
Pallado-Catalyzed Cascade Synthesis of 2-Alkoxyquinolines from 1,3-Butadiynamides


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. General Procedure for the Synthesis of 2-Alkoxyquinolines 6a–b, f, j (Condition A)
3.2. General Procedure for the Synthesis of 2-Alkoxyquinolines 6c–e, g-i and 7a–b (Condition B)
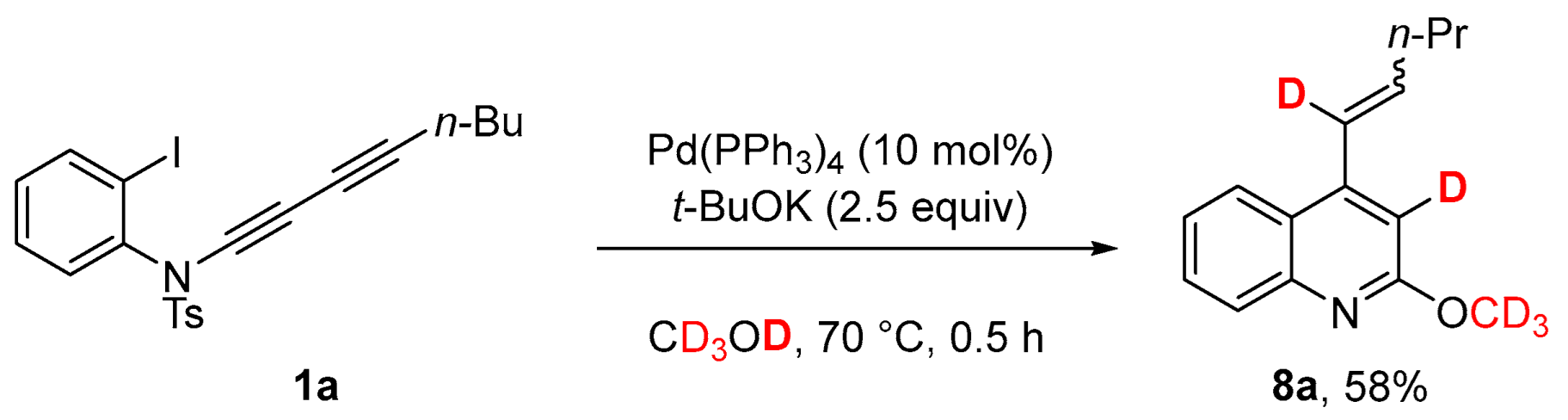
3.3. Deuterium Labeling Experiments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Saxena, A.; Majee, S.; Ray, D.; Saha, B. Inhibition of Cancer Cells by Quinoline-Based Compounds: A Review with Mechanistic Insights. Bioorg. Med. Chem. 2024, 103, 117681. [Google Scholar] [CrossRef]
- Zhao, Y.-Q.; Li, X.; Guo, H.-Y.; Shen, Q.-K.; Quan, Z.-S.; Luan, T. Application of Quinoline Ring in Structural Modification of Natural Products. Molecules 2023, 28, 6478. [Google Scholar] [CrossRef]
- Michael, J.P. Quinoline, Quinazoline and Acridone Alkaloids. Nat. Prod. Rep. 2008, 25, 166–187. [Google Scholar] [CrossRef]
- Gorka, A.P.; de Dios, A.; Roepe, P.D. Quinoline Drug–Heme Interactions and Implications for Antimalarial Cytostatic versus Cytocidal Activities. J. Med. Chem. 2013, 56, 5231–5246. [Google Scholar] [CrossRef]
- Negi, A.; Perveen, S.; Gupta, R.; Singh, P.P.; Sharma, R. Unraveling Dilemmas and Lacunae in the Escalating Drug Resistance of Mycobacterium tuberculosis to Bedaquiline, Delamanid, and Pretomanid. J. Med. Chem. 2024, 67, 2264–2286. [Google Scholar] [CrossRef]
- Kusuma, B.R.; Khandelwal, A.; Gu, W.; Brown, D.; Liu, W.; Vielhauer, G.; Holzbeierlein, J.; Blagg, B.S.J. Synthesis and Biological Evaluation of Coumarin Replacements of Novobiocin as Hsp90 Inhibitors. Bioorg. Med. Chem. 2014, 22, 1441–1449. [Google Scholar] [CrossRef]
- Hayashi, H.; Miwa, Y.; Miki, I.; Ichikawa, S.; Yoda, N.; Ishii, A.; Kono, M.; Suzuki, F. 5-HT3 Receptors Antagonists. 1. New Quinoline Derivatives. J. Med. Chem. 1992, 35, 4893–4902. [Google Scholar] [CrossRef]
- Kalajian, T.A.; Cannella, J.A.; Vasudevan, A.; Mizelle, J.; Rendon, L.F.; Nozari, A.; Ortega, R. An Overview of Local Anesthetics in Over-the counter Products. Pain Pract. 2024, 24, 364–373. [Google Scholar] [CrossRef]
- Abel, U.; Schlüter, T.; Schulz, A.; Hambruch, E.; Steeneck, C.; Hornberger, M.; Hoffmann, T.; Perovic-Ottstadt, S.; Kinzel, O.; Burnet, M.; et al. Synthesis and Pharmacological Validation of a Novel Series of Non-steroidal FXR Agonists. Bioorg. Med. Chem. Lett. 2010, 20, 4911–4917. [Google Scholar] [CrossRef]
- Kidwai, M.; Sapra, P.; Misra, P.; Saxena, R.K.; Singh, M. Microwave Assisted Solid Support Synthesis of Novel 1,2,4-Triazolo [3,4-b]-1,3,4-thiadiazepines as Potent Antimicrobial Agents. Bioorg. Med. Chem. 2001, 9, 217–220. [Google Scholar] [CrossRef]
