Abstract
Copper squarate is a metal–organic framework with an oxo-carbonic anion organic linker and a doubly charged metal mode. Its structure features large channels that facilitate the adsorption of relatively small molecules. This study focuses on exploring the potential of adsorbing small pollutants, primarily greenhouse gases, with additional investigations conducted on larger pollutants. The objective is to comprehend the efficacy of this new material in single and multiple molecular adsorption processes using theoretical methods based on density functional theory. Furthermore, we find that the molecular adsorption energies range from 3.4 KJ∙mol−1 to 63.32 KJ∙mol−1 depending on the size and number of adsorbed molecules. An exception is noted with an unfavorable adsorption energy value of 47.94 KJ∙mol−1 for 4-nitrophenol. More importantly, we demonstrate that water exerts an inhibitory effect on the adsorption of these pollutants, distinguishing copper squarate as a rare MOF with hydrophilic properties. The Connolly surface was estimated to give a more accurate idea of the volume and surface accessibility of copper squarate. Finally, using Monte Carlo simulations, we present a study of adsorption isotherms for individual molecules and molecules mixed with water. Our results point out that copper squarate is an efficient adsorbent for small molecular pollutants and greenhouse gases.
1. Introduction
Nowadays, scientists around the world are actively seeking new porous materials to tackle the pollution crisis caused by urbanization and emissions from motor vehicles and industry [1,2]. Extensive research has been conducted, resulting in the publication of in-depth studies on air emissions and the various types of pollutants present in our atmosphere, such as carbon dioxide (CO2), the main contributor to the greenhouse effect, which is largely responsible for global warming. Additionally, methane (CH4), tropospheric ozone (O3), dinitrogen monoxide (N2O), nitrogen trifluoride (NF3), and sulfur hexafluoride (SF6) are critical trace gases that contribute to climate change. These six compounds are among the main greenhouse gases identified in the Kyoto Protocol [3,4,5,6].
In addition, the combustion of coal, fossil fuels, or biomass also releases air pollutants, such as sulfur dioxide (SO2) and volatile organic compounds (VOCs), which contribute significantly to industrial and vehicle exhaust pollution. At the same time, VOCs produce a variety of pollutants, including polycyclic aromatic hydrocarbons (PAHs) derivatives such as NPAH nitrates. This category includes, for example, 4-nitrophenol (4NP), which is considered one of the most hazardous pollutants in our daily lives [7,8,9,10,11,12,13,14,15]. Nitrophenols are classified as priority pollutants by the U.S. Environmental Protection Agency [16]. Due to their high toxicity, atmospheric pollutants have considerable negative effects on human health, wildlife, and global warming. For this reason, emphasis has been placed on experimental and theoretical studies concerning the application of separation, adsorption, gas capture and storage, and selective catalysis as potential solutions [17,18,19,20,21,22].
Recently, metal–organic frameworks (MOFs) have been in great demand for the capture and storage of gaseous pollutants due to their extraordinary porosity, surface area, crystallinity, controllable structure, and chemical separation capabilities [23,24,25]. Consequently, a plethora of work has been carried out on various materials, such as zeolitic imidazolate frameworks (ZIFs), clay minerals, lignite, anthracites, and more, to adsorb pollutants [26,27,28,29,30]. The presence of water has also been considered, providing valuable information on its impact on the adsorption process [31,32,33].
In this paper, we will focus on a type of MOF [34] that has not yet been explored for adsorption or capture processes: the copper squarate (C-S) metal–organic framework. The crystal structure of this material was first analyzed and described in detail by Dinnebier et al. [35,36]. C-S was subsequently investigated by Colmenero et al. [37] using theoretical solid-state methods based on density functional theory (DFT). This study firmly established the crystal structure of copper squarate, previously determined using a restricted rigid-body Rietveld refinement, which discovered that it exhibits the phenomenon of negative linear compressibility due to the presence of the empty channel structural motif. Colmenero et al. demonstrated that other squarate MOFs, such as zinc [36] and uranyl squarates [38], also exhibit negative linear compressibility. In fact, Colmenero [39] has shown that oxo-carbonic acids in the solid state exhibit extremely anomalous mechanical behavior, and recently, Qiu et al. [40] have revealed that lead and barium squarates exhibit zero linear compressibility and thermal expansion effects over a wide range of pressures. The presence of empty spaces inside these structures and their large pore size have led us to believe in the viability of these materials for the capture and storage of hazardous gases. As far as we know, squarate MOFs have only been recently studied for molecular adsorption applications. Namely, the use of cobalt squarate MOF has recently been investigated for CO2/N2 separation by Zhang et al. [41].
The aim of our work is to characterize the behavior of squarate materials in molecular adsorption applications, which have not yet been explored, using solid simulation techniques. Specifically, we will investigate the ability of copper squarate to absorb a series of different classes of gaseous pollutants in the presence and absence of water. We will closely examine its response to these pollutants, determining its preferred adsorption sites, energies, and mechanisms, as well as its adsorption isotherms. Unfortunately, to our knowledge, no experimental or theoretical studies have been published on adsorption in copper squarate. The present work is therefore the first to provide theoretical results that will serve as a starting point for further experiments. The exploration of new materials for the adsorption of contaminants is crucial to counter the wave of pollution caused by urbanization that we are currently experiencing.
2. Results and Discussion
The copper squarate crystal structure was re-optimized, and the results obtained are fairly consistent with those obtained by Colmenero et al. [36]. The powder X-ray diffraction (PXRD) patterns of copper squarate were derived from the computed and experimental structures using the program REFLEX included in the Materials Studio program suite [42]. The results are displayed in Figure S1 of the Supplementary Materials and, as can be seen, they are consistent. Because of the small size of the unit cell, it was doubled along the axis to facilitate the adsorption of the pollutant, as was conducted in previous works studying the adsorption of similar molecules in materials like zeolite [43]. The calculations were then continued with a series of optimizations of the double unit cell containing the pollutant gases. These gases were introduced inside using the Adsorption Locator tool of Materials Studio [42] in order to conduct the adsorption processes inside this material. The results are presented in Table 1, where the lattice parameters of the crystal unit and the double cell of copper squarate are provided, as well as the parameters of the cells distorted due to the capture of one or more pollutants and water molecules. The optimized geometries of different species are compared with the corresponding experimental data in Table S1 of the supporting information. The optimized structure of copper squarate is shown in Figure 1. The optimized copper squarate unit and double cell can be downloaded from the Supplementary Information (Table S2).

Table 1.
Structural parameters of copper squarate (C-S) are in the pure form and contain adsorbed pollutant molecules and water separately. The experimental unit cell parameters are from Ref. [36].
