
Indigo Carmine Binding to Cu(II) in Aqueous Solution and Solid State: Full Structural Characterization Using NMR, FTIR and UV/Vis Spectroscopies and DFT Calculations




Abstract

1. Introduction
2. Results and Discussion
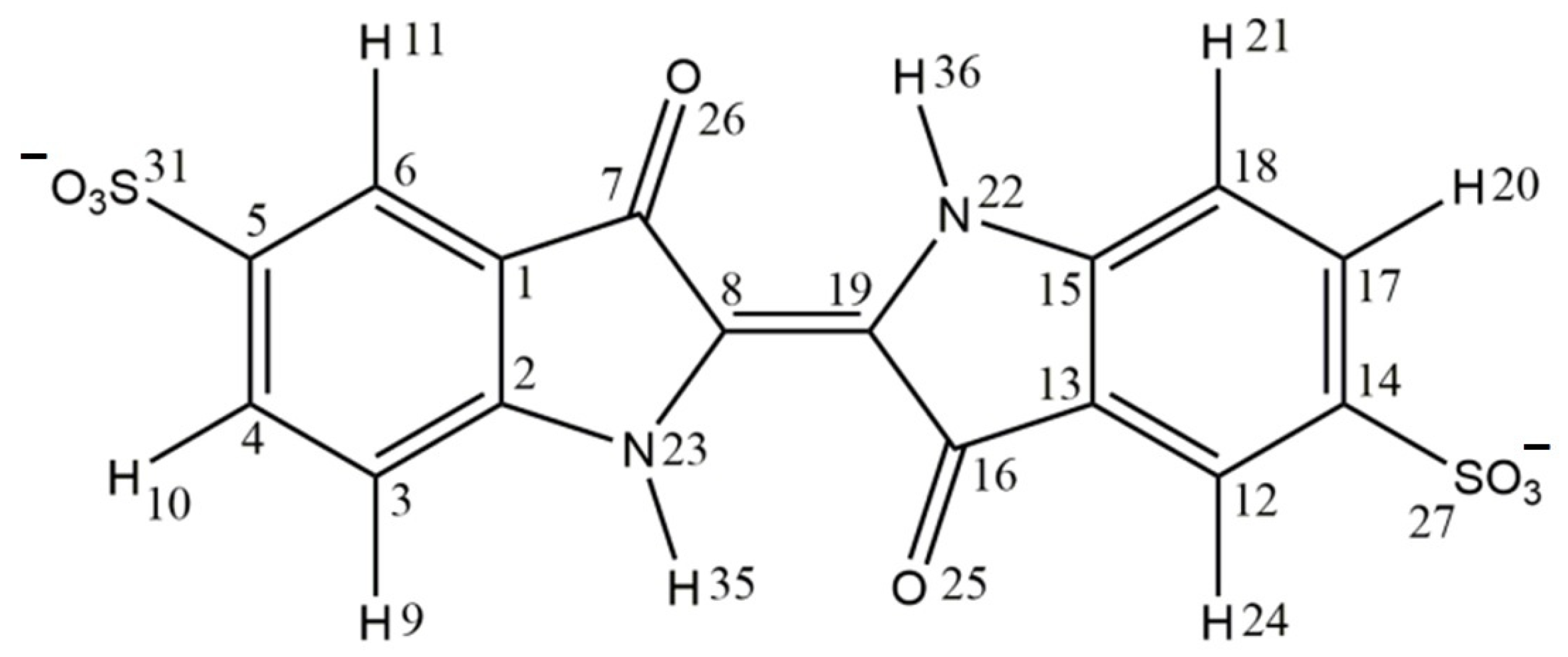
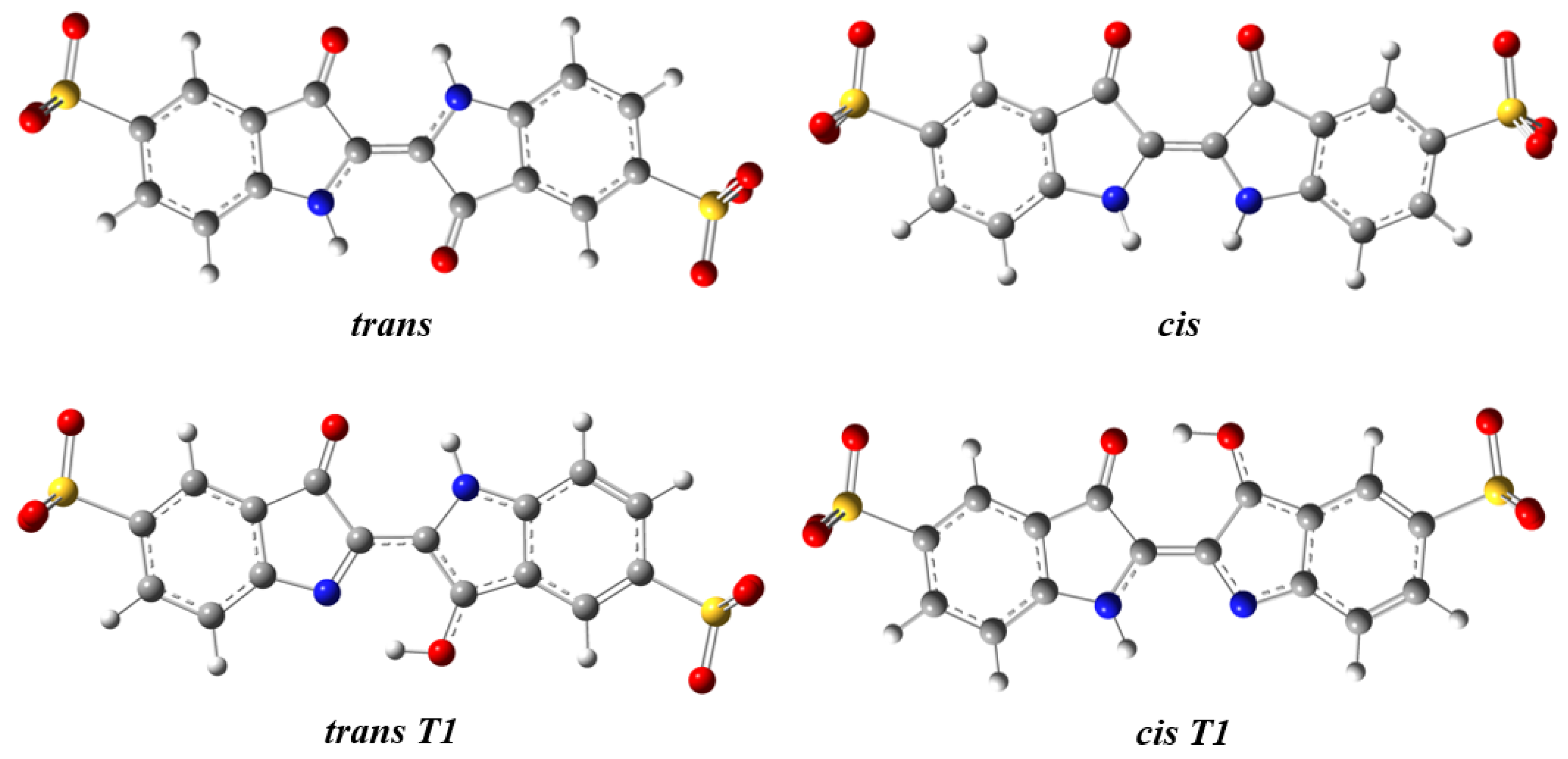
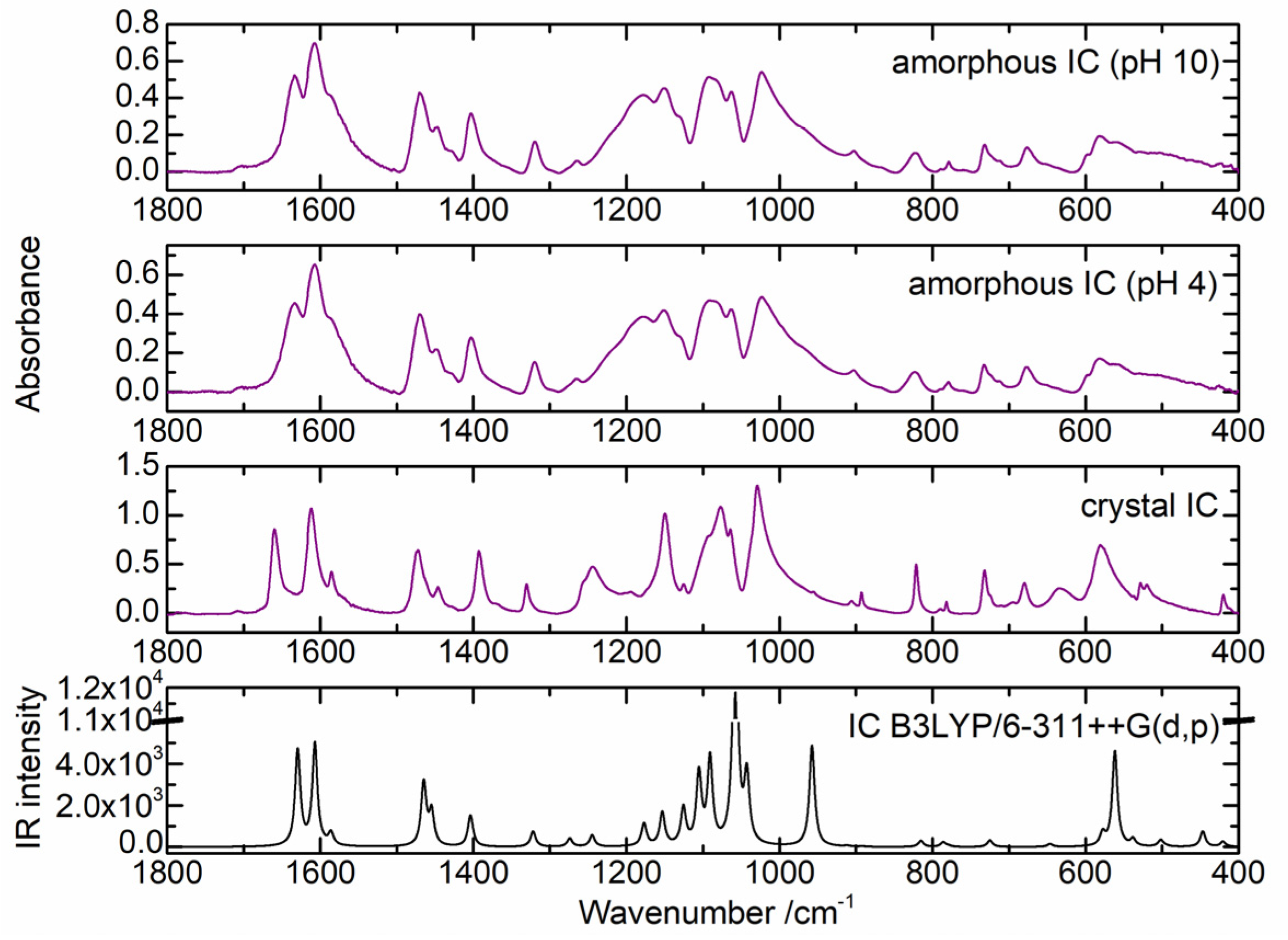
2.1. Structure and Energetics of the Indigo Carmine Molecule
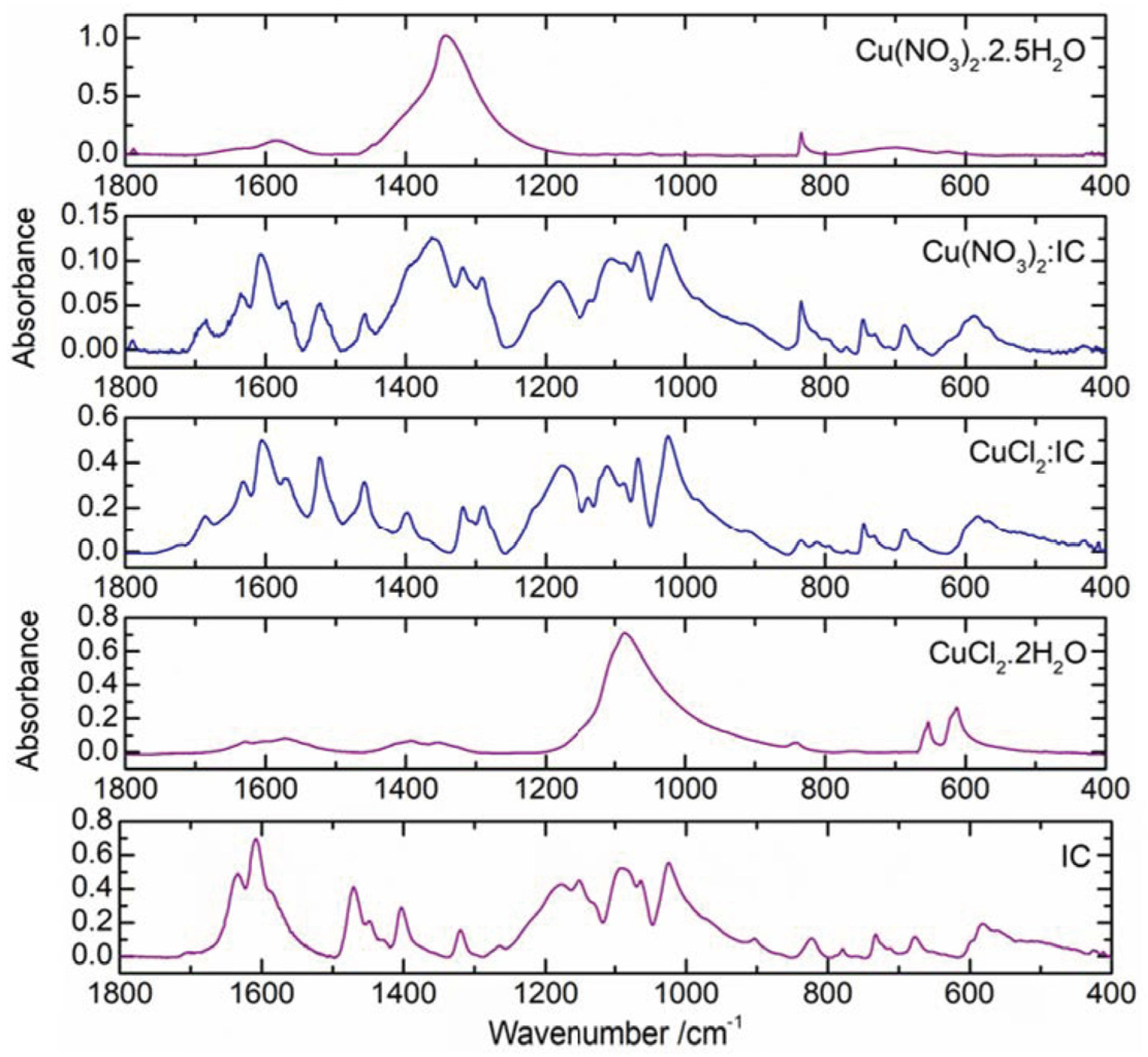
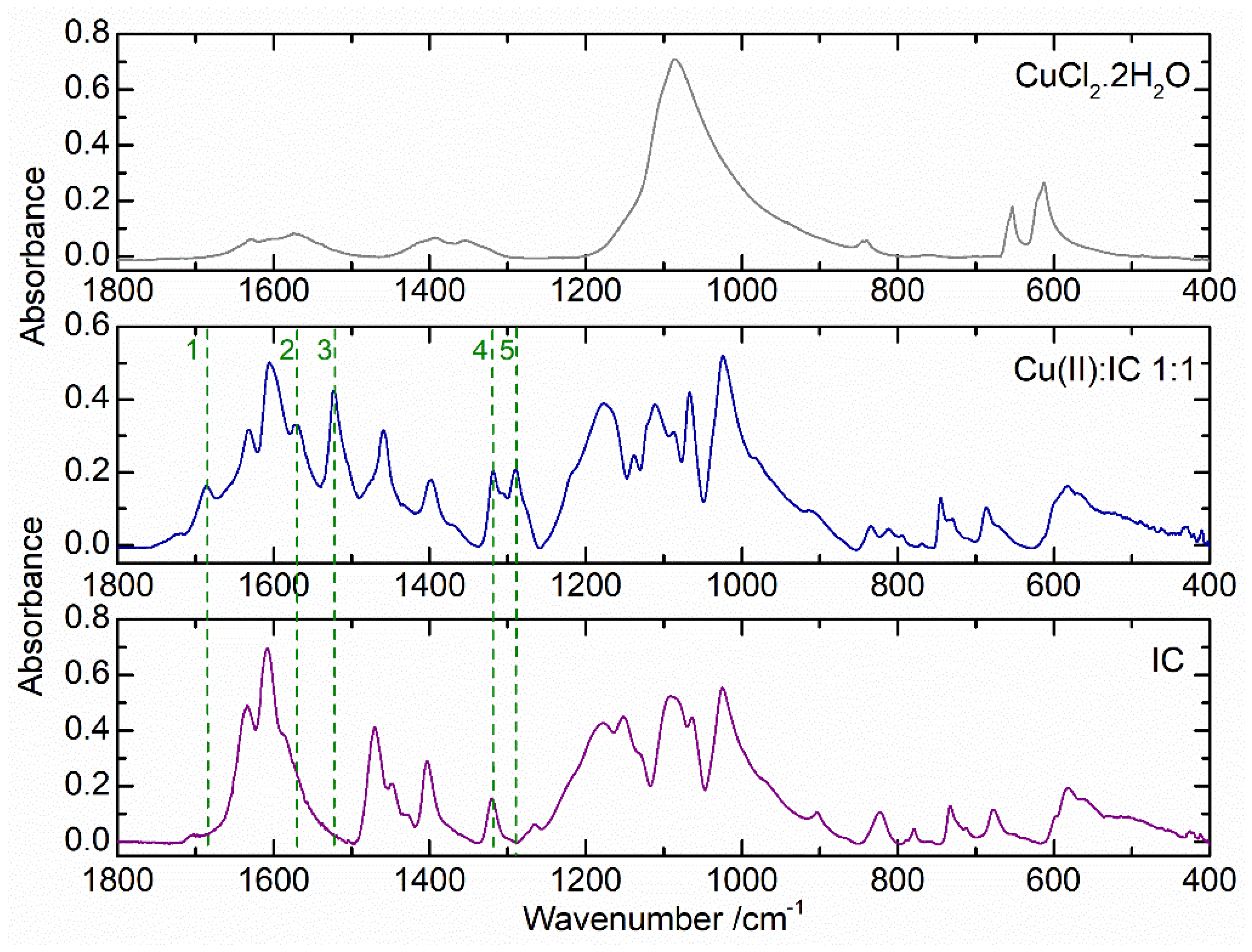
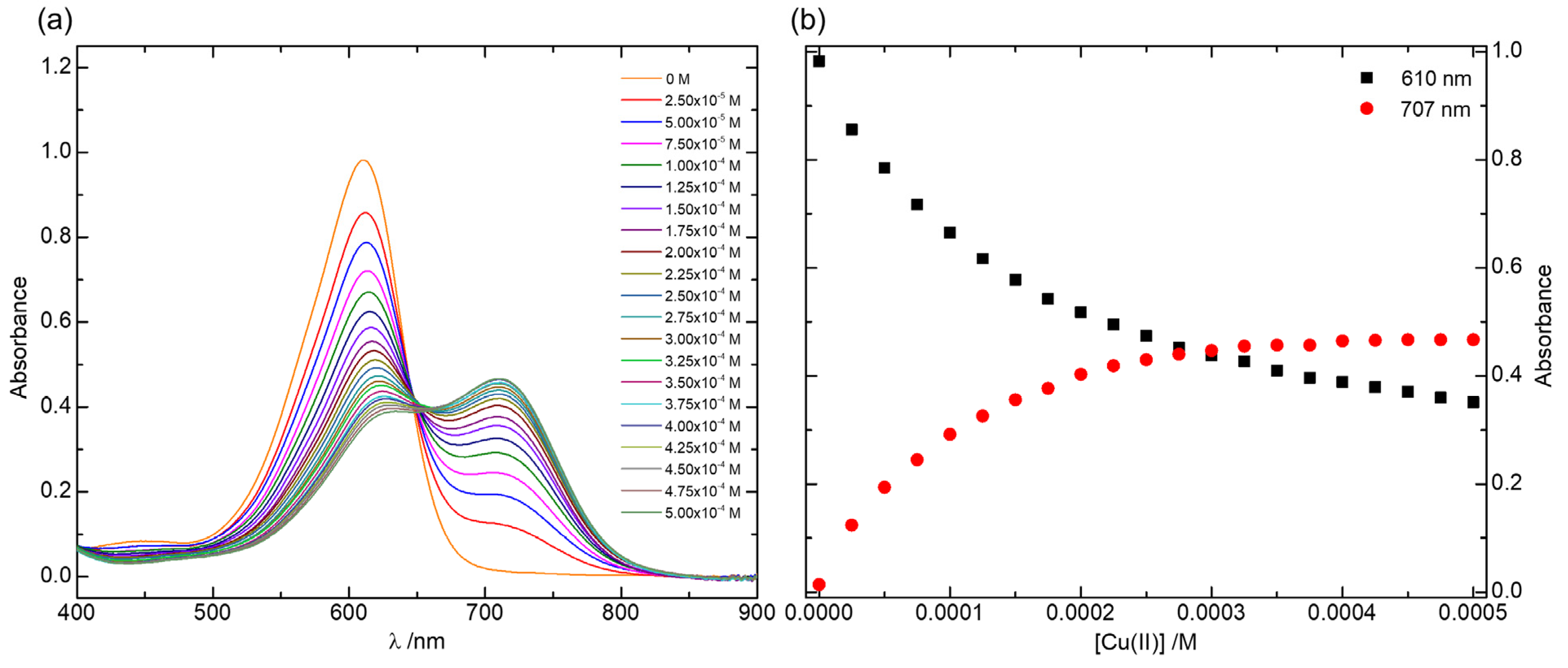
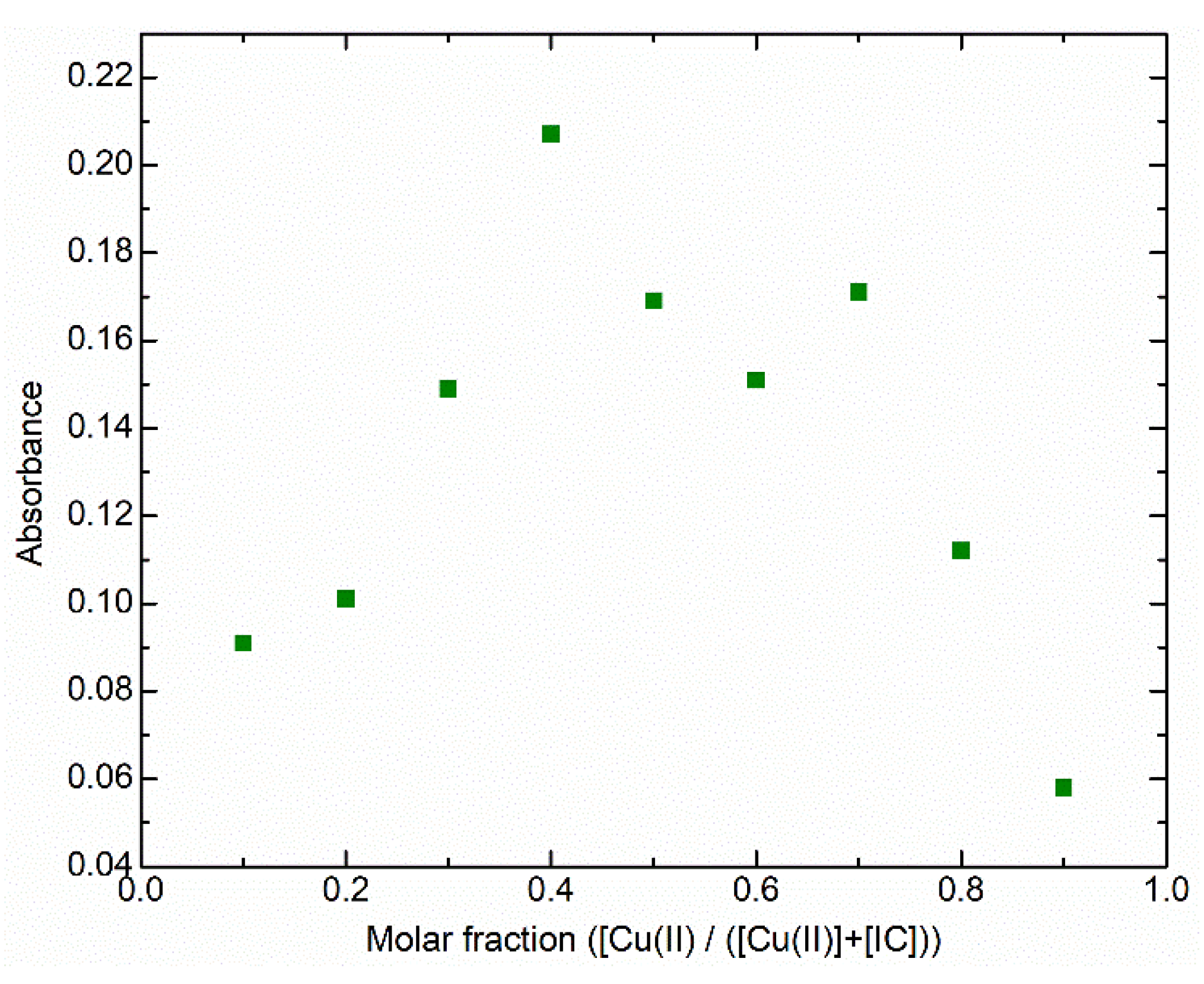
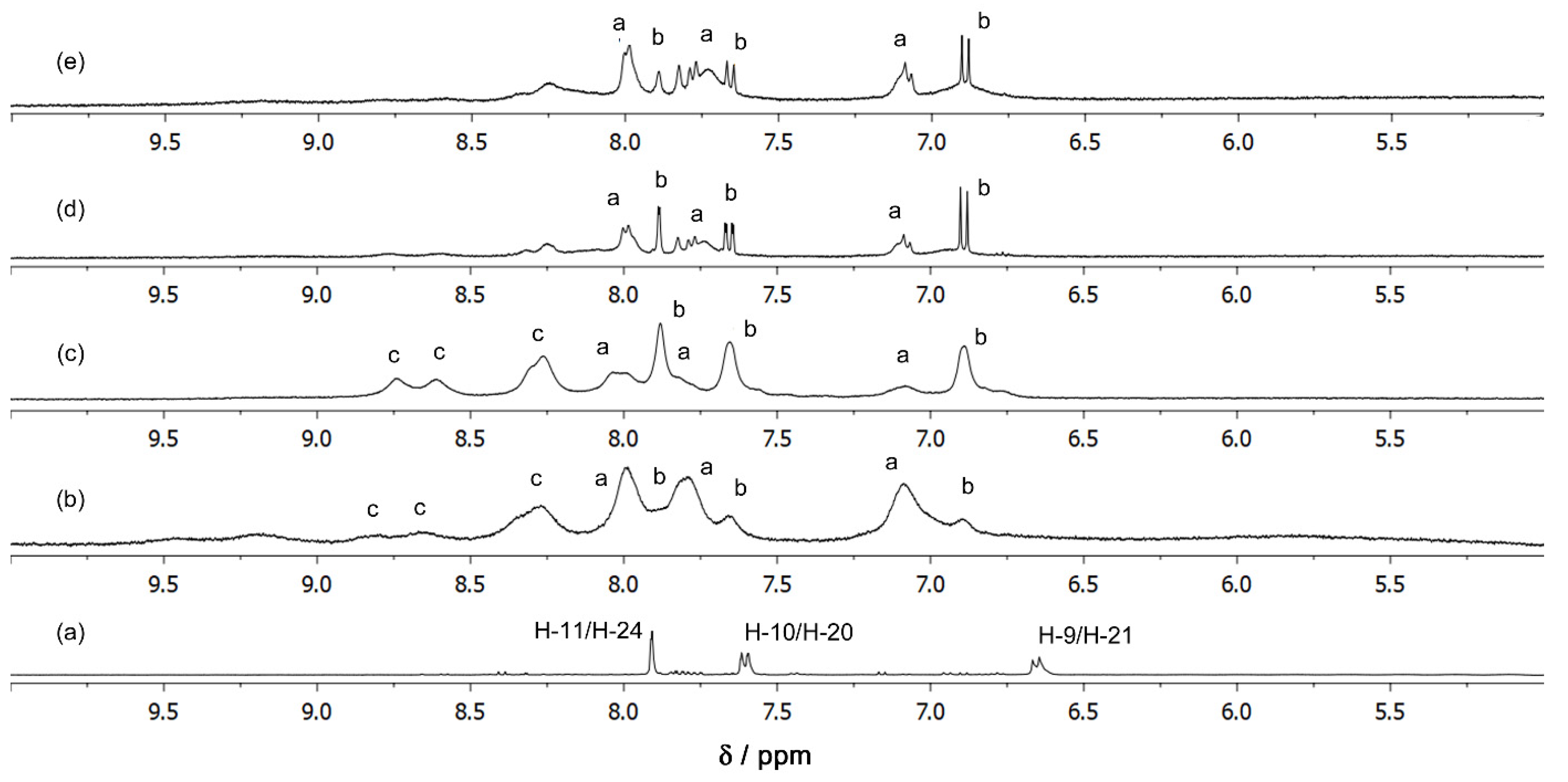
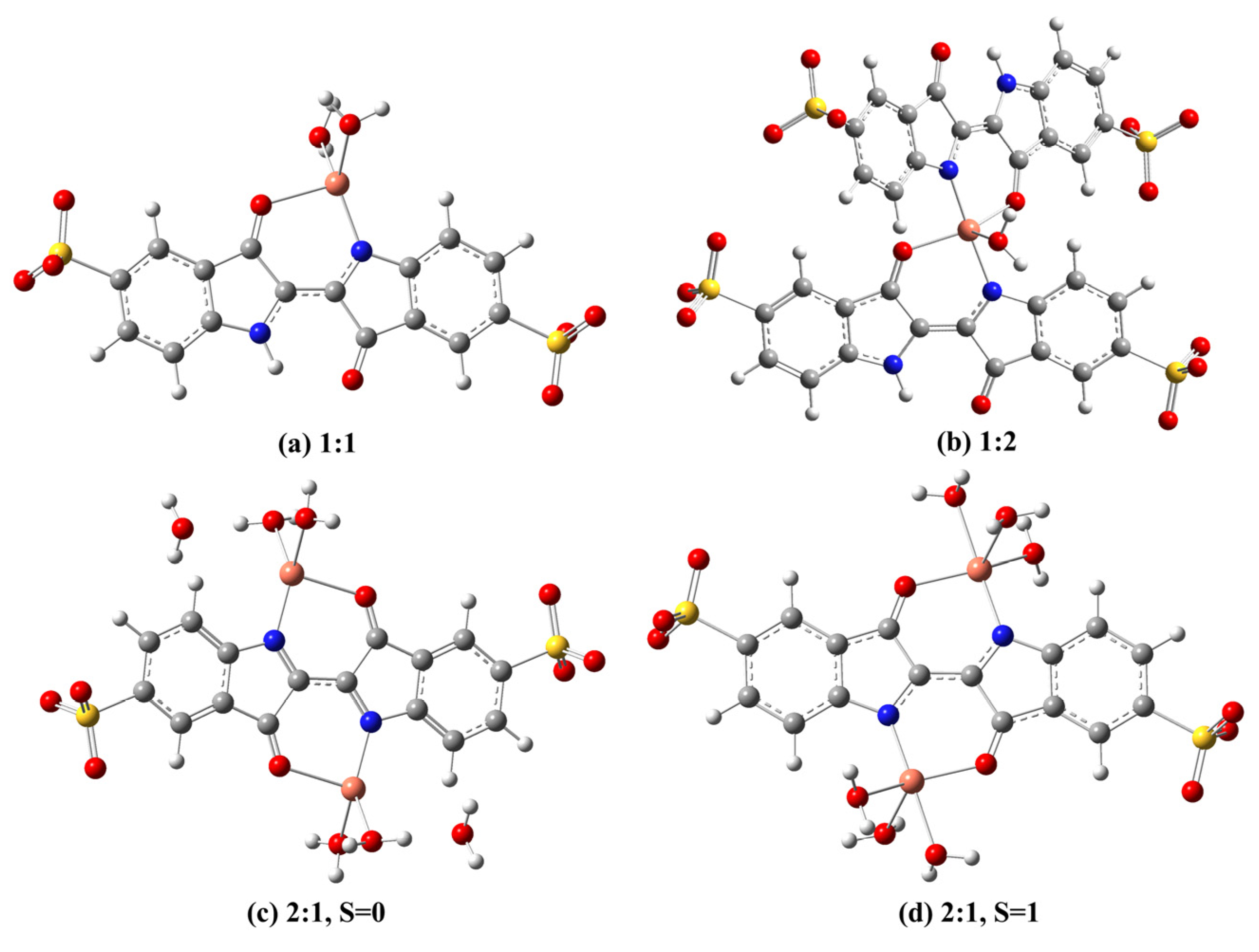
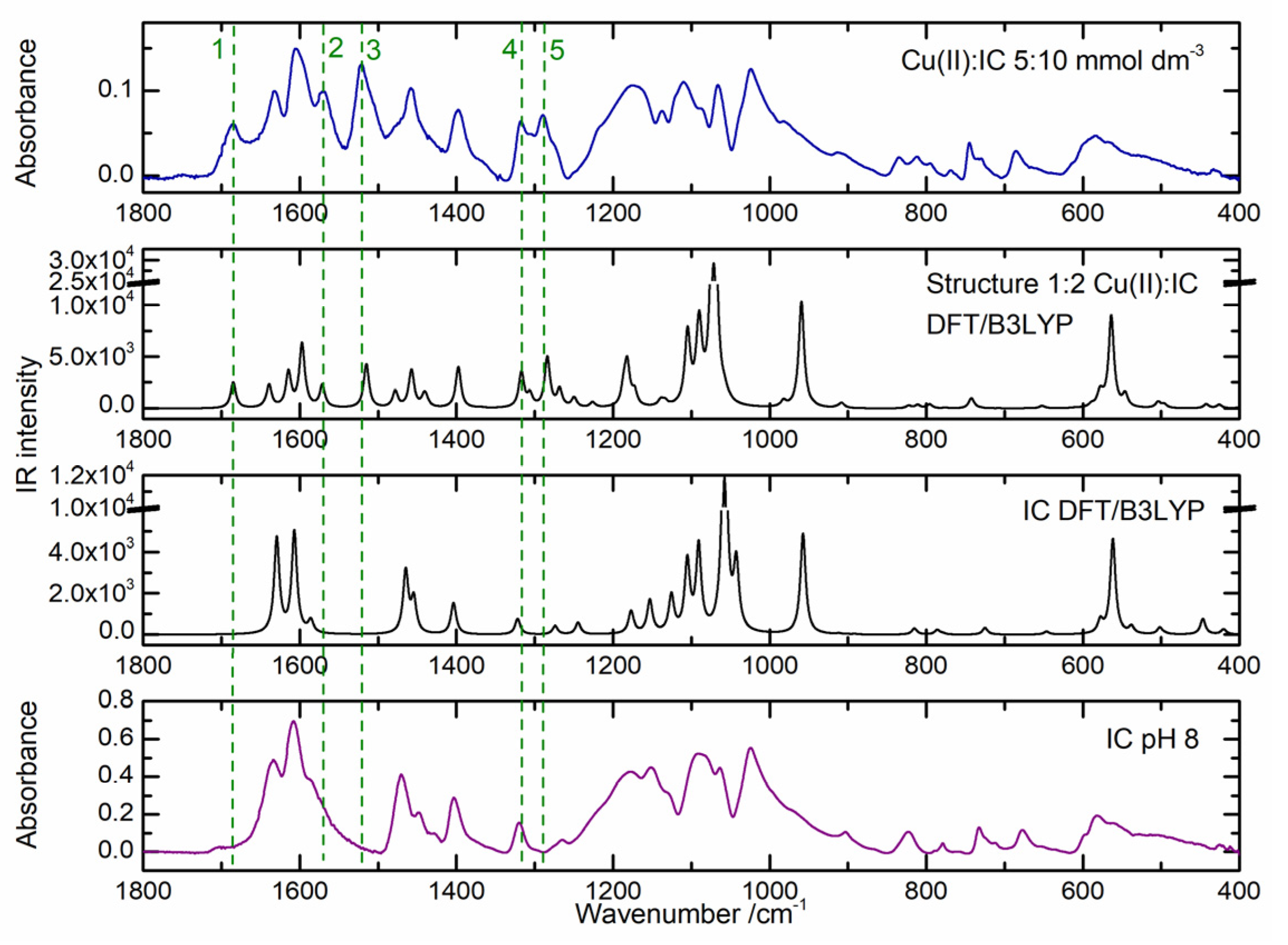
2.2. Cu(II) Complexes of Indigo Carmine
3. Materials and Methods
3.1. Materials and Preparation of Samples
3.2. Instrumentation
3.3. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Massoumi, A.; Tavallali, H. Kinetic Spectrophotometric Determination of Vanadium by Catalytic Effect on the Indigo Carmine-Bromate Reaction. Anal. Lett. 1998, 31, 193–206. [Google Scholar] [CrossRef]
- Gemeay, A.H.; Mansour, I.A.; El-Sharkawy, R.G.; Azhi, A.B. Kinetics and mechanism of the heterogeneous catalyzed oxidative degradation of indigo carmine. J. Mol. Catal. A Chem. 2003, 193, 109–120. [Google Scholar] [CrossRef]
- Shadi, I.T.; Chowdhry, B.Z.; Snowden, M.J.; Withnall, R. Analysis of the conversion of indigo into indigo carmine dye using SERRS. Chem. Commun. 2004, 12, 1436–1437. [Google Scholar] [CrossRef] [PubMed]
