

[Fe(µ2-OH)6]3− Linked Fe3O Triads: Mössbauer Evidence for Trigonal µ3-O2− or µ3-OH− Groups in Bridged versus Unbridged Complexes

 , , and
, , and
Abstract

1. Introduction
2. Results
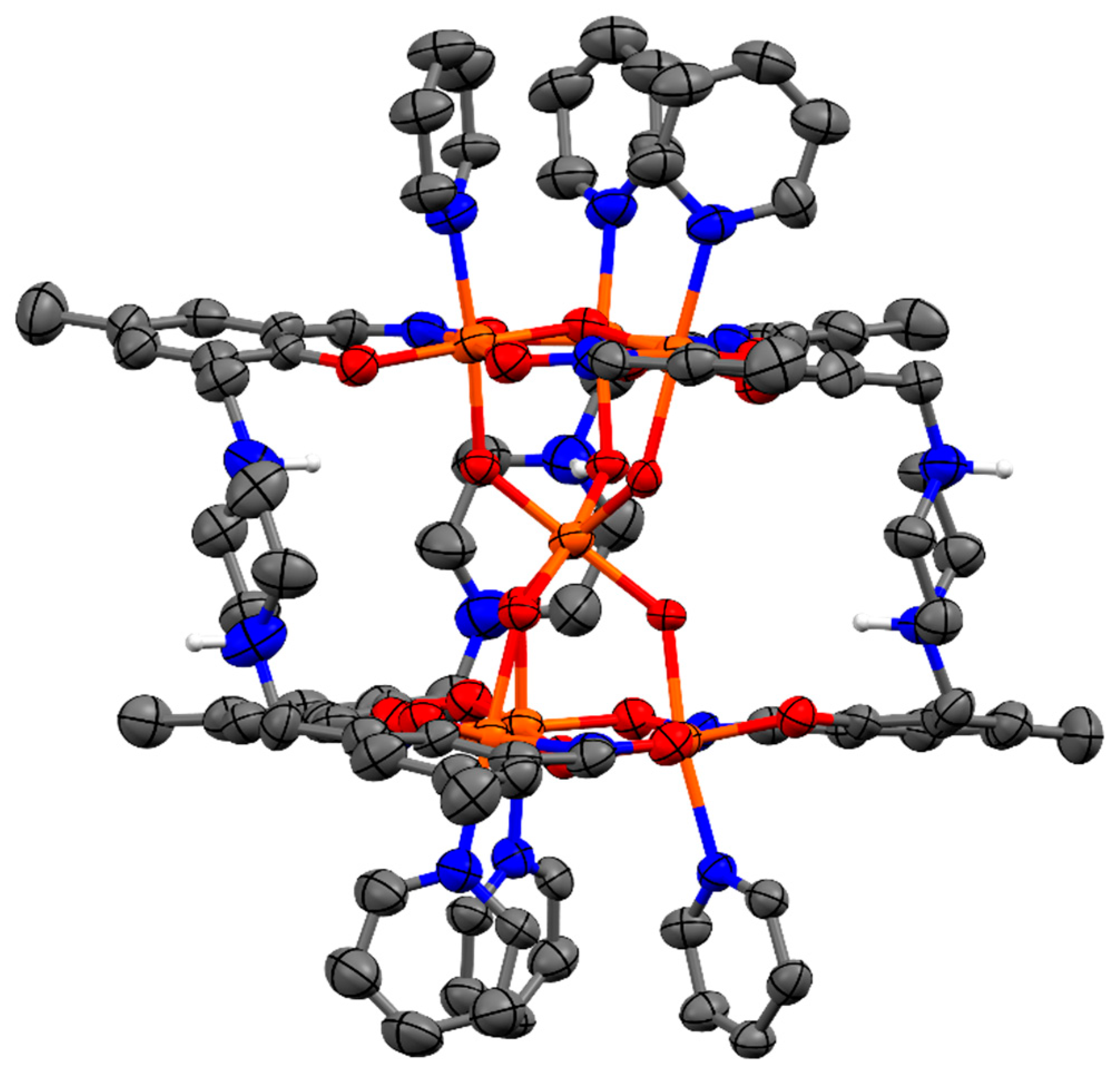
2.1. Discussion of the Crystal Structure of the Fe7 Complex of a ‘Single-Headed’ Derivatised Salicylaldoxime (C1)
2.2. Discussion of the Crystal Structures of Fe7 Complexes of Linked/’Double-Headed’ Derivatised Salicylaldoximes
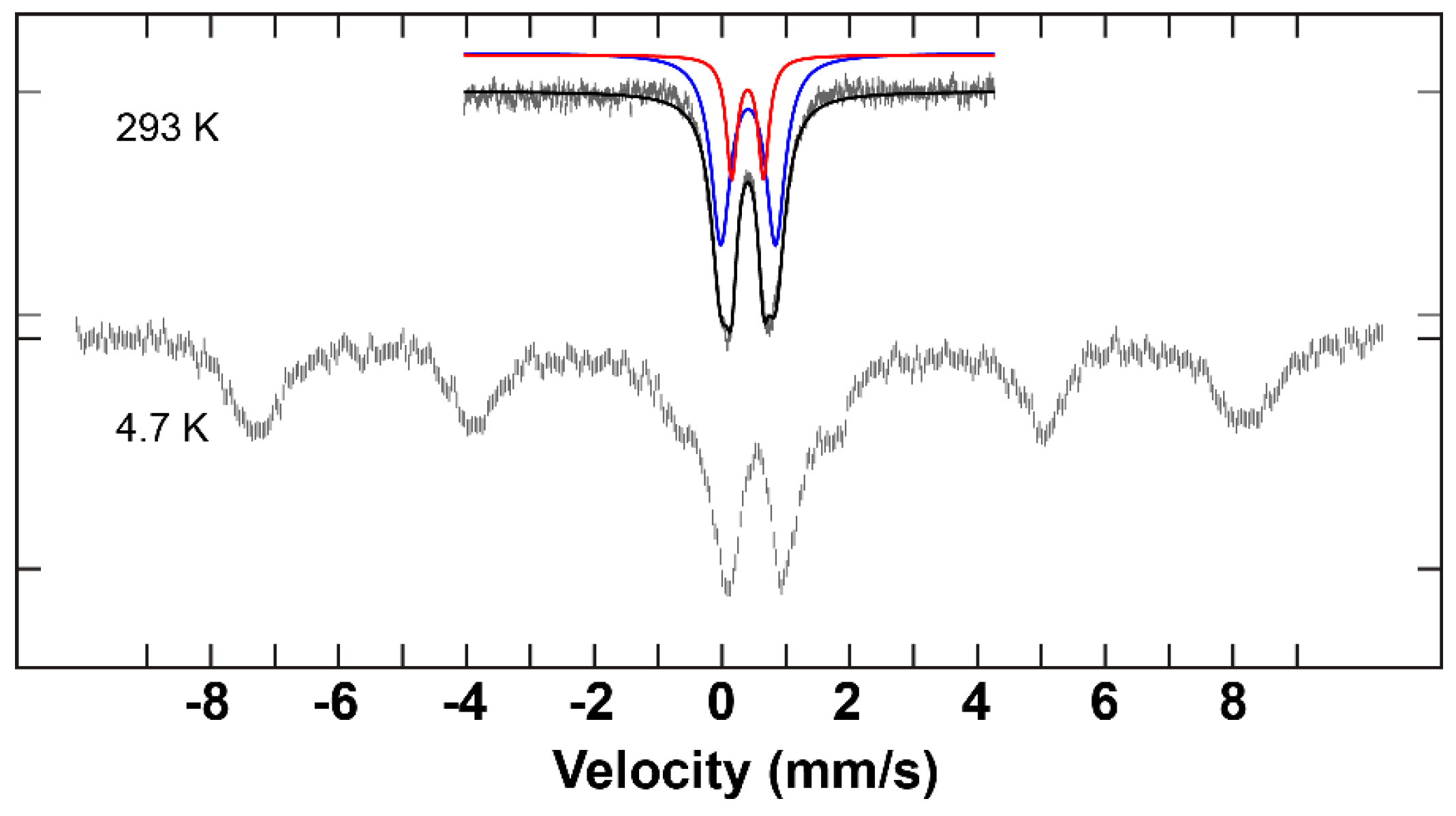
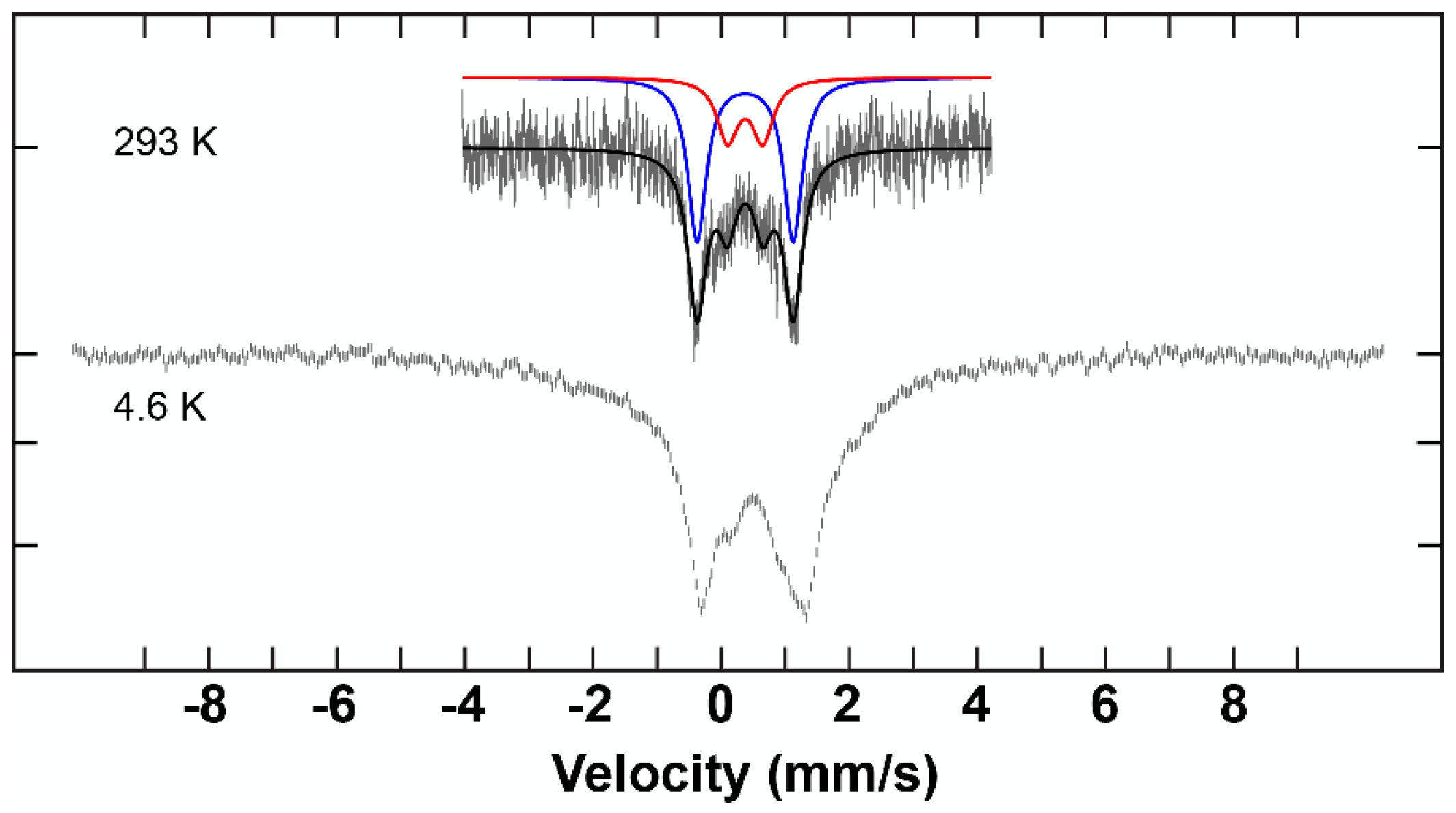
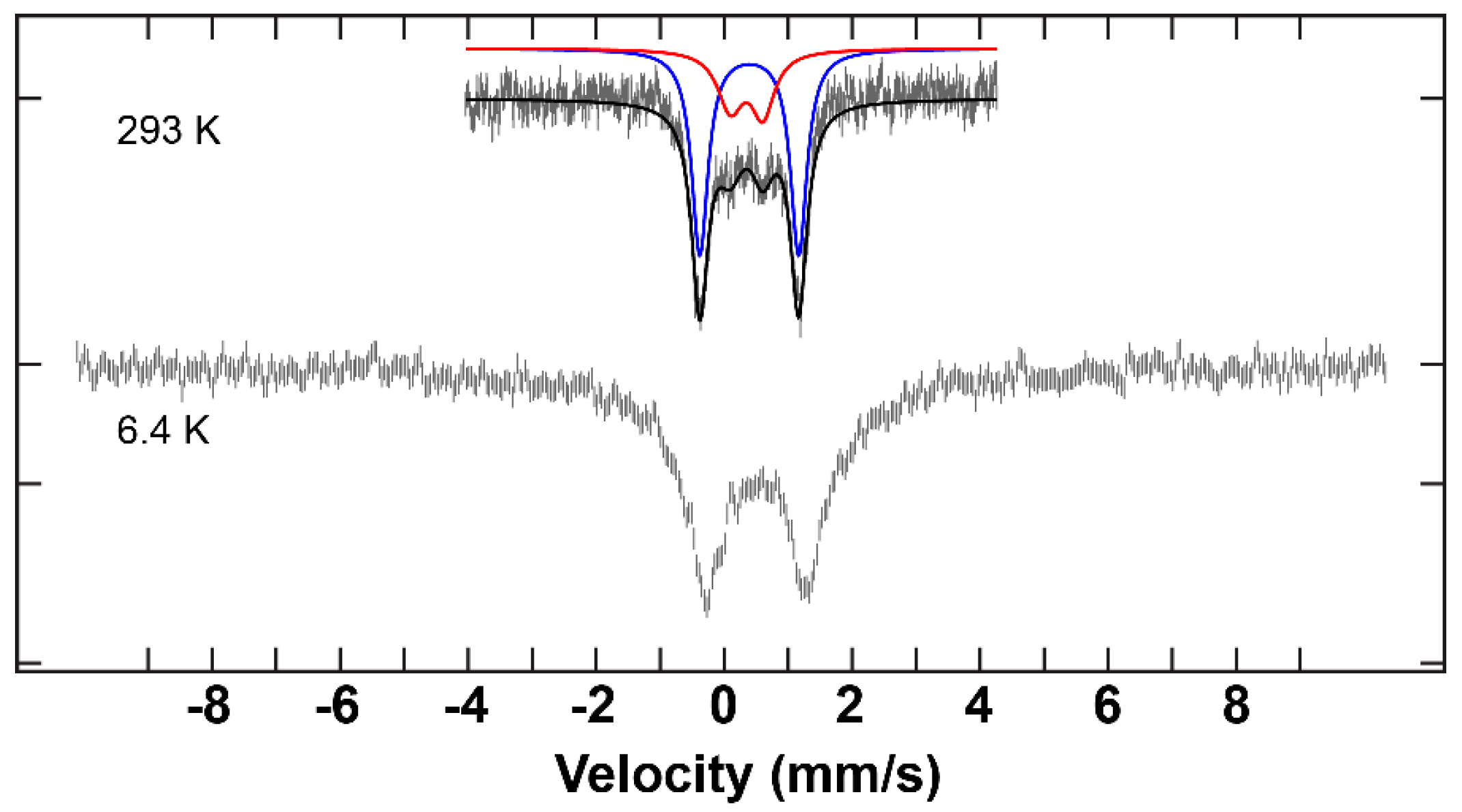
2.3. Mössbauer Results and Discussion
3. Materials and Methods
3.1. Synthesis of Ligands H4L2 and H4L3
3.1.1. L2a (Precursor for H4L2): 3,3′-[1,4-Piperazinediylbis(methylene)]bis [2-hydroxy-5-methylbenzaldehyde]
3.1.2. H4L2: 3,3′-[1,4-Piperazinediylbis(methylene)]bis [2-hydroxy-5-methylbenzaldehyde oxime]
3.1.3. L3a (Precursor for H4L3): 3,3′-[1,4-Phenylenebis[methylene(methylimino)methylene]]bis [2-hydroxy-5-methylbenzaldehyde]
3.1.4. H4L3: 3,3′-[1,4-Phenylenebis[methylene(methylimino)methylene]]bis [2-hydroxy-5-methylbenzaldehyde oxime]
3.2. Synthesis of Metal Complexes C1–C3
3.2.1. [. Fe7(µ3-OH)2(µ2-OH)6(H2L1-2H)5(H2L1-H)1(pyr)6]·(BF4)2·(H2O)8 (pyr)2 (C1·2BF4·8H2O·2pyr)
3.2.2. [Fe7O2(H4L2-2H)3(OH)6(pyr)6)]·(BF4)4·(H2O)7·PF6·(pyr)2 (C2·4BF4·7H2O·PF6·2pyr)
3.2.3. [Fe7O2(H4L3-2H)2(H4L3-3H)(OH)6(pyr)6)]·(PF6)4·(H2O)7 (C3·4PF6·7H2O)
3.3. X-ray Structure Determination
3.4. Mössbauer Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C1 | C2 | C3 | |
|---|---|---|---|
| Formula | B2C162F8Fe7H230N24O26 | B4C136F22Fe7H151N26O27P | C114H123Fe7N18O22 |
| CCDC | 2331487 | 2331488 | 2331489 |
| FW (g mol−1) | 3494.26 | 3464.98 | 2488.25 |
| T (K) | 153 | 153 | 100 |
| Crystal system | trigonal | triclinic | monoclinic |
| Space group | |||
| a (Å) | 22.558 (5) | 17.1685 (11) | 15.949 (3) |
| b (Å) | 22.558 (5) | 17.3485 (11) | 25.627 (5) |
| c (Å) | 33.105 (5) | 29.534 (2) | 18.611 (4) |
| α (°) | 90.000 (5) | 86.396 (6) | 90 |
| β (°) | 90.000 (5) | 76.716 (5) | 107.11 (3) |
| γ (°) | 120.000 (5) | 60.913 (4) | 90 |
| V (Å3) | 14589 (7) | 7469.2 (9) | 7270 (3) |
| Z (Z′) | 3 (0.167) | 2 (1) | 2 (0.5) |
| ρcalc (g cm−3) | 1.193 | 1.541 | 1.137 |
| μ (mm−1) | 4.665 | 6.32 | 0.74 |
| F (000) | 5526 | 3560 | 2582 |
