Abstract
Monoamine oxidase inhibitors (MAOIs) have been crucial in the search for anti-neurodegenerative medications and continued to be a vital source of molecular and mechanistic diversity. Therefore, the search for selective MAOIs is one of the main areas of current drug development. To increase the effectiveness and safety of treating Parkinson’s disease, new scaffolds for reversible MAO-B inhibitors are being developed. A total of 24 pyridazinobenzylpiperidine derivatives were synthesized and evaluated for MAO. Most of the compounds showed a higher inhibition of MAO-B than of MAO-A. Compound S5 most potently inhibited MAO-B with an IC50 value of 0.203 μM, followed by S16 (IC50 = 0.979 μM). In contrast, all compounds showed weak MAO-A inhibition. Among them, S15 most potently inhibited MAO-A with an IC50 value of 3.691 μM, followed by S5 (IC50 = 3.857 μM). Compound S5 had the highest selectivity index (SI) value of 19.04 for MAO-B compared with MAO-A. Compound S5 (3-Cl) showed greater MAO-B inhibition than the other derivatives with substituents of -Cl > -OCH3 > -F > -CN > -CH3 > -Br at the 3-position. However, the 2- and 4-position showed low MAO-B inhibition, except S16 (2-CN). In addition, compounds containing two or more substituents exhibited low MAO-B inhibition. In the kinetic study, the Ki values of S5 and S16 for MAO-B were 0.155 ± 0.050 and 0.721 ± 0.074 μM, respectively, with competitive reversible-type inhibition. Additionally, in the PAMPA, both lead compounds demonstrated blood–brain barrier penetration. Furthermore, stability was