- Green, F.J. The Sigma Aldrich Handbook of Stains, Dyes and Indicators; Aldrich Chemical: St. Louis, MI, USA, 1990; p. 403. [Google Scholar]
- Jenkins, C.L. Textile dyes are potential hazards. Arch. Environ. Health 1978, 40, 256–263. [Google Scholar]
- Jeffords, D.L.; Lange, P.H.; Wolf, W.C. Severe hypertensive reaction to indigo carmine. Urology 1977, 9, 180–181. [Google Scholar] [CrossRef]
- Ikeda, K.; Sannohe, Y.; Araki, S.; Inutsuka, S. Intra-arterial Dye Method with Vasomotors (PIAD Method) Applied for the Endoscopic Diagnosis of Gastric Cancer and the Side Effects of Indigo Carmine. Endoscopy 1982, 14, 119–123. [Google Scholar] [CrossRef]
- Amchova, P.; Kotolova, H.; Ruda-Kucerova, J. Health safety issues of synthetic food colorants. J. Regul. Toxicol. Pharmacol. 2015, 73, 914–922. [Google Scholar] [CrossRef] [PubMed]
- European Food Safety Authority. Follow-up of the re-evaluation of indigo carmine (E 132) as a food additive. EFSA J. 2023, 21, e08103. [Google Scholar]
- Bhatt, D.; Vyas, K.; Singh, S.; John, P.J.; Soni, I.P. Sunset Yellow induced biochemical and histopathological alterations in rat brain sub-regions. Acta Histochem. 2024, 126, 152155. [Google Scholar] [CrossRef]
- Kohno, Y.; Kitamura, S.; Yamada, T.; Sugihara, K.; Ohta, S. Production of superoxide radical in reductive metabolism of a synthetic food-coloring agent, indigocarmine, and related compounds. Life Sci. 2005, 77, 601–614. [Google Scholar] [CrossRef]