| Crystal size (mm) | 0.2 × 0.2 × 0.2 | 0.33 × 0.23 × 0.21 | 0.2 × 0.2 × 0.2 |
| Radiation | CuKα (λ = 1.54178) | CuKα (λ = 1.54178) | Synchrotron (λ = 0.71073) |
| 2Θ (°) | 13.128 to 144.218 | 13.174 to 130.18 | 4.302 to 49.426 |
| Index ranges | −27 ≤ h ≤ 26, −27 ≤ k ≤ 24, −25 ≤ l ≤ 38 | −20 ≤ h ≤ 20, −18 ≤ k ≤ 20, −34 ≤ l ≤ 34 | −18 ≤ h ≤ 17, 0 ≤ k ≤ 30, 0 ≤ l ≤ 21 |
| Reflections collected | 43942 | 96150 | 43481 |
| Independent reflections | 6222 [Rint = 0.0783, Rsigma = 0.0531] | 24944 [Rint = 0.1440, Rsigma = 0.1731] | 12205 [Rint = 0.0487, Rsigma = 0.0418] |
| Data/restraints/parameters | 6222/78/357 | 24944/1523/1559 | 12205/931/861 |
| Goodness-of-fit on F2 | 1.094 | 1.194 | 1.079 |
| Final R indexes [I ≥ 2σ(I)] | R1 = 0.0868, wR2 = 0.2465 | R1 = 0.1448, wR2 = 0.4009 | R1 = 0.0784, wR2 = 0.2466 |
| Final R indexes [all data] | R1 = 0.0985, wR2 = 0.2723 | R1 = 0.2433, wR2 = 0.4638 | R1 = 0.0935, wR2 = 0.2615 |
| Residual density (e− Å−3) | 1.62/−1.20 | 0.98/−0.61 | 0.58/−0.32 |
References
- Cohen, I.A. Metal-metal interactions in metalloporphyrins, metalloproteins and metalloenzymes. Struct. Bond. 1980, 40, 1–37. [Google Scholar]
- Vincent, J.B.; Olivier-Lilley, G.L.; Averill, B.A. Proteins containing oxo-bridged dinuclear iron centers: A bioinorganic perspective. Chem. Rev. 1990, 90, 1447–1467. [Google Scholar] [CrossRef]
- Horn, A.; Neves, A.; Bortoluzzi, A.J.; Drago, V.; Ortiz, W.A. Crystal structure and magnetic properties of a new tetranuclear iron(III) complex with asymmetric iron coordination as a model for polynuclear iron proteins. Inorg. Chem. Commun. 2001, 4, 173–176. [Google Scholar] [CrossRef]
- Högbom, M.; Nordlund, P. A protein carboxylate coordinated oxo-centered tri-nuclear iron complex with possible implications for ferritin mineralization. FEBS Lett. 2004, 567, 179–182. [Google Scholar] [CrossRef]
- Faiella, M.; Andreozzi, C.; Martin de Rosales, R.T.; Pavone, V.; Maglio, O.; Nastri, F.; DeGrado, W.F.; Lombardi, A. An artificial di-iron oxo-protein with phenol oxidase activity. Nat. Chem. Biol. 2009, 5, 882–884. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, M.L.C.; Pastore, A.J.; Davidson, V.L. Diversity of structures and functions of oxo-bridged non-heme diiron proteins. Arch. Biochem. Biophys. 2021, 705, 108917. [Google Scholar]