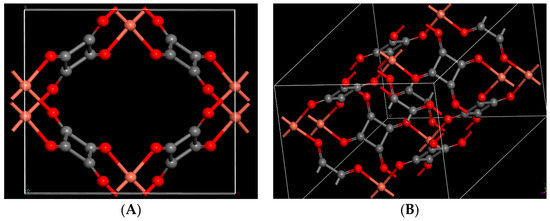
Figure 1.
Optimized crystal structure of copper squarate: (A) single unit cell; (B) double unit cell along edge. (Color code: Red: Oxygen/Orange: Copper/Gray: Carbon).
2.1. Crystal Bulk Structure and Analysis of Connolly Free Volume and Surface Area Properties
In this section, we employ another feature of Materials Studio 22.0 [42], the “Atoms Volume and Surfaces” to approximate the Connolly surface of copper squarate and the pollutants separately using the probe radius. This allows us to thoroughly explore the cavity and surface features of the internal complex structure after the adsorption process [44] and to obtain the occupied and free volumes, as well as surface area (Table 2). These parameters are then used to calculate the pore volume using the expression below [45]:
Table 2.
Connolly free volume, surface area of supercell sizes, and density of copper squarate.
From Table 2, we notice a good agreement between the experimental density (2.427 ) and the theoretical value (2.263 ), showing the accuracy of the DFT-D3 method in describing unit cell structures in crystalline materials. The total pore volume of copper squarate (1 × 1 × 2 cell) is expected to be 0.0958 , i.e., 23% of the total cell volume reported in Table 1. This value is close to the experimental one (24.6%) approximated from available data (density and cell volume) in the work of Colmenero et al. [36] considering the (1 × 1 × 2) cell, as well as using the theoretical cell free volume. All these calculations use a probe radius of 1.0 Å and a grid interval of 0.6 Å [45].
Moreover, we employed the same grid interval of 0.6 Å for the Connolly surface parameters to estimate the volume and surface area of other adsorbed species. Meanwhile, we varied the probe radius from one molecule to another due to the electronic density of those molecules [32,45]. For H2O, SO2, N2O, and CH4, we use a probe radius of 1.37 Å due to their similar electronic cloud characteristic except for CH4. The corresponding results were (18.99 Å3/34.79 Å2), (40.48 Å3/58.08 Å2), (28.87 Å3/50.42 Å2), and (27.47 Å3/46.18 Å2), respectively. In the case of 4NP, a slightly larger probe radius of 1.4 Å was employed to achieve a result of (115 Å3/138.02 Å2). Finally, for the more electronegative molecules CO2, O3, NF3, SF6, CF4, and 2NP, a probe radius of 1.7 Å was chosen, considering their higher electron density due to hindrance, resulting in (32.28 Å3/51.37 Å2), (31.25 Å3/50.37 Å2), (40.71 Å3/60.59 Å2), (74.81 Å3/89.45 Å2), (50.04 Å3/69.05 Å2), and (110.49 Å3/134.58 Å2), respectively. These results indicate the ability of C-S to adsorb the pollutants listed, except 2NP and 4NP, which have almost the same Connolly surface and volume as the filter, potentially causing repulsive disturbances.
To delve deeper into the understanding of C-S capacity as a filter and elucidate the outcomes of the Connolly analysis, we employed the DFT-D3 veracity method. This method allows us to ascertain the maximum quantity of adsorbed pollutants within the bulk. Table 3 shows the structural parameters of the co-adsorption process.
Table 3.
The structural parameters of copper squarate containing a co-adsorbed mixture of a pollutant with water.
2.2. Adsorption Analysis
Achieving optimal DFT performance in this study involves a hierarchical approach. First, we locate the most stable configuration in conformational space, as was previously described using the Adsorption Locator module with COMPASS III force field [46], by dropping the molecule at various pressures employing the Metropolis algorithm. The DFT geometry is then optimized for the most favorable configuration. (Figure 2) shows the single final adsorption structure of pollutants and water in C-S and (Table 4) gives the energy data for the adsorption process. Negative adsorption energies indicate favorable capture of gases [31].
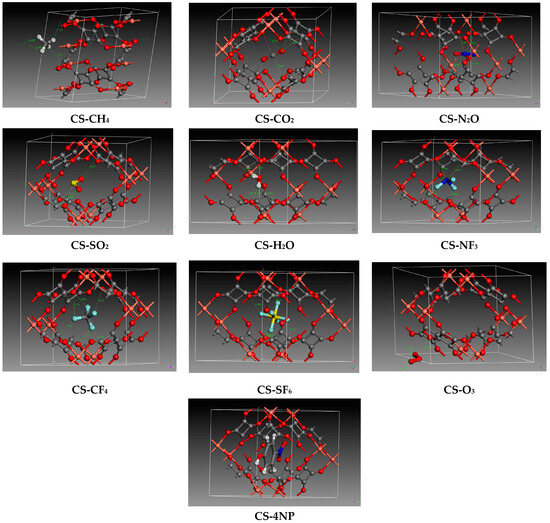
Figure 2.
Zoomed interactions of optimized copper squarate containing single pollutants or water. (Color code: Red: Oxygen/Orange: Copper/Gray: Carbon/White: Hydrogen/Royal blue: nitrogen/Yellow: Sulfur/Light blue: Fluorine).
Table 4.
Adsorption and co-adsorption energies in copper squarate calculated with CASTEP.
2.2.1. Pollutants and Water Adsorption
The results indicate favorable adsorption of CO2, N2O, SO2, and H2O molecules into C-S using force field loading, with up to five, one, one, and eighteen species per double unit cell, respectively. The accuracy of these findings was validated by CASTEP calculations, revealing adsorption energies of −18.93, −63.32, −38.04, and −51.16 , respectively, for a single captured molecule. This suggests the existence of electrostatic interactions, as illustrated in the zoomed images in Figure 2. For all four species, we observe a short consistent interaction involving C-S oxygen with C (CO2), N (N2O), S (SO2), and strong hydrogen [47] bound with H (H2O), comprising a bound distance of 2.811, 2.954, 2.775, and 1.966 Å, respectively. In addition, a weaker interaction is evident with C (C-S), interacting with O (CO2), N (N2O), O (SO2), and O (H2O) at distances ranging from 2.847–3.080 Å.
In Table 4, the calculated energies are CH4 (−219.678 eV), CO2 (−1016.651 eV), O3 (−1285.930 eV), SF6 (−4198.029 eV), NF3 (−2218.200 eV), CF4 (−2751.691 eV), N2O (−979.812 eV), SO2 (−1165.160 eV), H2O (−463.290 eV), 4NP (−2574.182 eV), and C-S (−28,157.254 eV), referring to the number of adsorbed pollutant and water molecules.