- Şensoy, E. Determination of the effects of sunset yellow on mouse liver and pancreas using histological methods. Toxicol. Res. 2024, 13, tfae070. [Google Scholar] [CrossRef]
- Jana, G.; Sing, S.; Das, A.; Basu, A. Interaction of food colorant indigo carmine with human and bovine serum albumins: A multispectroscopic, calorimetric, and theoretical investigation. Int. J. Biol. Macromol. 2024, 259, 129143. [Google Scholar] [CrossRef] [PubMed]
- Salas-Peregrin, J.M.; Suarez-Varela, J. Synthesis, characterization and termal behaviour of some metal indigodisulphonates. J. Therm. Anal. 1984, 29, 515–521. [Google Scholar] [CrossRef]
- Zanoni, T.B.; Cardoso, A.A.; Zanoni, M.V.B.; Ferreira, A.A.P. Exploratory study on sequestration of some essential metals by indigo carmine food dye. Braz. J. Pharm. Sci. 2010, 46, 723–730. [Google Scholar] [CrossRef]
- Haleim, I.A.; Abbo, M. Formation and stability of nickel (II), iron (II) and copper (II) chelates of indigoid. Int. J. Basic Appl. Chem. Sci. 2014, 4, 1–9. [Google Scholar]
- Tavallali, H.; Deilamy-Rad, G.; Moaddeli, A.; Asghari, K. Indigo Carmine-Cu complex probe exhibiting dual colorimetric/fluorimetric sensing for selective determination of mono hydrogen phosphate ion and its logic behavior. Spectrochim. Acta A Mol. Biomol. 2017, 183, 319–331. [Google Scholar] [CrossRef] [PubMed]
- El-sayed, B.A.; Ibrahim, I.A.; Mohamed, W.A.A.; Ahmed, M.A.M. Synthesis and characterization of crystalline nano TiO2 and ZnO and their effects on the photodegradation od indigo carmine dye. Int. J. Adv. Eng. Nano Technol. 2015, 12, 2347–6389. [Google Scholar]