- Weighardt, K.; Pohl, K.; Jibril, I.; Huttner, G. Hydrolysis products of the monomeric amine complex (C6H15N3)FeCl3: The structure of the octameric iron(III) cation of {[(C6H15N3)6Fe8(µ3-O)2(µ2-OH)12]Br7(H2O)}Br·8H2O. Angew. Chem. Int. Ed. 1984, 23, 77–78. [Google Scholar] [CrossRef]
- Barra, A.L.; Debrunner, P.; Gatteschi, D.; Schulz, C.E.; Sessoli, R. Superparamagnetic-like behavior in an octanuclear iron cluster. Europhys. Lett. 1996, 35, 133–138. [Google Scholar] [CrossRef]
- Jones, P.L.; Jeffery, J.C.; McCleverty, J.A.; Ward, M.D. Dinuclear and tetranuclear oxo-bridged iron(III) complexes of the ambidentate ligand 3-(2-pyridyl)pyrazole. Polyhedron 1997, 16, 1567–1571. [Google Scholar] [CrossRef]
- Aromi, G.; Brechin, E.K. Synthesis of 3d metallic single-molecule magnets. Struct. Bond. 2006, 122, 1–67. [Google Scholar] [CrossRef]
- Rodriguez, E.; Gich, M.; Roig, A.; Molins, E.; Nedelko, N.; Slawska-Waniewska, A.; Szewczyk, A. Investigations of the stability of {[(tacn)6Fe8(μ3-O)2(μ2-OH)12]Br7(H2O)}Br·8H2O (Fe8) cluster in aqueous solution by spectroscopic and magnetic methods. Polyhedron 2006, 25, 113–118. [Google Scholar] [CrossRef]
- Boudalis, A.K.; Sanakis, Y.; Raptopoulou, C.P.; Terzis, A.; Tuchagues, J.-P.; Perlepes, S.P. A trinuclear cluster containing the {Fe3(μ3-O)}7+ core: Structural, magnetic and spectroscopic (IR, Moessbauer, EPR) studies. Polyhedron 2005, 24, 1540–1548. [Google Scholar] [CrossRef]
- Raptopoulou, C.P.; Sanakis, Y.; Boudalis, A.K.; Psycharis, V. Salicylaldoxime (H2salox) in iron(III) carboxylate chemistry: Synthesis, X-ray crystal structure, spectroscopic characterization and magnetic behavior of trinuclear oxo-centered complexes. Polyhedron 2005, 24, 711–721. [Google Scholar] [CrossRef]
- Murray, K.S. Binuclear oxo-bridged iron(III) complexes. Coord. Chem. Rev. 1974, 12, 1–35. [Google Scholar] [CrossRef]
- Shiemke, A.K.; Loehr, T.M.; Sanders-Loehr, J. Resonance Raman study of oxyhemerythrin and hydroxomethemerythrin. Evidence for hydrogen bonding of ligands to the iron-oxygen-iron center. J. Am. Chem. Soc. 1986, 108, 2437–2443. [Google Scholar] [CrossRef] [PubMed]
- Doi, K.; Antanaitis, B.C.; Aisen, P. The binuclear iron centers of uteroferrin and the purple acid phosphatases. Struct. Bond. 1988, 70, 1–26. [Google Scholar]
- Que, L., Jr.; True, A.E. Dinuclear iron- and manganese-oxo sites in biology. Prog. Inorg. Chem. 1990, 38, 97–200. [Google Scholar] [CrossRef]