demonstrated by the 2V5Z-S5 complex by pi–pi stacking with Tyr398 and Tyr326. These results suggest that S5 and S16 are potent, reversible, selective MAO-B inhibitors that can be used as potential agents for the treatment of neurological disorders.
1. Introduction
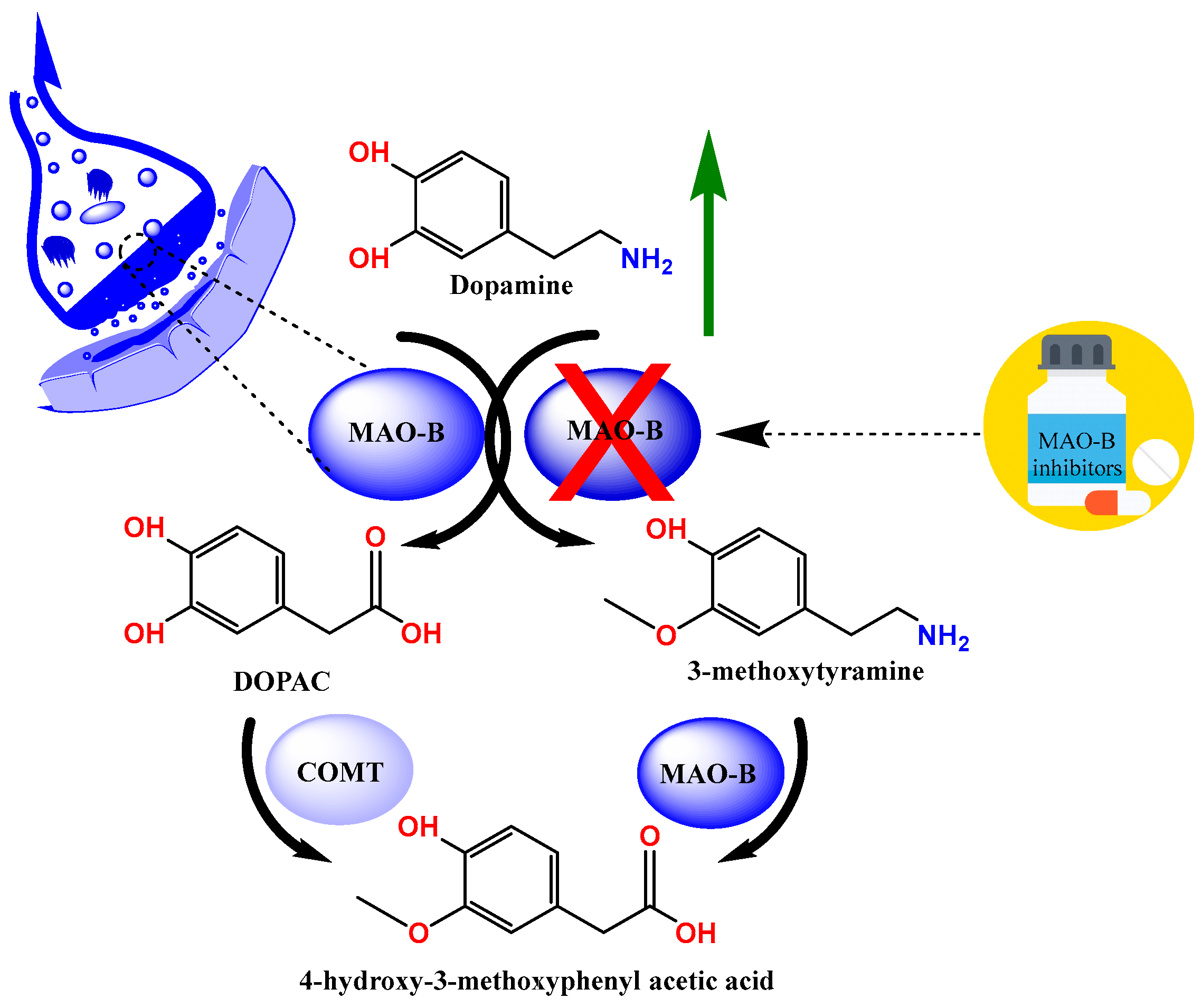
Parkinson’s disease (PD) is a neurodegenerative disease that is particularly common among the elderly and is characterized by the death of dopaminergic neurons in the substantia nigra pars compacta [1,2]. The typical motor features of PD (bradykinesia, stiffness, and tremor) are caused by a subsequent dopamine shortage; however, non-dopamine neurodegeneration results in additional non-motor symptoms that manifest at different times [3]. The pathogenesis of the disease is characterized by elevated acetylcholine and low dopamine levels; therefore, treatment strategies aimed at increasing dopamine levels are recommended. One such strategy is the inhibition of monoamine oxidases (MAOs), which are responsible for the metabolism of endogenous amines (Figure 1) [4,5,6,7].
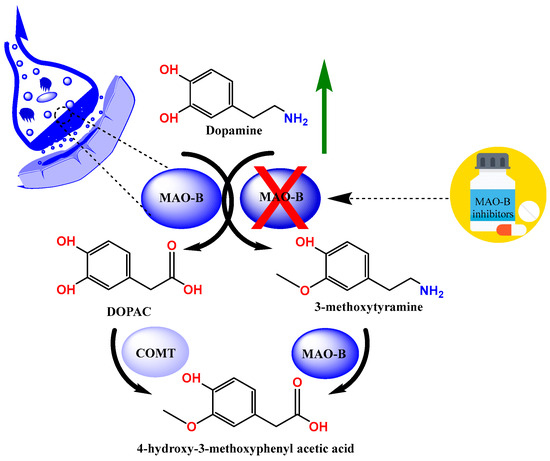
Figure 1.
The metabolism of dopamine.
MAO inhibitors (MAOIs) have been crucial in the search for anti-neurodegenerative medications and continue to be a vital source of molecular and mechanistic diversity [7]. Therefore, the search for selective MAOIs is one of the main areas of current drug development. To increase the effectiveness and safety of treating PD, new scaffolds for reversible human MAO-B (MAO-B) inhibitors are being developed [8,9,10,11].