- Adel, M.; Ahmed, M.A.; Mohamed, A.A. Effective removal of indigo carmine dye from wastewaters by adsorption onto mesoporous magnesium ferrite nanoparticles. Environ. Nanotechnol. Monit. Manag. 2021, 16, 100550. [Google Scholar] [CrossRef]
- Job, P. Formation and stability of inorganic complexes in solution. Ann. Chim. 1928, 9, 113–134. [Google Scholar]
- Renny, J.S.; Tomasevich, L.L.; Tallmadge, E.H.; Collum, D.B. Method of Continuous Variations: Applications of Job Plots to the Study of Molecular Associations in Organometallic Chemistry. Angew. Chem. Int. Ed. 2013, 52, 2–18. [Google Scholar] [CrossRef]
- Gil, V.M.S.; Oliveira, N.C. On the use of the method of continous variations. J. Chem. Educ. 1990, 67, 473–478. [Google Scholar] [CrossRef]
- Freitas, A.R.; Silva, M.; Ramos, M.L.; Justino, L.L.G.; Fonseca, S.M.; Barsan, M.M.; Brett, C.M.A.; Silva, M.R.; Burrows, H.D. Synthesis, structure, and spectral and electrochemical properties of chromium (III) tris-(8-hydroxyquinolinate). Dalton Trans. 2015, 44, 11491–11503. [Google Scholar] [CrossRef] [PubMed]
- Veríssimo, L.M.P.; Ramos, M.L.; Justino, L.L.G.; Burrows, H.D.; Cruz, P.F.; Cabral, A.M.T.D.P.V.; Veiga, F.J.B.; Esteso, M.A.; Ribeiro, A.C.F. The structure and diffusion behaviour of the 1: 1 copper (II) complex of ethambutol in aqueous solution. J. Mol. Liq. 2018, 262, 63–70. [Google Scholar] [CrossRef]