- Kurtz, D.M., Jr. Oxo- and hydroxo-bridged diiron complexes: A chemical perspective on a biological unit. Chem. Rev. 1990, 90, 585–606. [Google Scholar] [CrossRef]
- Nordlund, P.; Sjoeberg, B.M.; Eklund, H. Three-dimensional structure of the free radical protein of ribonucleotide reductase. Nature 1990, 345, 593–598. [Google Scholar] [CrossRef]
- Holmes, M.A.; Le Trong, I.; Turley, S.; Sieker, L.C.; Stenkamp, R.E. Structures of deoxy and oxy hemerythrin at 2.0 Å resolution. J. Mol. Biol. 1991, 218, 583–593. [Google Scholar] [CrossRef]
- Wilkins, R.G. Binuclear iron centers in proteins. Chem. Soc. Rev. 1992, 21, 171–178. [Google Scholar] [CrossRef]
- Solomon, E.I.; Zhang, Y. The electronic structures of active sites in non-heme iron enzymes. Acc. Chem. Res. 1992, 25, 343–352. [Google Scholar] [CrossRef]
- Mason, K.; Chang, J.; Prescimone, A.; Garlatti, E.; Carretta, S.; Tasker, P.A.; Brechin, E.K. Linking [MIII3] triangles with “double-headed” phenolic oximes. Dalton Trans. 2012, 41, 8777–8785. [Google Scholar] [CrossRef]
- Thorpe, J.M.; Beddoes, R.L.; Collison, D.; Garner, C.D.; Helliwell, M.; Holmes, J.M.; Tasker, P.A. Surface coordination chemistry: Corrosion inhibition by tetranuclear cluster formation of iron with salicylaldoxime. Angew. Chem. Int. Ed. 1999, 38, 1119–1121. [Google Scholar] [CrossRef]
- Gass, I.A.; Milios, C.J.; Collins, A.; White, F.J.; Budd, L.; Parsons, S.; Murrie, M.; Perlepes, S.P.; Brechin, E.K. Polymetallic clusters of iron(III) with derivatized salicylaldoximes. Dalton Trans. 2008, 2043–2053. [Google Scholar] [CrossRef] [PubMed]
- Mason, K.; Gass, I.A.; Parsons, S.; Collins, A.; White, F.J.; Slawin, A.M.Z.; Brechin, E.K.; Tasker, P.A. Building Fe(III) clusters with derivatised salicylaldoximes. Dalton Trans. 2010, 39, 2727–2734. [Google Scholar] [CrossRef] [PubMed]
- Mason, K.; Chang, J.; Garlatti, E.; Prescimone, A.; Yoshii, S.; Nojiri, H.; Schnack, J.; Tasker, P.A.; Carretta, S.; Brechin, E.K. Linking [FeIII3] triangles with “double-headed” phenolic oximes. Chem. Commun. 2011, 47, 6018–6020. [Google Scholar] [CrossRef] [PubMed]
- Holynska, M.; Clerac, R.; Langer, T.; Poettgen, R.; Dehnen, S. Selective syntheses, structures and magnetic properties of Fe(III) complexes of different nuclearities. Polyhedron 2013, 52, 1425–1430. [Google Scholar] [CrossRef]
