200 | 3.05 ± 1.31 | 7.14 ± 0.35 | 19.61 ± 0.62 | >400 |
| 5 | 2.16 ± 0.30 | 0.79 ± 0.13 | 39.67 ± 4.58 | 40.3 ± 11.14 | 0.60 ± 0.05 | 14.36 ± 0.45 | 145.57 ± 7.88 |
| 6 | 3.09 ± 0.28 | 0.59 ± 0.10 | >200 | 34.59 ± 8.01 | 1.58 ± 0.07 | 23.91 ± 5.73 | 239.27 ± 31.35 |
| 7 | 0.86 ± 0.03 | 0.43 ± 0.03 | 81.57 ± 7.43 | 28.38 ± 13.05 | 0.85 ± 0.02 | 11.44 ± 4.42 | 224.38 ± 27.18 |
| 8 | 177.84 ± 4.38 | >200 | >200 | >200 | 186.79 ± 48.65 | >200 | 189.84 ± 22.09 |
| 9 | 135.31 ± 32.29 | 87.68 ± 15.27 | >200 | >200 | 50.50 ± 0.31 | 125.54 ± 7.08 | >400 |
| 10 | >200 | 59.29 ± 4.56 | >200 | >200 | 63.00 ± 4.08 | 114.75 ± 0.55 | >400 |
| 11 | >200 | >200 | >200 | >200 | >200 | >200 | >400 |
| 12 | >200 | >200 | >200 | >200 | >200 | >200 | >400 |
| 13 | >200 | 139.10 ± 11.31 | >200 | >200 | 178.68 ± 5.94 | >200 | >400 |
| 14 | >200 | >200 | >200 | >200 | >200 | >200 | >400 |
| 15 | 93.92 ± 2.92 | 31.70 ± 1.54 | 94.91 ± 0.50 | 174.80 ± 16.64 | 69.17 ± 4.04 | >200 | 89.91 ± 28.08 |
| 16 | >200 | >200 | >200 | >200 | >200 | >200 | >400 |
| 17 | >200 | >200 | >200 | >200 | >200 | >200 | >400 |
| 18 | >200 | >200 | >200 | >200 | >200 | >200 | >400 |
| 19 | >200 | >200 | >200 | >200 | >200 | >200 | 47.50 ± 15.3 |
| 20 | 194.01 ± 12.77 | 173.81 ± 2.47 | >200 | >200 | >200 | >200 | 301.32 ± 34.94 |
| 21 | 186.68 ± 6.63 | >200 | >200 | >200 | >200 | >200 | >400 |
| 22 | 124.84 ± 23.42 | 117.63 ± 5.70 | 146.00 ± 7.17 | 178.53 ± 3.62 | 141.56 ± 10.15 | 160.56 ± 2.18 | 178.03 ± 45.72 |
| 23 | 129.78 ± 4.93 | 70.91 ± 3.73 | 139.89 ± 5.53 | 167.79 ± 10.78 | 122.54 ± 8.76 | 198.81 ± 0.86 | >400 |
| 24 | >200 | >200 | >200 | >200 | >200 | >200 | 218.27 ± 50.82 |
| 25 | 173.81 ± 12.3 | 162.38 ± 15.77 | >200 | >200 | 187.21 ± 17.4 | >200 | >400 |
| 26 | 187.84 ± 15.83 | >200 | >200 | >200 | >200 | >200 | >400 |
| 27 | 187.87 ± 14.88 | 112.23 ± 6.59 | 164.29 ± 11.74 | >200 | 144.41 ± 7.36 | >200 | >400 |
| 28 | 190.39 ± 12.09 | >200 | >200 | >200 | >200 | >200 | >400 |
| 29 | >200 | >200 | >200 | >200 | >200 | >200 | >400 |
| a CPT | 4.41 ± 1.11 | 12.07 ± 5.74 | 20.41 ± 3.70 | 1.71 ± 0.47 | 16.96 ± 3.82 | 2.11 + 2.05 | 11.11 ± 3.43 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, G.; Wang, Y.; Zhu, M.; Yi, J.; Ma, J.; Yang, B.; Sun, W.; Dai, F.; Yin, J.; Zeng, G. Inhibition of DNA Topoisomerase Ι by Flavonoids and Polyacetylenes Isolated from Bidens pilosa L. Molecules 2024, 29, 3547. https://doi.org/10.3390/molecules29153547
Zeng G, Wang Y, Zhu M, Yi J, Ma J, Yang B, Sun W, Dai F, Yin J, Zeng G. Inhibition of DNA Topoisomerase Ι by Flavonoids and Polyacetylenes Isolated from Bidens pilosa L. Molecules. 2024; 29(15):3547. https://doi.org/10.3390/molecules29153547
Chicago/Turabian StyleZeng, Guiyuan, Yinyue Wang, Meihua Zhu, Jumei Yi, Junjie Ma, Bijuan Yang, Weiqing Sun, Fang Dai, Junlin Yin, and Guangzhi Zeng. 2024. "Inhibition of DNA Topoisomerase Ι by Flavonoids and Polyacetylenes Isolated from Bidens pilosa L." Molecules 29, no. 15: 3547. https://doi.org/10.3390/molecules29153547
APA StyleZeng, G., Wang, Y., Zhu, M., Yi, J., Ma, J., Yang, B., Sun, W., Dai, F., Yin, J., & Zeng, G. (2024). Inhibition of DNA Topoisomerase Ι by Flavonoids and Polyacetylenes Isolated from Bidens pilosa L. Molecules, 29(15), 3547. https://doi.org/10.3390/molecules29153547