- Pires, A.S.; Batista, J.; Murtinho, D.; Nogueira, C.; Karamysheva, A.; Ramos, M.L.; Milne, B.F.; Tavares, N.T.; Gonçalves, J.; Gonçalves, A.C.; et al. Synthesis, characterization and evaluation of the antibacterial and antitumor activity of halogenated salen copper (II) complexes derived from camphoric acid. Appl. Organomet. Chem. 2020, 34, e5569. [Google Scholar] [CrossRef]
- Hartman, J.R.; Vachet, R.W.; Pearson, W.; Wheat, R.J.; Callahan, J.H. A comparison of the gas, solution, and solid state coordination environments for the copper (II) complexes of a series of aminopyridine ligands of varying coordination number. Inorganica Chim. Acta 2003, 343, 119–132. [Google Scholar] [CrossRef]
- Hathaway, B.J. A new look at the stereochemistry and electronic properties of complexes of the copper (II) ion. Struct Bond. 1984, 57, 55–118. [Google Scholar]
- Halcrow, M.A. Jahn–Teller distortions in transition metal compounds, and their importance in functional molecular and inorganic materials. Chem. Soc. Rev. 2013, 42, 1784–1795. [Google Scholar] [CrossRef] [PubMed]
- Vicente, R.; Ribas, J.; Alvarez, S.; Seguí, A.; Solans, X.; Verdaguer, M. Synthesis, X-ray diffraction structure, magnetic properties, and MO analysis of a binuclear (µ-tetrathiooxalato) copper (II) complex, (AsPh4)2[(C3OS4)CuC2S4Cu(C3OS4)]. Inorg. Chem. 1987, 26, 4004–4009. [Google Scholar] [CrossRef]