- Wenzel, M.; Forgan, R.S.; Faure, A.; Mason, K.; Tasker, P.A.; Piligkos, S.; Brechin, E.K.; Plieger, P.G. A New Polynuclear Coordination Type for (Salicylaldoxime)copper(II) Complexes: Structure and Magnetic Properties of an (Oxime)Cu6 Cluster. Eur. J. Inorg. Chem. 2009, 2009, 4613–4617. [Google Scholar] [CrossRef]
- De Silva, D.N.T.; Dais, T.N.; Jameson, G.B.; Cutler, D.J.; Brechin, E.K.; Davies, C.G.; Jameson, G.N.L.; Plieger, P.G. Synthesis and Characterization of Symmetrically versus Unsymmetrically Proton-Bridged Hexa-Iron Clusters. ACS Omega 2021, 6, 16661–16669. [Google Scholar] [CrossRef]
- Aldred, R.; Johnston, R.; Levin, D.; Neilan, J. Magnesium-mediated ortho-specific formylation and formaldoximation of phenols. J. Chem. Soc. Perkin Trans. 1994, 1, 1823–1831. [Google Scholar] [CrossRef]
- Stevens, J.R.; Plieger, P.G. Anion-driven conformation control and enhanced sulfate binding utilising aryl linked salicylaldoxime dicopper helicates. Dalton Trans. 2011, 40, 12235–12241. [Google Scholar] [CrossRef] [PubMed]
- Forgan, R.S.; Davidson, J.E.; Galbraith, S.G.; Henderson, D.K.; Parsons, S.; Tasker, P.A.; White, F.J. Transport of metal salts by zwitterionic ligands; simple but highly efficient salicylaldoxime extractants. Chem. Commun. 2008, 4049–4051. [Google Scholar] [CrossRef] [PubMed]
- Steiner, T. Reviews: The hydrogen bond in the solid state. Angew. Chem. Int. Ed. 2002, 41, 48–76. [Google Scholar] [CrossRef]
- Mössbauer Spectroscopy of Environmental Materials and Their Industrial Utilization; Murad, E., Cashion, J., Eds.; Springer: New York, NY, USA, 2004; p. 440. [Google Scholar]
- Wang, Q.; Wilson, C.; Blake, A.J.; Collinson, S.R.; Tasker, P.A.; Schroeder, M. The one-pot halomethylation of 5-substituted salicylaldehydes as convenient precursors for the preparation of heteroditopic ligands for the binding of metal salts. Tetrahedron Lett. 2006, 47, 8983–8987. [Google Scholar] [CrossRef]
- Plieger, P.G.; Tasker, P.A.; Galbraith, S.G. Zwitterionic macrocyclic metal sulfate extractants containing 3-dialkylaminomethylsalicylaldimine units. Dalton Trans. 2004, 313–318. [Google Scholar] [CrossRef]
- CrystalClear; 1.4.0; Rigaku Americas Corporation: The Woodlands, TX, USA, 2005.
- PROCESS-AUTO; Rigaku Corporation: Tokyo, Japan, 1998.