Generally, metal–organic frameworks (MOFs) are known to be unstable in humid environments. Zeolite has been highlighted in the literature for its hydrophobic nature [48,49,50]. However, recent experimental studies have identified exceptions, such as ZIF-8 and ZIF-90, which show stability in the presence of water [51,52]. In our study, we theoretically demonstrate that C-S exhibits stability in the presence of water, which is consistent with the hydrophilic behavior observed in some MOFs. In particular, we show that O-H bond lengths in water approach 0.966 Å, which closely matches the experimental value of 0.98 Å [52].
Methane is a non-polar molecule and has been the subject of previous adsorption studies, particularly on zeolites [45]. In our study, different quantities of CH4 ranging from one to four were absorbed and optimized to identify the most favorable insertion sites. Our results are consistent with those of a previous study in which four methane molecules were observed inside the ZIF-4 cell [32]. By analyzing the modeled structures, we observed that all CH4 molecules are in close proximity to the C-S, involving a hydrogen interaction with the oxygen atom of C-S, with a bonding distance of 2.737 Å, as illustrated in (Figure 2). This interaction represents a limiting case of hydrogen bonding, characterized by a weak donor to a strong acceptor due to their substantial contribution to dispersion [47]. This contributes to system stabilization, resulting in negative adsorption values ranging from −19.42 to −23.02 for one and four CH4 uptake molecules, respectively.
From Table 4, we can observe high adsorption energies of −30.51, −50.05, and −26.94 for the adsorption of one, three, and five O3 molecules, respectively, including a strong uptake within the bulk. This is attributed to the reactive nature and high polarity of ozone due to the presence of oxygen atoms. Capture involves intermolecular Van der Waals forces, mainly OO3-OC-S with an average bond length of 2.8 Å for all the optimized structures. In this case, the charged fluctuation of the oxygen and the electronic density leads to a sensitive instantaneous dipole moment, creating a type of London dispersion.
The vacuum inside the cavity allows the capture of one molecule with fluorine atoms due to the steric effect of the electronic density of the halogen, causing a repulsion with the oxo-carbonic anion of the copper squarate. We observed that the adsorption energy is −14.12 for NF3, while it decreases consistently for CF4 and SF6, with values of −3.40 and −3.78 , respectively. We deduce that as the number of fluorine atoms increases, the adsorption energy decreases. Additionally, we notice that when the central atom is nitrogen, the adsorption energy is higher, as seen with N2O. The optimized lengths are shown in Figure 2, where it can be seen that the carbon of C-S interacts with the fluorine of CF4 and NF3 at 2.7 and 2.8 Å, while a shorter value of 1.98 Å is noted with SF6 because its octahedral form minimizes the distance between the pollutant and the bulk.
A positive adsorption energy of +47.94 was noted for 4NP (Table 4), indicating unfavorable capture of the aromatic pollutant by the C-S. This is in contradiction with the adsorption locator calculation, which allows the uptake of one molecule. This implies the lack of accuracy of the force field and the foremost importance of using the DFT-D3 method to complement and achieve a reliable calculation.
2.2.2. Co-Adsorption of Pollutants and Water
In this section, we study the effect of a humid environment on the adsorption inside copper squarate bulk. The adsorption energy and interaction energy are calculated using Equations (2) and (3), respectively, and the results are presented in Table 4.
According to Table 4, the co-adsorption of the pollutant with water gives negative adsorption energy, revealing favorable adsorption energy, except for 4NP. The latter has a positive adsorption value in both dry and humid environments, which can be caused by the unavailability of sufficient space on the pores due to its large size and radius.
For CH4, SO2, and CO2, the adsorption energies increased in the presence of water to reach −33.11, −43.48, and −40.39 , respectively. This implies the same interaction as above between the pollutant and the bulk. In addition, weak hydrogen bonds are formed between the water and the pollutant, which further increases the adsorption energy. The same trend is noted for molecules with fluorine atoms, where the values of the mixed adsorption energy increased to reach −26.83, −41.31, and −6.186 for NF3, CF4, and SF6, respectively. The important impact on the adsorption energetic value is due to the multiple interactions caused by the reactive sites of these types of pollutants. In contrast, for ozone and dinitrogen monoxide, the adsorption energy decreases slightly but remains favorable to the adsorption process. Most of the interaction energies are positive, indicating repulsion between the species and far adsorption between the small pollutants and the water inside the pore. This is due to the electron density, as depicted in Figure 3.
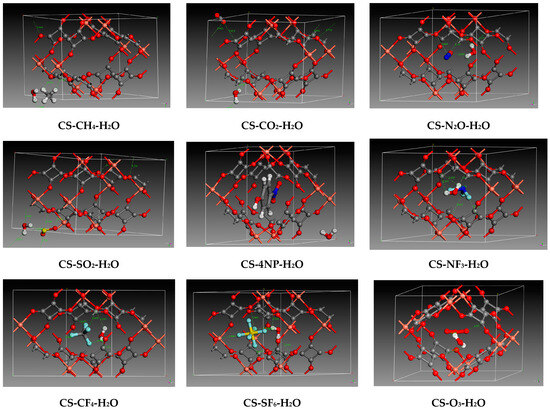
Figure 3.
Zoomed interactions of optimized copper squarate containing co-adsorbed pollutants with water. (Color code: Red: Oxygen/Orange: Copper/Gray: Carbon/White: Hydrogen/Royal blue: nitrogen/Yellow: Sulfur/Light blue: Fluorine).
Finally, the adsorption of 4NP in the presence of water does not counterbalance capture towards a favorable process. In previous work on nitrophenol on activated carbon bulk under humid conditions, the authors observed a possibility of adsorption by water as it moved to place itself between 4NP and the surface bulk, creating hydrogen bonds and electrostatic interactions. This suggests a lack of space inside the C-S pore, which can create repulsion due to the high charge density of water, 4NP, and C-S cell, as can be seen in Figure 3 where water is placed in the cell extremity, not inside. We also noticed this in the previous section on the Connolly free volume, where we noted that 4NP and 2NP have almost the same volume. The interaction energy here is negative due to the hydrogen bonding of the aromatic compound to the copper squarate.
These fluctuations in no way interfere with copper squarate’s favorable ability to capture these small pollutants. On the contrary, they positively highlight the fact that the adsorbent can capture them in both dry and humid states, without the presence of water playing a counterbalancing role. It is important to remember that the small pollutants, as well as water, are energetically stable separately in the C-S.