- Chaudhuri, P.; Oder, K.; Wieghardt, K.; Gehring, S.; Haase, W.; Number, B.; Weiss, J. Moderately strong intramolecular magnetic exchange interaction between the copper (II) ions separated by 11.25. Å in [L2Cu2(OH2)2(µ-terephthalato)](ClO4)2 (L = 1,4,7-trimethyl-1,4,7-triazacyclononane). J. Am. Chem. Soc. 1988, 110, 3657–3658. [Google Scholar] [CrossRef]
- Felthouse, T.R.; Duesler, E.N.; Hendrickson, D.N. Magnetic exchange interactions in copper (II) dimers bridged by aromatic diamines. Crystal and molecular structure of µ-benzidine-bis (2,2′,2″-triaminotriethylamine) dicopper (II) nitrate. J. Am. Chem. Soc. 1978, 100, 618–619. [Google Scholar] [CrossRef]
- Ipatov, I.; Cordova, F.; Doriol, L.J.; Casida, M.E. Excited-state spin-contamination in time-dependent density-functional theory for molecules with open-shell ground states. J. Mol. Struct. Theochem 2009, 914, 60–73. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis set for molecular calculations. I. second row atoms, z=11-18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Justino, L.L.G.; Reva, I.; Fausto, R. Thermally and vibrationally induced conformational isomerizations, infrared spectra, and photochemistry of gallic acid in low-temperature matrices. J. Chem. Phys. 2016, 145, 014304. [Google Scholar] [CrossRef]
- Justino, L.L.G.; Braz, S.; Ramos, M.L. Spectroscopic and DFT Study of Alizarin Red S Complexes of Ga(III) in Semi-Aqueous Solution. Photochem 2023, 3, 61–81. [Google Scholar] [CrossRef]