- Wasa, M.; Chan, K.S.L.; Zhang, X.-G.; He, J.; Miura, M.; Yu, J.-Q. Ligand-Enabled Methylene C(sp3)-H Bond Activation with a Pd(II) Catalyst. J. Am. Chem. Soc. 2012, 134, 18570–18572. [Google Scholar] [CrossRef]
- He, J.; Li, S.; Deng, Y.; Fu, H.; Laforteza, B.N.; Spangler, J.E.; Homs, A.; Yu, J.-Q. Ligand-Controlled C(sp3)-H Arylation and Olefination in Synthesis of Unnatural Chiral α-Amino Acids. Science 2014, 343, 1216–1220. [Google Scholar] [CrossRef]
- Jia, W.-L.; Fernandez-Ibanez, M.A. Ligand-Enabled γ-C(sp3)-H Acetoxylation of Triflyl-Protected Amines. Eur. J. Org. Chem. 2018, 2018, 6088–6091. [Google Scholar] [CrossRef]
- Broch, S.; Aboab, B.; Anizon, F.; Moreau, P. Synthesis and In Vitro Antiproliferative Activities of Quinoline Derivatives. Eur. J. Med. Chem. 2010, 45, 1657–1662. [Google Scholar] [CrossRef]
- Cherng, Y.-J. Efficient Nucleophilic Substitution Reactions of Quinolyl and Isoquinolyl Halides with Nucleophiles under Focused Microwave Irradiation. Tetrahedron 2002, 58, 1125–1129. [Google Scholar] [CrossRef]
- Morel, A.F.; Larghi, E.L.; Selvero, M.M. Mild, Efficient and Selective Silver Carbonate Mediated O-Alkylation of 4-Hydroxy-2-quinolones: Synthesis of 2,4-Dialkoxyquinolines. Synlett 2005, 2005, 2755–2758. [Google Scholar] [CrossRef]
- Lu, Z.; Li, Y.; Ru, Y.; Yang, S.; Hao, C.; Zuo, M.; Jiao, R.; Wu, W.; Zhou, Y.; Yao, H.; et al. Coordination Effect Enabled Palladium-Catalyzed Regioselective O-Alkylation of 2-Pyridones. Chem. Commun. 2022, 58, 1215–1218. [Google Scholar] [CrossRef]
- Cadena, M.; Villatoro, R.S.; Gupta, J.S.; Phillips, C.; Allen, J.B.; Arman, H.D.; Wherritt, D.J.; Clanton, N.A.; Ruchelman, A.L.; Simmons, E.M.; et al. Pd-Catalyzed Chemoselective O-Benzylation of Ambident 2-Quinolone Nucleophiles. ACS Catal. 2022, 12, 10199–10206. [Google Scholar] [CrossRef]
- Silva, V.L.M.; Silva, A.M.S. Palladium-Catalyzed Synthesis and Transformation of Quinolones. Molecules 2019, 24, 228. [Google Scholar] [CrossRef]
- Hong, W.P.; Shin, I.; Lim, H.N. Recent Advances in One-Pot Modular Synthesis of 2-Quinolones. Molecules 2020, 25, 5450. [Google Scholar] [CrossRef]
- Nedolya, N.A.; Brandsma, L.; Trofimov, B.A. An Efficient Synthesis of 2-Trimethylsilyloxy-4-neopentyl quinoline via 3-Lithio-1-tert-butylallene and Phenyl Isocyanate. Mendeleev Commun. 1997, 7, 92. [Google Scholar] [CrossRef]
- Huang, Y.-N.; Li, Y.-L.; Li, J.; Deng, J. Beyond a Protecting Reagent: DMAP-Catalyzed Cyclization of Boc-Anhydride with 2-Alkenylanilines. J. Org. Chem. 2016, 81, 4645–4653. [Google Scholar] [CrossRef]
- Witulski, B.; Stengel, T. N-Functionalized 1-Alkynylamides: New Building Blocks for Transition Metal Mediated Inter- and Intramolecular [2+2+1] Cycloadditions. Angew. Chem. Int. Ed. 1998, 37, 489–492. [Google Scholar] [CrossRef]
- Iftikhar, R.; Mazhar, A.; Iqbal, M.S.; Khan, F.Z.; Askary, S.H.; Sibtain, H. Ring Forming Transformation of Ynamides via Cycloaddition. RSC. Adv. 2023, 13, 10715–10756. [Google Scholar] [CrossRef]