The main geometric parameters of the co-adsorption (water/pollutant) were analyzed and are reported in Table 5 above. The results indicate that there is no significant change in the bond distance and valence angle between them compared with the isolated adsorbed form. This is due to the presence of more free space, allowing more water molecules to be captured, confirming the observation that the interaction energy is positive. The presence of additional adsorbed molecules can lead to an increase in distortion due to space limitations. This phenomenon has already been observed in solid simulations, notably in studies of methane, where the angle of increased from 109° to 114° [45].
Table 5.
Main geometrical parameters for the optimized co-adsorption (1 X −1 H2O) structures (distances in Å and angles in degrees) on copper squarate X = (CH4, CO2, O3, SF6, NF3, CF4, N2O, and SO2).
2.3. Sorption Isotherms
The isothermal sorption of pollutants and water in singular and mixed forms was studied at 298 K using the “sorption isotherm” module implemented in Materials Studio 22.0 [42]. This study is based on random moves to perform energy changes for the new conformation. This is by measuring the chemical potential, expressed as the gas fugacity with pressure inside the cell bulk. The results are shown in Figure 4, indicating the average adsorption capacity of gas molecules by the copper squarate.
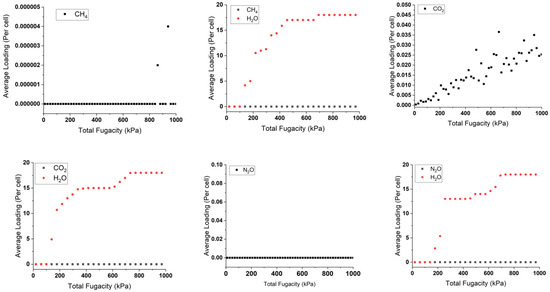
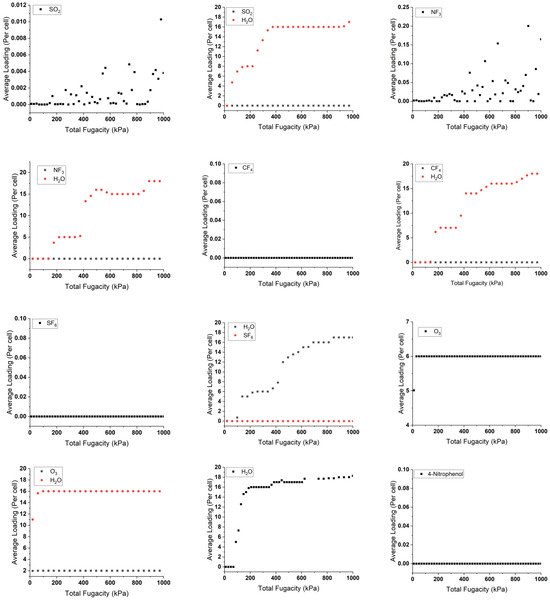
Figure 4.
Sorption isotherms of single and mixture adsorbed pollutants with and without water at 298 K.
By analyzing the adsorbed singular graphs in Figure 4, we observe that most of the pollutants present a consistent loading profile with molecules inserted around zero or zero per the considered unit cell in low- or high-pressure variations. For example, N2O shows zero loaded molecules, while the adsorption study shows high negative adsorption energy of −63.32 kJ/mol, indicating a favorable capture. This aligns also with the Connolly studies, which show available space inside copper squarate. These particular results contradict the precise results obtained by the DFT-D3 method. These strange results can be attributed to the limitations of the force field used in this C-S model, which includes pressure and is unable to accurately describe the unusual properties of the material, in particular its negative linear compressibility [36], highlighting the necessity of complementing it with ab initio calculations. In contrast, water is well described, showing an uptake loading of 15 molecules per cell, which is in good agreement with CASTEP calculations. This agreement may indicate the established hydrophilic nature of the solid. Similarly, ozone molecules show comparable behavior due to their common nature with water.
In the presence of water, the line graph representing the pollutant does not change. There are two reasons for this. The first reason is the same as when they are loaded individually. The second reason may be due to the hydrophilicity of water, which dominates the pollutants and competes with them, reducing their loading capacity in the cell. This phenomenon is evident with ozone, which loads around three molecules instead of six in a dry environment. This is due to the stronger hydrogen bonds between the water and the copper squarate to the intermolecular interactions between the pollutants and the bulk.
3. Quantum Computational Methods
All calculations for the solid-state compounds considered in this work were performed with CASTEP code [53] using theoretical methods based on DFT using norm-conserving pseudopotentials and plane wave basis sets [54]. Due to the large pore size of MOFs, which contain hundreds of atoms or more, quantum mechanical methods such as post-HF are impractical for such large systems [55]. The geometry optimizations were performed employing the Broyden–Fletcher–Goldfarb–Shanno scheme (BFGS) [55,56]. We treated the exchange-correlation energy term using the generalized gradient approximation (GGA) with Perdew–Burke–Ernzerhof (PBE) density functionals [57]. Since the pollutant molecules are packed together inside the unit cell of copper squarate, their description requires the inclusion of Van der Waals forces. For this purpose, Grimme’s empirical dispersion correction method (DFT-D3) for dispersive vdW interactions was used in calculations [58,59]. Customized atom pseudopotentials [60] generated with the CASTEP code and a plane wave kinetic energy cut-off of 1000 eV were utilized. Convergence criteria were set at 2 × 10−5 eV/atom for energy, 0.05 eV/Å for interatomic forces, 0.1 GPa for maximum stress, and 0.002 Å for displacements. Atomic forces and charges were evaluated at the minimum in potential energy surface to predict the harmonic vibrational infrared spectrum using density functional perturbation theory [61,62].
After a full geometry optimization of all CH4, CO2, O3, SF6, NF3, CF4, N2O, SO2, 4NP, water, and copper squarate, the most stable adsorption sites of the pollutants inside the solid material were determined in order to minimize the first adsorption energy. The adsorption sites and mechanisms within the copper squarate were determined by Monte Carlo simulations [63], implemented in the Adsorption Locator module using COMPASS III [46] force field in Materials Studio 22.0 [42] code. Simulations involve a fraction of 10,000 steps following a number of cycles of 3 with 15,000 steps per cycle. Subsequent to the initial results, Density Functional Theory (DFT) calculations were conducted to optimize performance by adjusting the generic force fields within the Adsorption Locator module. More specifically, it was already proved that force fields such as COMPASS III [46] and Dreiding [64] tend to overestimate the number of molecules adsorbed in simulations [65], as was demonstrated in previous studies involving ZIF-4 or ZIF-6 [32,45].