- Miertus, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects. J. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Jaquemin, D.; Perpète, E.A.; Scuseria, G.E.; Ciofini, I.; Adamo, C. TD-DFT performance for the visible absorption spectra of organic dyes: Conventional versus long-range hybrids. J. Chem. Theory Comput. 2008, 4, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Ariyarathna, I.R. Electronic structure analysis and DFT benchmarking of Rydberg-type alkali-metal-crown ether, -cryptand, and -adamanzane complexes. Phys. Chem. Chem. Phys. 2024, 26, 16989–16997. [Google Scholar] [CrossRef] [PubMed]
- Frish, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | Symmetry | ΔG298k | P(%)298K |
|---|---|---|---|
| trans | Ci | 0.0 | 100.0 |
| cis | C2 | 37.0 | 0.0 |
| trans T1 | C1 | 50.8 | 0.0 |
| cis T1 | C1 | 73.4 | 0.0 |
| Bond Lengths (Å) | Angles (Degrees) | ||
|---|---|---|---|
| O26⋯H36 | 2.305 | H35-N23-C8 | 123.2 |
| H35-N23 | 1.011 | N23-C8-C19 | 126.1 |
| N23-C8 | 1.378 | C7-C8-C19 | 125.8 |
| C8-C19 | 1.361 | C8-C7-26 | 125.0 |
| C7-C8 | 1.494 | C7-C8-C19-C16 | 180.0 |
| C1-C7 | 1.464 | N23-C8-C7-C1 | 0.0 |
| C7-O26 | 1.231 | C8-C7-O26-H36 | 0.0 |
| C1-C2 | 1.412 | C19-C8-N23-H35 | 0.0 |
| C2-C3 | 1.396 | C3-C4-C5-C6 | 0.0 |
| δ1H RMN (exp.) a | ||||
|---|---|---|---|---|
| Free Ligand | Complex a | Complex b | Complex c | |
| H-36 | - b | - b | - b | - b |
| H-11/H-34 | 7.90 | 7.98 | 7.89 | 8.84 |
| H-10/H-20 | 7.58 | 7.80 | 7.64 | 8.67 |
| H-9/H-21 | 6.66 | 7.08 | 6.90 | 8.26 |
| Element a | Weight % | Atomic % | Calc. wt. % 1:2 Structure | Calc. wt. % 2:3 Structure |