- Kabsch, W. XDS. Acta Crystallogr. Sect. D 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. Sect. D 2011, 67, 235–242. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef]
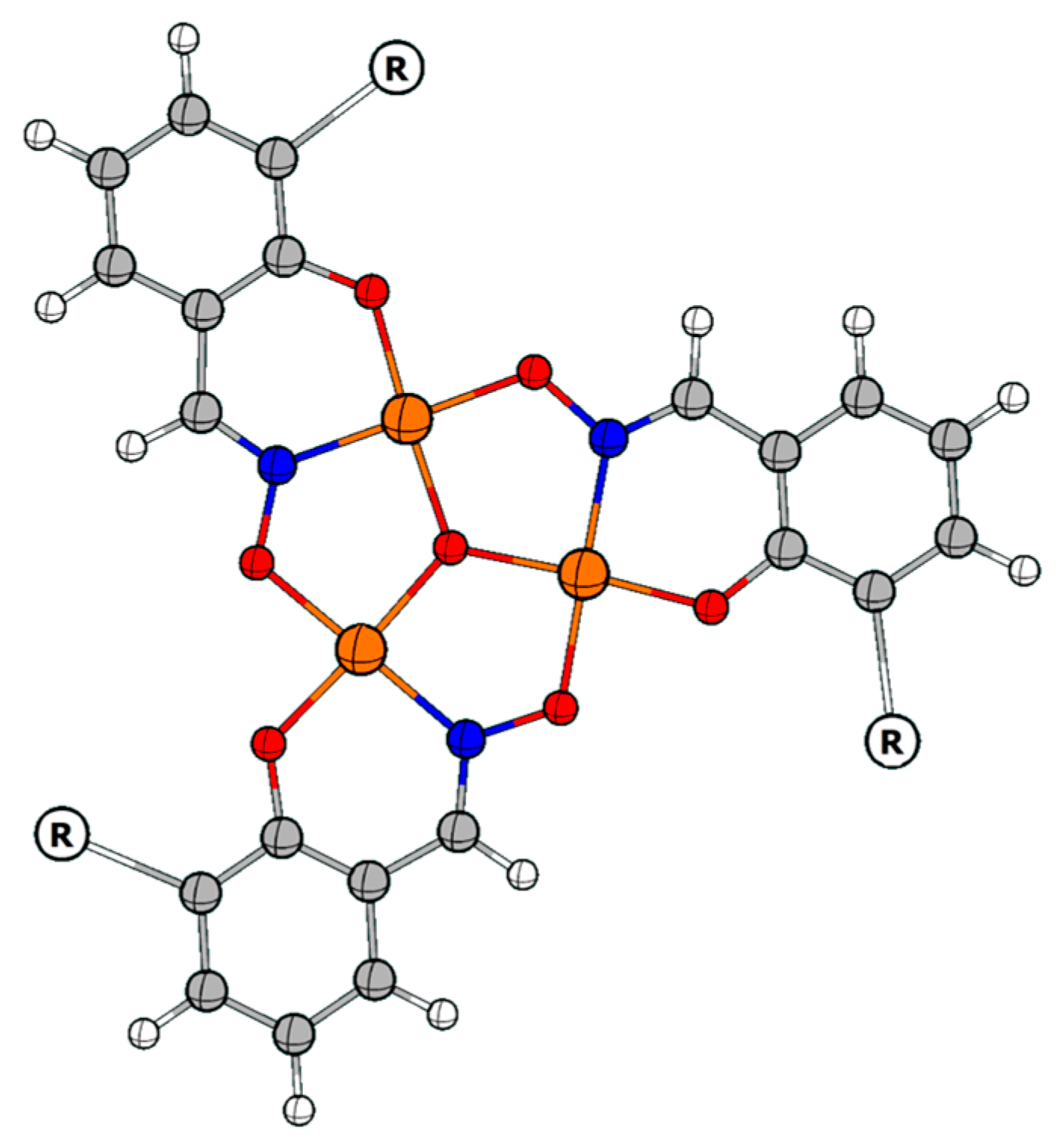
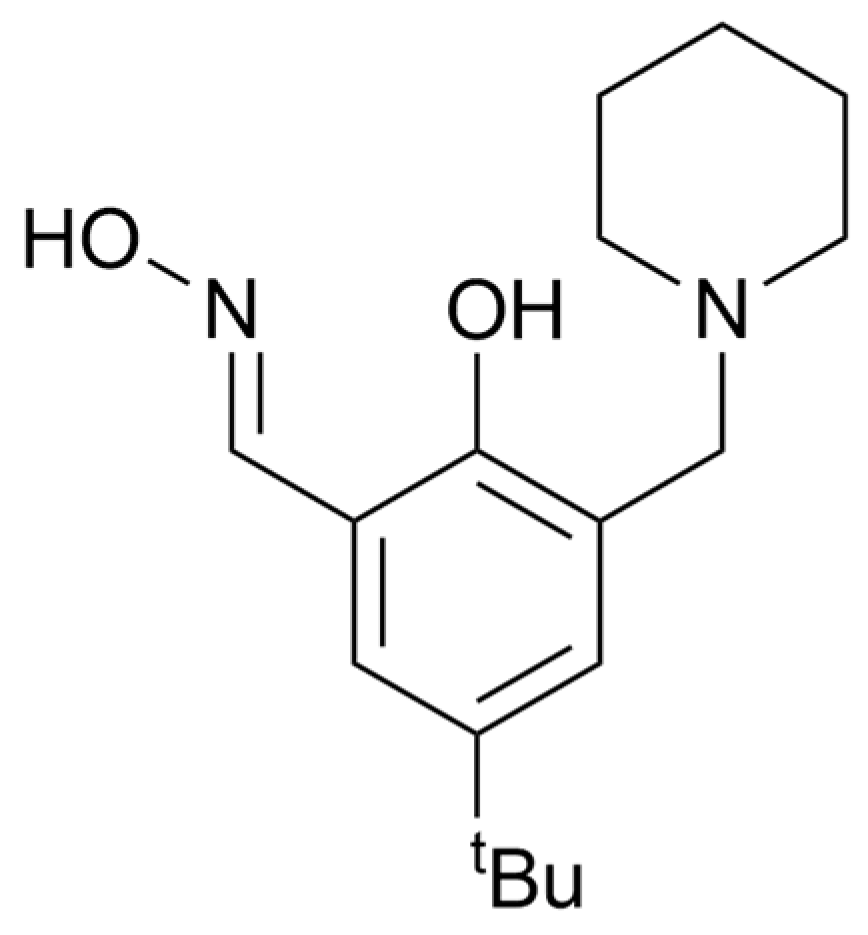
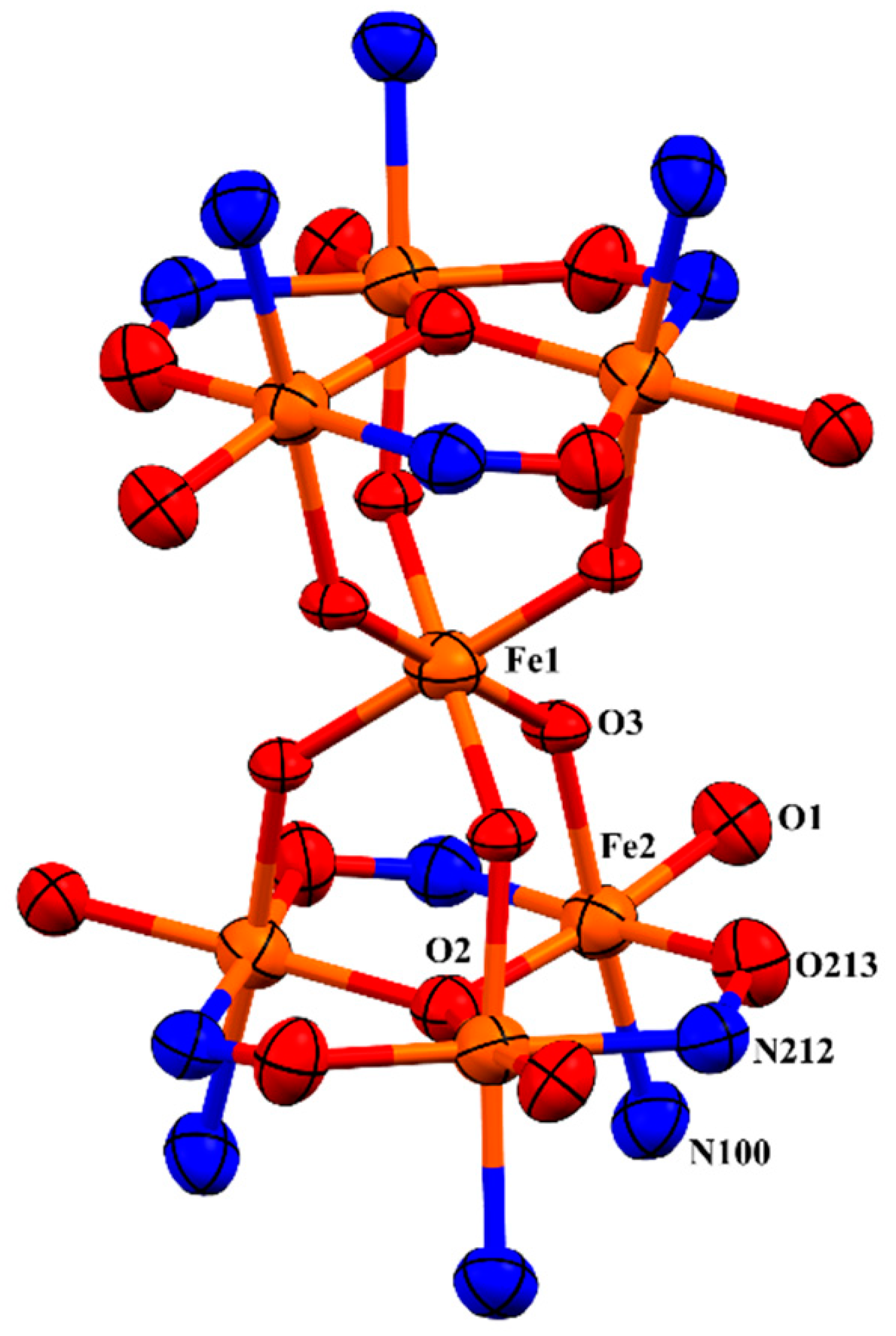
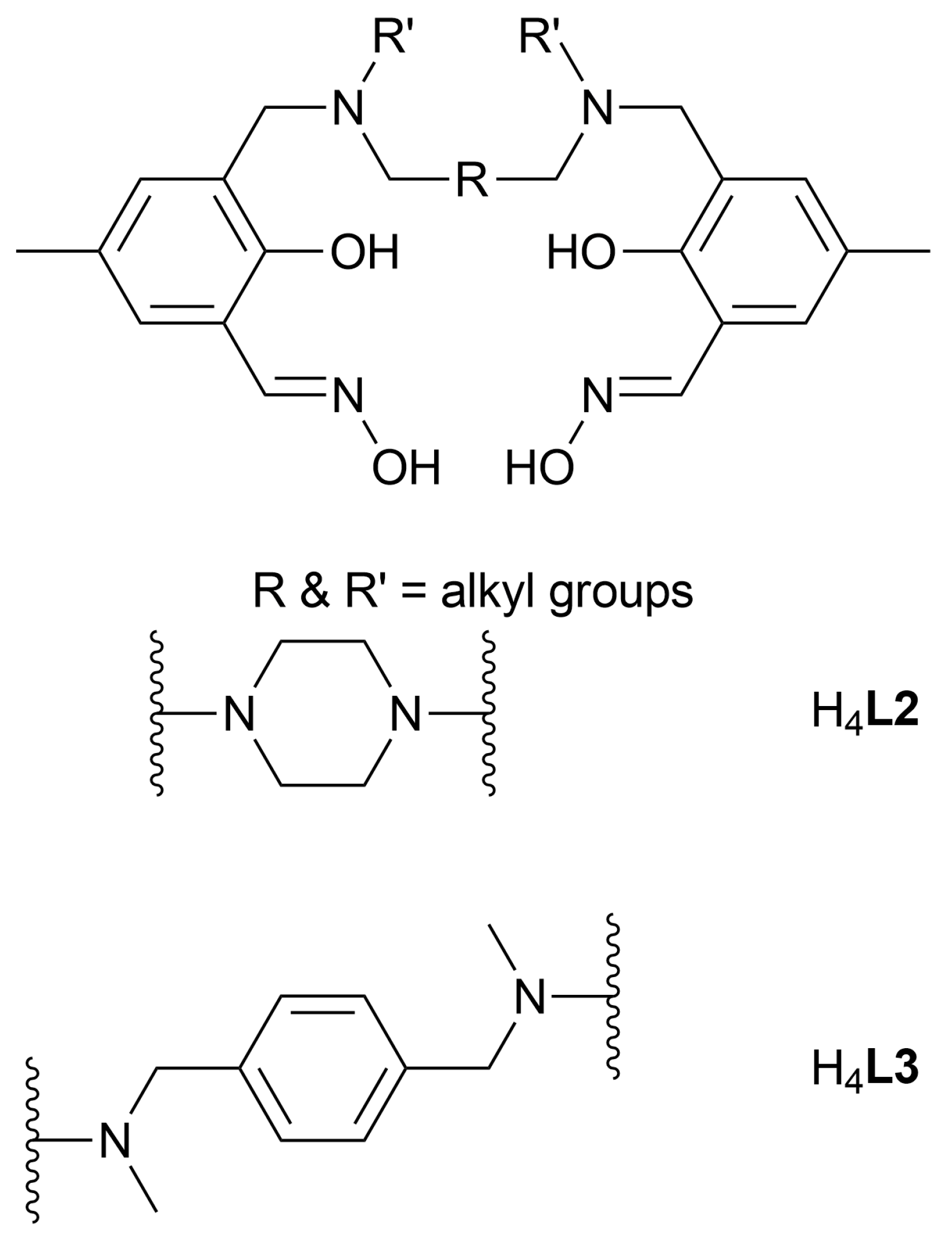
| Atoms | Length (Å) | Atoms | Length (Å) |
|---|---|---|---|
| Fe2–O2 | 1.9287 (11) | Fe2–N100 | 2.208 (4) |
| Fe2–O213 | 1.959 (3) | Fe2–O3 | 2.043 (3) |
| Fe2–O1 | 1.939 (3) | Fe1–O3 | 1.920 (2) |
| Fe2–N212 | 2.124 (3) | O213–N212 | 1.378 (6) |
| Atoms | Angle (°) | Atoms | Angle (°) |
| N100–Fe2–O2 | 94.9 (2) | O1–Fe2–O2 | 172.6 (2) |
| N100–Fe2–O213 | 90.1 (2) | O213–Fe2–N212 | 176.5 (2) |
| N100–Fe2–O1 | 88.1 (2) | O1–Fe2–O213 | 91.5 (2) |
| N100–Fe2–N212 | 90.0 (2) | O213–Fe2–O2 | 95.2 (2) |
| O3–Fe2–O1 | 87.0 (2) | O2–Fe2–N212 | 88.3 (2) |
| O3–Fe2–O2 | 89.7 (2) | N212–Fe2–O1 | 85.1 (2) |
| O3–Fe2–N212 | 87.2 (2) | Fe1–O3–Fe2 | 133.8 (2) |
| Fe2–O2–Fe2 * | 117.4 (2) | O3–Fe1–O3 * | 89.8 (2) |
| N100–Fe2–O3 | 174.5 (2) |