The adsorption process occurs through chemical or physical binding, facilitated by the attractive forces between the solid cavity of the copper squarate and the pollutants, in both dry and humid conditions. The adsorption energies were calculated according to equations [66]:
In these equations, refers to the isolated adsorption energy for each individual species () and denotes the mixed adsorption energy for species (). stands for the total energy of copper squarate (C-S) with gas molecules included, corresponds to the energies of pure C-S, and is the energy of the respective pollutants. In addition, , , represent the quantities of loaded molecules. The assessment of pollutants and water is conducted using a 10 Å supercell. A negative adsorption energy implies favorable adsorption.
The interaction energies between the co-adsorbed pollutants on the C-S material were calculated with the help of the energetic database obtained from DFT calculations using equation [45]:
where represents the total energy of the solid material after adsorption of the n pollutants by copper squarate bulk, and is the sum of the total energy of the previously adsorbed species calculated separately.
The Grand Canonical Monte Carlo (GCMC) method [67] implemented in the Sorption module of Materials Studio 22.0 [42] was used to determine adsorption isotherms with the Metropolis algorithm and COMPASS III. The gas molecules were randomly loaded at a fixed temperature of 298 K into the fixed-volume copper squarate structure based on the gas fugacity. The simulations involve 100,000 equilibration steps followed by 1,000,000 Monte Carlo steps, assuming periodic boundary conditions. Long-range interactions were treated using the Ewald and group summation method with an Ewald accuracy of 0.0001 kcal/mol and a cut-off distance of 15.5 Å. The results are presented as a line graph (N = f (µ)), where f is the density of the number of loaded molecules N, expressed as a function of chemical potential.
4. Conclusions
In this work, we focus our interest on a new kind of MOF, namely copper squarate, whose adsorption behavior has not been explored in previous studies. For this reason, the adsorption of a series of small pollutants and some aromatic compounds was investigated using DFT calculations in a dry and humid environment. As a first observation, we note that small pollutants such as CH4, CO2, O3, SF6, NF3, CF4, N2O, and SO2 favor the adsorption by different amounts depending on their chemical composition.
Squarate demonstrates excellent stability in the presence of the considered pollutants, exhibiting a favorable capture of the latter that results in a negative adsorption energy value. This phenomenon can be attributed to the existence of electrostatic interaction forces between pollutants and squarate. These interactions are particularly pronounced at short distances, occurring between the oxygen of squarate and the carbon atoms, nitrogen, and sulfur in the molecules CO2, N2O, SO2, and H2O, respectively. The presence of other weak parallel interactions further reinforces the binding of these molecules of the C-S. Additionally, the non-polarity of CH4 gas makes it a favorable insert within squarate, accentuated by a hydrogen bond. Finally, the dipole nature of O3 imparts characteristic properties, enabling it to react very selectively as an electrophile. These characteristic properties enable them to react very selectively as an electrophile. This characteristic facilitates its strong adsorption with squarate, driven by the intermolecular forces of Van der Waals and London dispersion.
Pollutants with higher volume and surface density are adsorbed in smaller quantities; for example, molecules with fluorine o nitrogen atoms. Indeed, adsorption in NF3, CF4, and SF6 molecules is influenced by the number of fluorine atoms used and the adapted geometry. The smaller this number, the more the adsorption process is promoted. Furthermore, the more it adopts a complex structure, the more the interactions between pollutants and squarate are minimized. In contrast to all previous gases, nitrophenols are not able to be adsorbed in this bulk because of the small pore size.
The adsorption energy of the molecule containing nitrogen as N2O in a dry environment is revelaed to be the highest one (−63.02), even if the maximal insertion is one molecule per cell compared to four insert molecules of CH4 with a maximum value of −23.02. This indicates the strong interaction involved in the first adsorption. Globally, the addition of water does not interfere with the adsorption, keeping the adsorption energy negative.
The co-adoption of CH4, SO2, and CO2 with H2O implies an increase in adsorption energy. The same trend is observed for molecules containing fluorine atoms, such as NF3, CF4, and SF6. This impact on adsorption energy values is attributed to multiple interactions caused by the reactive sites of these pollutants, as well as the formation of weak bonds between water molecules.
In a humid environment, the same order is found for O3 and N2O molecules as in the dry state, with a very slight decrease in adsorption energies. Meanwhile, 4NP is an exception to the other pollutants studied. On the other hand, it is characterized by an unfavorable capture on C-S in both humid and dry environments, resulting in positive adsorption energy. This is attributed to its large size and the unavailability of sufficient space in the pore. However, C-S exhibits a high capacity to insert water inside its cavity, revealing its hydrophilic nature. This behavior stands in contrast to enormous MOFs well-documented in the literature for their highly hydrophobic characteristics. Such hydrophilicity is particularly advantageous, especially considering that gas molecules are typically present in humid conditions, allowing us to regard C-S as a revolutionary MOF for adsorption, making it a novel target in this field.
The number of molecules fixed in the C-S decreases from the dry to the wet state, indicating that water molecules compete with pollutants and form strong hydrogen bonds with the squarate, compared to the interactions between pollutants and the bulk.
In future work, it is recommended to simulate a molecular work with a reduced part of copper squarate to understand deeper what is happening inside the cell cavity and comprehend with better precision to adsorption nature and the type of electrostatic interaction, including Van der Waals or hydrogen bonds in both dry and humid environments, as well as to determine the factors that govern this adsorption phenomenon. In addition, to complete this, a molecular dynamics (MD) trajectory will be helpful to understand dynamic adsorption.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules29133140/s1, Figure S1: Comparison between the Powder X-ray diffraction patterns derived from the computed and experimental structures [35]; Table S1: Comparison between the theoretical data on the geometry of molecules and the experimental data obtained from NIST database [68]; Table S2: The reported calculated structure in *.cif format, used in the DFT-PBE calculations.
Author Contributions
Conceptualization, C.A., N.G., V.T., F.C. and D.H.; Methodology, C.A.; Validation, V.T., F.C. and D.H.; Formal analysis, C.A. and D.H.; Investigation, C.A. and N.G.; Resources, V.T.; Writing—original draft, C.A.; Writing—review and editing, N.G., V.T., F.C. and D.H.; Supervision, N.G., V.T., F.C. and D.H.; Project administration, V.T.; Funding acquisition, V.T. and D.H. All authors have read and agreed to the published version of the manuscript.
Funding
The authors acknowledge the Ministerio de Ciencia, Innovación y Universidades of Spain through the grants PID2020-112887GB-I00 and PID2020-113084GB-I00/AEI/10.13039/501100011033.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in the study are included in the article and Supplementary Materials; further inquiries can be directed to the corresponding authors.