|---|---|---|---|---|
| Carbon | 30.9 | 44.7 | 35.6 | 37.6 |
| Nitrogen | 7.5 | 9.4 | 5.6 | 5.5 |
| Oxygen | 28.1 | 30.5 | 27.3 | 27.1 |
| Sodium | 6.9 | 5.2 | 9.2 | 9.0 |
| Sulfur | 9.9 | 5.4 | 12.9 | 12.5 |
| Copper | 15.8 | 4.3 | 6.4 | 8.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Braz, S.; Justino, L.L.G.; Ramos, M.L.; Fausto, R. Indigo Carmine Binding to Cu(II) in Aqueous Solution and Solid State: Full Structural Characterization Using NMR, FTIR and UV/Vis Spectroscopies and DFT Calculations. Molecules 2024, 29, 3223. https://doi.org/10.3390/molecules29133223
Braz S, Justino LLG, Ramos ML, Fausto R. Indigo Carmine Binding to Cu(II) in Aqueous Solution and Solid State: Full Structural Characterization Using NMR, FTIR and UV/Vis Spectroscopies and DFT Calculations. Molecules. 2024; 29(13):3223. https://doi.org/10.3390/molecules29133223
Chicago/Turabian StyleBraz, Sofia, Licínia L. G. Justino, M. Luísa Ramos, and Rui Fausto. 2024. "Indigo Carmine Binding to Cu(II) in Aqueous Solution and Solid State: Full Structural Characterization Using NMR, FTIR and UV/Vis Spectroscopies and DFT Calculations" Molecules 29, no. 13: 3223. https://doi.org/10.3390/molecules29133223
APA StyleBraz, S., Justino, L. L. G., Ramos, M. L., & Fausto, R. (2024). Indigo Carmine Binding to Cu(II) in Aqueous Solution and Solid State: Full Structural Characterization Using NMR, FTIR and UV/Vis Spectroscopies and DFT Calculations. Molecules, 29(13), 3223. https://doi.org/10.3390/molecules29133223