| C2 | C3 | |
|---|---|---|
| Atoms | Length (Å) | Length (Å) |
| Fetri–µ3O | 1.881 (7)–1.952 (7) | 1.913 (4)–1.936 (3) |
| Fetri–µ2OH | 1.997 (6)–2.034 (6) | 2.036 (3)–2.045 (3) |
| Femid–µ2OH | 1.911 (6)–1.969 (6) | 1.964 (3)–1.972 (3) |
| Feplane–µ3O | 0.338–0.376 | 0.330 |
| Femid–µ3O | 3.447 (7)–3.491 (8) | 3.512 (4) |
| µ3O–µ3O | 6.938 (10) | 7.024 (4) |
| Atoms | Angle (°) | Angle (°) |
| Fetri–µ3O–Fetri | 114.6 (3)–119.2 (4) | 116.2 (2)–118.0 (2) |
| Fetri–µ2OH–Femid | 131.6 (4)–136.3 (4) | 134.6 (6)–135.3 (6) |
| µ3O–Femid–µ3O | 178.2 (2) | 180 |
| Complex | δ (mm/s) | ΔEQ (mm/s) | ΓL (mm/s) | ΓR (mm/s) | I (%) |
|---|---|---|---|---|---|
| C1 | 0.41 | 0.86 | 0.37 | 0.37 | 77.8 |
| 0.40 | 0.50 | 0.21 | 0.21 | 28.9 | |
| C2 | 0.40 | 1.50 | 0.35 | 0.35 | 70 |
| 0.40 | 0.55 | 0.35 ± 0.15 | 0.35 ± 0.15 | 30 | |
| C3 | 0.40 | 1.55 | 0.30 | 0.30 | 75 |
| 0.35 | 0.45 | 0.35 | 0.35 | 26 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Silva, D.N.T.; Dais, T.N.; Jameson, G.B.; Davies, C.G.; Jameson, G.N.L.; Plieger, P.G. [Fe(µ2-OH)6]3− Linked Fe3O Triads: Mössbauer Evidence for Trigonal µ3-O2− or µ3-OH− Groups in Bridged versus Unbridged Complexes. Molecules 2024, 29, 3218. https://doi.org/10.3390/molecules29133218