Acknowledgments
This project has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No. 872081. The authors acknowledge the Ministerio de Ciencia, Innovación y Universidades of Spain through the grants PID2020-112887GB-I00 and PID2020-113084GB-I00. The authors acknowledge the CTI (CSIC) and CESGA and the “Red Española de Computación” for the grant RES-AECT-2022-3-0006 and computing facilities.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- De Lotto, R. Assessment of Development and Regeneration Urban Projects: Cultural and Operational Implications in Metropolization Context. Int. J. Energy Environ. 2008, 2, 25–34. [Google Scholar]
- Molina, M.J.; Molina, L.T. Megacities and Atmospheric Pollution. J. Air Waste Manag. Assoc. 2004, 54, 644–680. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Jin, Y.; Lu, J. Experimental Study on Adsorption of SO2 and DCM from Air Pollutants by Modified Biochar. Biomass Convers. Biorefin 2023, 1–15. [Google Scholar] [CrossRef]
- Zhang, J.; Zhou, J.Z.; Xu, Z.P.; Li, Y.; Cao, T.; Zhao, J.; Ruan, X.; Liu, Q.; Qian, G. Decomposition of Potent Greenhouse Gas Sulfur Hexafluoride (SF6) by Kirschsteinite-Dominant Stainless Steel Slag. Environ. Sci. Technol. 2014, 48, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Zheng, X.; Dong, H.; Wang, R.; Mei, B.; Zhu, J. A 3-Year Record of N2O and CH4 Emissions from a Sandy Loam Paddy during Rice Seasons as Affected by Different Nitrogen Application Rates. Agric. Ecosyst. Environ. 2012, 152, 1–9. [Google Scholar] [CrossRef]
- Ehhalt, D.; Prather, M.; Dentener, F.; Derwent, R.; Dlugokencky, E.; Holland, E.; Isaksen, I.; Katima, J.; Kirchhoff, V.; Matson, P.; et al. Atmospheric Chemistry and Greenhouse Gases. In Climate change 2001: The Scientific Basis; Intergovernmental Panel on Climate Change: Geneva, Switzerland, 2001. [Google Scholar]
- Arani, M.H.; Jaafarzadeh, N.; Moslemzadeh, M.; Ghalhari, M.R.; Arani, S.B.; Mohammadzadeh, M. Dispersion of NO2 and SO2 Pollutants in the Rolling Industry with AERMOD Model: A Case Study to Assess Human Health Risk. J. Environ. Health Sci. Eng. 2021, 19, 1287–1298. [Google Scholar] [CrossRef]
- García-Berríos, Z.I.; Arce, R. Photodegradation Mechanisms of 1-Nitropyrene, an Environmental Pollutant: The Effect of Organic Solvents, Water, Oxygen, Phenols, and Polycyclic Aromatics on the Destruction and Product Yields. J. Phys. Chem. A 2012, 116, 3662–3664. [Google Scholar] [CrossRef][Green Version]
- Wang, H.L.; Nie, L.; Li, J.; Wang, Y.F.; Wang, G.; Wang, J.H.; Hao, Z.P. Characterization and Assessment of Volatile Organic Compounds (VOCs) Emissions from Typical Industries. Chin. Sci. Bull. 2013, 58, 724–730. [Google Scholar] [CrossRef]
- Inomata, S.; Yamada, H.; Tanimoto, H. Investigation on VOC Emissions from Automobile Sources by Means of Online Mass Spectrometry. Curr. Pollut. Rep. 2016, 2, 188–199. [Google Scholar] [CrossRef]
- Lin, Y.; Qiu, X.; Ma, Y.; Ma, J.; Zheng, M.; Shao, M. Concentrations and Spatial Distribution of Polycyclic Aromatic Hydrocarbons (PAHs) and Nitrated PAHs (NPAHs) in the Atmosphere of North China, and the Transformation from PAHs to NPAHs. Environ. Pollut. 2015, 196, 164–170. [Google Scholar] [CrossRef]
- Keyte, I.J.; Albinet, A.; Harrison, R.M. On-Road Traffic Emissions of Polycyclic Aromatic Hydrocarbons and Their Oxy- and Nitro- Derivative Compounds Measured in Road Tunnel Environments. Sci. Total Environ. 2016, 566–567, 1131–1142. [Google Scholar] [CrossRef] [PubMed]
- Supong, A.; Bhomick, P.C.; Sinha, U.B.; Sinha, D. A Combined Experimental and Theoretical Investigation of the Adsorption of 4-Nitrophenol on Activated Biocarbon Using DFT Method. Korean J. Chem. Eng. 2019, 36, 2023–2034. [Google Scholar] [CrossRef]
- Ipek, I.; Kabay, N.; Yüksel, M. Separation of Bisphenol A and Phenol from Water by Polymer Adsorbents: Equilibrium and Kinetics Studies. J. Water Process Eng. 2017, 16, 206–211. [Google Scholar] [CrossRef]
- Plaza-Medina, E.F.; Rodríguez-Córdoba, W.; Morales-Cueto, R.; Peon, J. Primary Photochemistry of Nitrated Aromatic Compounds: Excited-State Dynamics and NO• Dissociation from 9-Nitroanthracene. J. Phys. Chem. A 2011, 115, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Bakovic, L.; Chen, A. Kinetics of the Electrochemical Oxidation of 2-Nitrophenol and 4-Nitrophenol Studied by in Situ UV Spectroscopy and Chemometrics. Electrochim. Acta 2007, 52, 6517–6524. [Google Scholar] [CrossRef]
- Bertinelli, L.; Camacho, C.; Zou, B. Carbon Capture and Storage and Transboundary Pollution: A Differential Game Approach. Eur. J. Oper. Res. 2014, 237, 721–728. [Google Scholar] [CrossRef]
- Shan, Y.; Du, J.; Zhang, Y.; Shan, W.; Shi, X.; Yu, Y.; Zhang, R.; Meng, X.; Xiao, F.S.; He, H. Selective Catalytic Reduction of NOx with NH3: Opportunities and Challenges of Cu-Based Small-Pore Zeolites. Natl. Sci. Rev. 2021, 8, nwab010. [Google Scholar] [CrossRef] [PubMed]
- Inomata, S.; Fushimi, A.; Sato, K.; Fujitani, Y.; Yamada, H. 4-Nitrophenol, 1-Nitropyrene, and 9-Nitroanthracene Emissions in Exhaust Particles from Diesel Vehicles with Different Exhaust Gas Treatments. Atmos. Environ. 2015, 110, 93–102. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, L.; Chen, L.; Han, C.; Akutagawa, T.; Endo, O.; Yamauchi, M.; Neroda, A.; Toriba, A.; Tang, N. Polycyclic Aromatic Hydrocarbons and Nitro-Polycyclic Aromatic Hydrocarbons in Five East Asian Cities: Seasonal Characteristics, Health Risks, and Yearly Variations. Environ. Pollut. 2021, 287. [Google Scholar] [CrossRef]