- Hu, Y.-C.; Zhao, Y.; Wan, B.; Chen, Q.-A. Reactivity of Ynamides in Catalytic Intermolecular Annulations. Chem. Soc. Rev. 2021, 50, 2582–2625. [Google Scholar] [CrossRef]
- Wang, X.-N.; Yeom, H.-S.; Fang, L.-C.; He, S.; Ma, Z.-X.; Kedrowski, B.L.; Hsung, R.P. Ynamides in Ring Forming Transformations. Acc. Chem. Res. 2014, 47, 560–578. [Google Scholar] [CrossRef]
- Evano, G.; Coste, A.; Jouvin, K. Ynamides: Versatile Tools in Organic Synthesis. Angew. Chem. In. Ed. 2010, 49, 2840–2859. [Google Scholar] [CrossRef]
- DeKorver, K.A.; Li, H.; Lohse, A.G.; Hayashi, R.; Lu, Z.; Zhang, Y.; Hsung, R.P. Ynamides: A Modern Functional Group for the New Millennium. Chem. Rev. 2010, 110, 5064–5106. [Google Scholar] [CrossRef]
- Lenko, I.; Alayrac, C.; Bożek, I.; Witulski, B. 1,3-Butadiynamides the Ethynylogous Ynamides: Synthesis, Properties and Applications in Heterocyclic Chemistry. Molecules 2023, 28, 4564. [Google Scholar] [CrossRef]
- Talbi, I.; Alayrac, C.; Lohier, J.-F.; Touil, S.; Witulski, B. Application of Ynamides in the Synthesis of 2-(Tosylamido)- and 2,5-Bis(tosylamido)thiophenes. Org. Lett. 2016, 18, 2656–2659. [Google Scholar] [CrossRef]
- Witulski, B.; Schweikert, T.; Schollmeyer, D.; Nemkovich, N.A. Synthesis and Molecular Properties of Donor-π-Spacer-Acceptor Ynamides with up to Four Conjugated Alkyne Units. Chem. Commun. 2010, 46, 2953–2955. [Google Scholar] [CrossRef]
- Lenko, I.; Mamontov, A.; Alayrac, C.; Legay, R.; Witulski, B. Media-Driven Pd-Catalyzed Reaction Cascades with 1,3-Diynamides Leading Selectively to Either Indoles or Quinolines. Angew. Chem. Int. Ed. 2021, 60, 22729–22734. [Google Scholar] [CrossRef]
- Witulski, B.; Alayrac, C.; Tevzadze-Saeftel, L. Palladium-Catalyzed Synthesis of 2-Aminoindoles by a Heteroanulation Reaction. Angew. Chem. Int. Ed. 2003, 42, 4257–4260. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lenko, I.; Mamontov, A.; Alayrac, C.; Witulski, B. Pallado-Catalyzed Cascade Synthesis of 2-Alkoxyquinolines from 1,3-Butadiynamides. Molecules 2024, 29, 3505. https://doi.org/10.3390/molecules29153505
Lenko I, Mamontov A, Alayrac C, Witulski B. Pallado-Catalyzed Cascade Synthesis of 2-Alkoxyquinolines from 1,3-Butadiynamides. Molecules. 2024; 29(15):3505. https://doi.org/10.3390/molecules29153505
Chicago/Turabian StyleLenko, Illia, Alexander Mamontov, Carole Alayrac, and Bernhard Witulski. 2024. "Pallado-Catalyzed Cascade Synthesis of 2-Alkoxyquinolines from 1,3-Butadiynamides" Molecules 29, no. 15: 3505. https://doi.org/10.3390/molecules29153505
APA StyleLenko, I., Mamontov, A., Alayrac, C., & Witulski, B. (2024). Pallado-Catalyzed Cascade Synthesis of 2-Alkoxyquinolines from 1,3-Butadiynamides. Molecules, 29(15), 3505. https://doi.org/10.3390/molecules29153505