De Silva DNT, Dais TN, Jameson GB, Davies CG, Jameson GNL, Plieger PG. [Fe(µ2-OH)6]3− Linked Fe3O Triads: Mössbauer Evidence for Trigonal µ3-O2− or µ3-OH− Groups in Bridged versus Unbridged Complexes. Molecules. 2024; 29(13):3218. https://doi.org/10.3390/molecules29133218
Chicago/Turabian StyleDe Silva, D. Nirosha T., Tyson N. Dais, Geoffrey B. Jameson, Casey G. Davies, Guy N. L. Jameson, and Paul G. Plieger. 2024. "[Fe(µ2-OH)6]3− Linked Fe3O Triads: Mössbauer Evidence for Trigonal µ3-O2− or µ3-OH− Groups in Bridged versus Unbridged Complexes" Molecules 29, no. 13: 3218. https://doi.org/10.3390/molecules29133218
APA StyleDe Silva, D. N. T., Dais, T. N., Jameson, G. B., Davies, C. G., Jameson, G. N. L., & Plieger, P. G. (2024). [Fe(µ2-OH)6]3− Linked Fe3O Triads: Mössbauer Evidence for Trigonal µ3-O2− or µ3-OH− Groups in Bridged versus Unbridged Complexes. Molecules, 29(13), 3218. https://doi.org/10.3390/molecules29133218