- Zeng, Y.; Zou, R.; Zhao, Y. Covalent Organic Frameworks for CO2 Capture. Adv. Mater. 2016, 28, 2855–2873. [Google Scholar] [CrossRef]
- Wang, S.M.; Mu, X.T.; Liu, H.R.; Zheng, S.T.; Yang, Q.Y. Pore-Structure Control in Metal–Organic Frameworks (MOFs) for Capture of the Greenhouse Gas SF6 with Record Separation. Angew. Chem. -Int. Ed. 2022, 61, e202207066. [Google Scholar] [CrossRef]
- Jang, Y.; Sekaran, B.; Singh, P.P.; Misra, R.; D’Souza, F. Accelerated Intramolecular Charge Transfer in Tetracyanobutadiene- and Expanded Tetracyanobutadiene-Incorporated Asymmetric Triphenylamine-Quinoxaline Push-Pull Conjugates. J. Phys. Chem. A 2023, 127, 4455–4462. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Chen, Y.; Ma, Y.; Bailey, J.; Wang, Z.; Lee, D.; Sheveleva, A.M.; Tuna, F.; McInnes, E.J.L.; Frogley, M.D.; et al. Control of the Pore Chemistry in Metal-Organic Frameworks for Efficient Adsorption of Benzene and Separation of Benzene/Cyclohexane. Chem 2023, 9, 739–754. [Google Scholar] [CrossRef]
- Zhang, S.; Yang, Q.; Liu, X.; Qu, X.; Wei, Q.; Xie, G.; Chen, S.; Gao, S. High-Energy Metal-Organic Frameworks (HE-MOFs): Synthesis, Structure and Energetic Performance. Coord. Chem. Rev. 2016, 307, 292–312. [Google Scholar] [CrossRef]
- Thi Thuy Huong, T.; Ngoc Thanh, P.; Thi Xuan Huynh, N.; Ngoc Son, D. Metal-Organic Frameworks: State-of-the-Art Material for Gas Capture and Storage. VNU J. Sci. Math. Phys. 2016, 32, 67–85. [Google Scholar]
- Paudel, H.P.; Shi, W.; Hopkinson, D.; Steckel, J.A.; Duan, Y. Computational Modelling of Adsorption and Diffusion Properties of CO2 and CH4 in ZIF-8 for Gas Separation Applications: A Density Functional Theory Approach. React. Chem. Eng. 2021, 6, 990–1001. [Google Scholar] [CrossRef]
- Miyazaki, R.; Nakatani, N.; Levchenko, S.V.; Yokoya, T.; Nakajima, K.; Hara, K.; Fukuoka, A.; Hasegawa, J.Y. DFT Mechanistic Study on the Complete Oxidation of Ethylene by the Silica-Supported Pt Catalyst: C=C Activation via the Ethylene Dioxide Intermediate. J. Phys. Chem. C 2019, 123, 12706–12715. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, J.; Zhang, C.; Li, Z.; Lu, B.; Zhu, J. Molecular Simulation of C2H4/CO2/N2/O2 Adsorption Characteristics in Lignite and Anthracite. AIP Adv. 2021, 11, 085205. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, J.; Zhang, Y.; Ma, J.; Huang, L.; Yu, S.; Chen, L.; Song, G.; Qiu, M.; Wang, X. Applications of Water-Stable Metal-Organic Frameworks in the Removal of Water Pollutants: A Review. Environ. Pollut. 2021, 291, 118076. [Google Scholar] [CrossRef]
- Adjal, C.; Timón, V.; Guechtouli, N.; Boussassi, R.; Hammoutène, D.; Senent, M.L. The Role of Water in the Adsorption of Nitro-Organic Pollutants on Activated Carbon. J. Phys. Chem. A 2023, 127, 8146–8158. [Google Scholar] [CrossRef]
- Timón, V.; Senent, M.L.; Hochlaf, M. Structural Single and Multiple Molecular Adsorption of CO2 and H2O in Zeolitic Imidazolate Framework (ZIF) Crystals. Microporous Mesoporous Mater. 2015, 218, 33–41. [Google Scholar] [CrossRef]
- Furukawa, H.; Gándara, F.; Zhang, Y.B.; Jiang, J.; Queen, W.L.; Hudson, M.R.; Yaghi, O.M. Water Adsorption in Porous Metal-Organic Frameworks and Related Materials. J. Am. Chem. Soc. 2014, 136, 4369–4381. [Google Scholar] [CrossRef]
- Guo, S.; Huang, X.; Situ, Y.; Huang, Q.; Guan, K.; Huang, J.; Wang, W.; Bai, X.; Liu, Z.; Wu, Y.; et al. Interpretable Machine-Learning and Big Data Mining to Predict Gas Diffusivity in Metal-Organic Frameworks. Adv. Sci. 2023, 10, 2301461. [Google Scholar] [CrossRef] [PubMed]
- Dinnebier, R.E.; Nuss, H.; Jansen, M. Anhydrous CuC4O4, a Channel Structure Solved from X-Ray Powder Diffraction Data. Z. Anorg. Allg. Chem. 2005, 631, 2328–2332. [Google Scholar] [CrossRef]
- Colmenero, F.; Lobato, Á.; Timón, V. Compressing the Channels in the Crystal Structure of Copper Squarate Metal-Organic Framework. Solids 2022, 3, 374–384. [Google Scholar] [CrossRef]
- Colmenero, F. Negative Linear Compressibility in Nanoporous Metal-Organic Frameworks Rationalized by the Empty Channel Structural Mechanism. Phys. Chem. Chem. Phys. 2021, 23, 8508–8524. [Google Scholar] [CrossRef]
- Colmenero, F.; Cobos, J.; Timón, V. Negative Linear Compressibility in Uranyl Squarate Monohydrate. J. Phys. Condens. Matter 2019, 31, 175701. [Google Scholar] [CrossRef]
- Colmenero, F. Addendum: Anomalous Mechanical Behavior of the Deltic, Squaric and Croconic Cyclic Oxocarbon Acids. Mater. Res. Express 2019, 6, 045610. [Google Scholar] [CrossRef]
- Qiu, W.; Zeng, Q.; Li, C.; Hao, J.; Li, Y. Theoretical Investigation of Zero Linear Compressibility on Metal Squarates MC4O4 (M = Pb and Ba). J. Phys. Chem. C 2023, 127, 9957–9963. [Google Scholar] [CrossRef]
- Zhang, L.; He, Z.; Liu, Y.; You, J.; Lin, L.; Jia, J.; Chen, S.; Hua, N.; Ma, L.A.; Ye, X.; et al. A Robust Squarate-Cobalt Metal-Organic Framework for CO2/N2 Separation. ACS Appl. Mater. Interfaces 2023, 15, 30394–30401. [Google Scholar] [CrossRef]
- BIOVIA. Dassault Systemes, Materials Studio 2022. Dassault Systemes: San Diego, CA, USA, 2022. [Google Scholar]
- Delgado, M.R. Structure and Stability of Gas Adsorption Complexes in Periodic Porous Solids as Studied by VTIR Spectroscopy: An Overview. Appl. Sci. 2020, 10, 8589. [Google Scholar] [CrossRef]
- Manak, M.; Jirkovsky, L.; Kolingerova, I. Interactive Analysis of Connolly Surfaces for Various Probes. Comput. Graph. Forum 2017, 36, 160–172. [Google Scholar] [CrossRef]
- El, N.; Timón, V.; Boussessi, R.; Dalbouha, S.; Senent, M.L. DFT Studies of Single and Multiple Molecular Adsorption of CH4, SF6 and H2O in Zeolitic-Imidazolate Framework (ZIF-4 and ZIF-6). Inorganica Chim Acta 2019, 490, 272–281. [Google Scholar] [CrossRef]
- Akkermans, R.L.C.; Spenley, N.A.; Robertson, S.H. COMPASS III: Automated Fitting Workflows and Extension to Ionic Liquids. Mol. Simul. 2021, 47, 540–551. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Gautam, R.; Steiner, T. The Weak Hydrogen Bond. In Structural Chemistry and Biology; Oxford University Press: Oxford, UK, 1999; ISBN 9780198509707. [Google Scholar]
- Park, K.S.; Ni, Z.; Cô, A.P.; Choi, J.Y.; Huang, R.; Uribe-Romo, F.J.; Chae, H.K.; O’keeffe, M.; Yaghi, O.M. Exceptional Chemical and Thermal Stability of Zeolitic Imidazolate Frameworks. Proc. Natl. Acad. Sci. USA 2006, 103, 10186–10191. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Tanaka, Y. A Simple Step toward Enhancing Hydrothermal Stability of ZIF-8. ACS Omega 2019, 4, 19905–19912. [Google Scholar] [CrossRef] [PubMed]
- Shieh, F.K.; Wang, S.C.; Leo, S.Y.; Wu, K.C.W. Water-Based Synthesis of Zeolitic Imidazolate Framework-90 (ZIF-90) with a Controllable Particle Size. Chem.-A Eur. J. 2013, 19, 11139–11142. [Google Scholar] [CrossRef]
- Nalaparaju, A.; Zhao, X.S.; Jiang, J.W. Molecular Understanding for the Adsorption of Water and Alcohols in Hydrophilic and Hydrophobic Zeolitic Metal-Organic Frameworks. J. Phys. Chem. C 2010, 114, 11542–11550. [Google Scholar] [CrossRef]
- Benedict, W.S.; Gailar, N.; Plyler, E.K. Rotation-Vibration Spectra of Deuterated Water Vapor. J. Chem. Phys. 1956, 24, 1139–1165. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.J.; Refson, K. MC Payne. First Princ. Methods Using Castep. Z. Für Krist. Cryst. Mater. 2005, 220, 567–570. [Google Scholar]
- Hai, G.; Wang, H. Theoretical Studies of Metal-Organic Frameworks: Calculation Methods and Applications in Catalysis, Gas Separation, and Energy Storage. Coord. Chem. Rev. 2022, 469, 214670. [Google Scholar] [CrossRef]
- Payne, M.C.; Teter, M.P.; Ailan, D.C.; Arias, T.A.; Joannopouios, J.D. Iterative Minimization Techniques for Ab Initio Total-Energy Calculations: Molecular Dynamics and Conjugate Gradients. Rev. Mod. Phys. 1992, 64, 1045. [Google Scholar] [CrossRef]
- Pfrommer, B.G.; Cô, M.; Louie, S.G.; Cohen, M.L. Relaxation of Crystals with the Quasi-Newton Method. J. Comput. Phys. 1997, 131, 233–240. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Semiempirical GGA-Type Density Functional Constructed with a Long-Range Dispersion Correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, S.; Moellmann, J.; Reckien, W.; Bredow, T.; Grimme, S. System-Dependent Dispersion Coefficients for the DFT-D3 Treatment of Adsorption Processes on Ionic Surfaces. ChemPhysChem 2011, 12, 3414–3420. [Google Scholar] [CrossRef] [PubMed]
- Troullier, N.; Martins, J.L. EfFicient Pseudopotentials for Plane-Wave Calculations-II. Phys. Rev. B 1991, 8, 1993. [Google Scholar] [CrossRef] [PubMed]
- Baroni, S.; De Gironcoli, S.; Corso, A.D.; Giannozzi, P. Phonons and Related Crystal Properties from Density-Functional Perturbation Theor. Rev. Mod. Phys. 2001, 73, 515. [Google Scholar] [CrossRef]
- Refson, K.; Tulip, P.R.; Clark, S.J. Variational Density-Functional Perturbation Theory for Dielectrics and Lattice Dynamics. Phys. Rev. B Condens. Matter Mater. Phys. 2006, 73, 155114. [Google Scholar] [CrossRef]
- Khaled, K.F. Monte Carlo Simulations of Corrosion Inhibition of Mild Steel in 0.5 M Sulphuric Acid by Some Green Corrosion Inhibitors. J. Solid State Electrochem. 2009, 13, 1743–1756. [Google Scholar] [CrossRef]
- Mayo, S.L.; Olafson, B.D.; Goddard, W.A., III. DREIDING: A Generic Force Field for Molecular Simulations. J. Phys. Chem. 1990, 94, 8897–8909. [Google Scholar] [CrossRef]
- Babarao, R.; Dai, S.; Jiang, D.E. Effect of Pore Topology and Accessibility on Gas Adsorption Capacity in Zeolitic-Imidazolate Frameworks: Bringing Molecular Simulation Close to Experiment. J. Phys. Chem. C 2011, 115, 8126–8135. [Google Scholar] [CrossRef]
- Gao, H.; Pishney, S.; Janik, M.J. First Principles Study on the Adsorption of CO2 and H2O on the K2CO3 (001) Surface. Surf. Sci. 2013, 609, 140–146. [Google Scholar] [CrossRef]
- Smit, B.; Frenkel, D. Understanding Molecular Simulation, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2023; Volume 1. [Google Scholar]
- NIST Computational Chemistry Comparison and Benchmark Database. NIST Standard Reference Database Number 101. Release 22, May 2022, Editor: Russell D. Johnson III. Available online: https://cccbdb.nist.gov/ (accessed on 23 May 2024).





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).