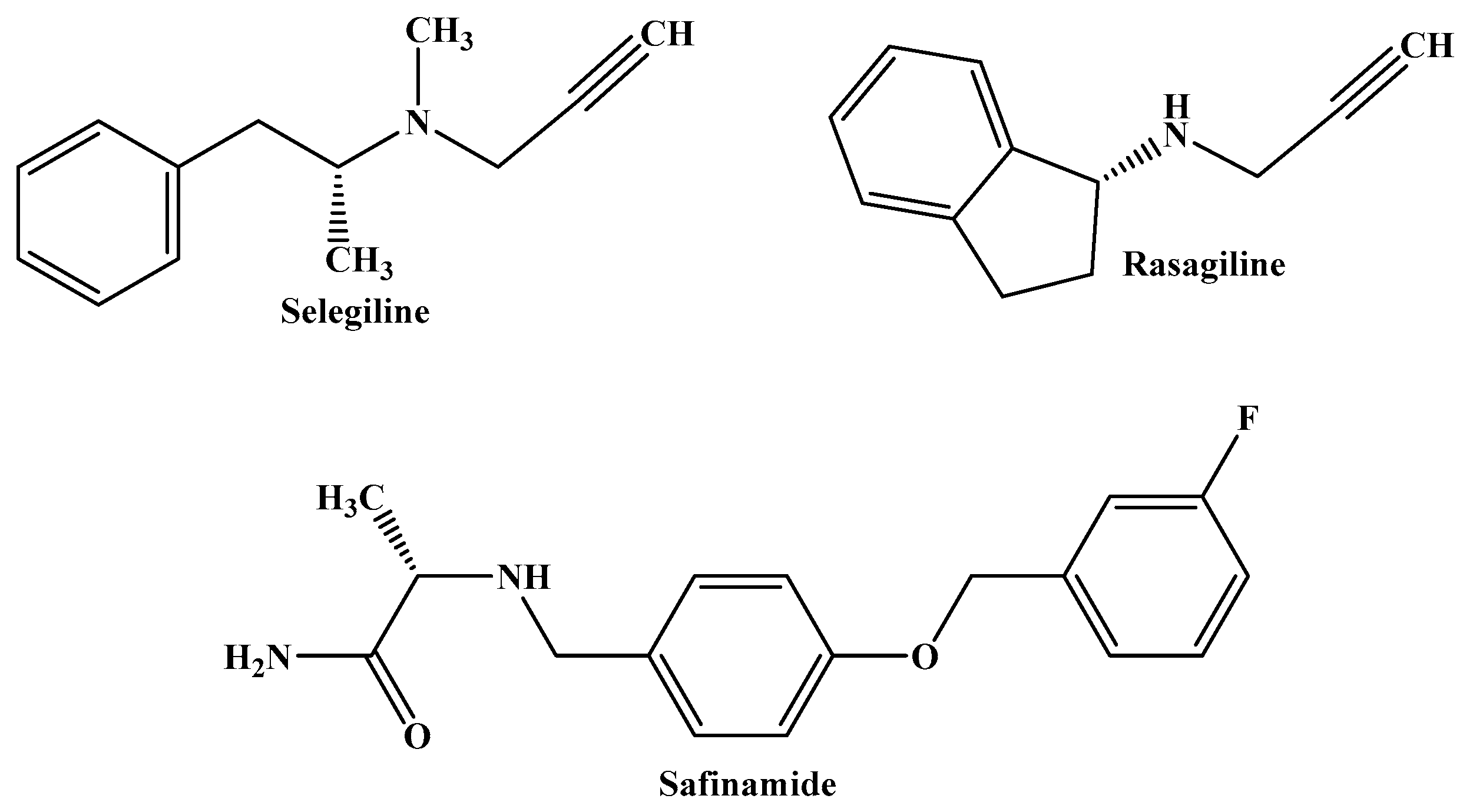
Selegiline and rasagiline are MAO-B inhibitors used to treat levodopa-induced motor fluctuations, advanced PD, and the loss of OFF time in patients. Safinamide, a third MAO-B inhibitor, is used as an adjuvant therapy or as a dopamine agonist in conjunction with levodopa, and it combines additional non-dopaminergic qualities that may be beneficial for patients with PD (Figure 2) [12,13,14].
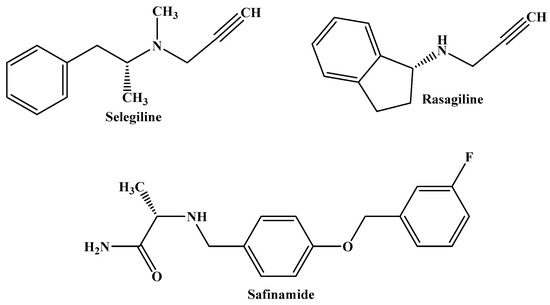
Figure 2.
The molecular structures of selegiline, rasagiline, and safinamide, which are MAO-B inhibitors currently used in the treatment of PD.
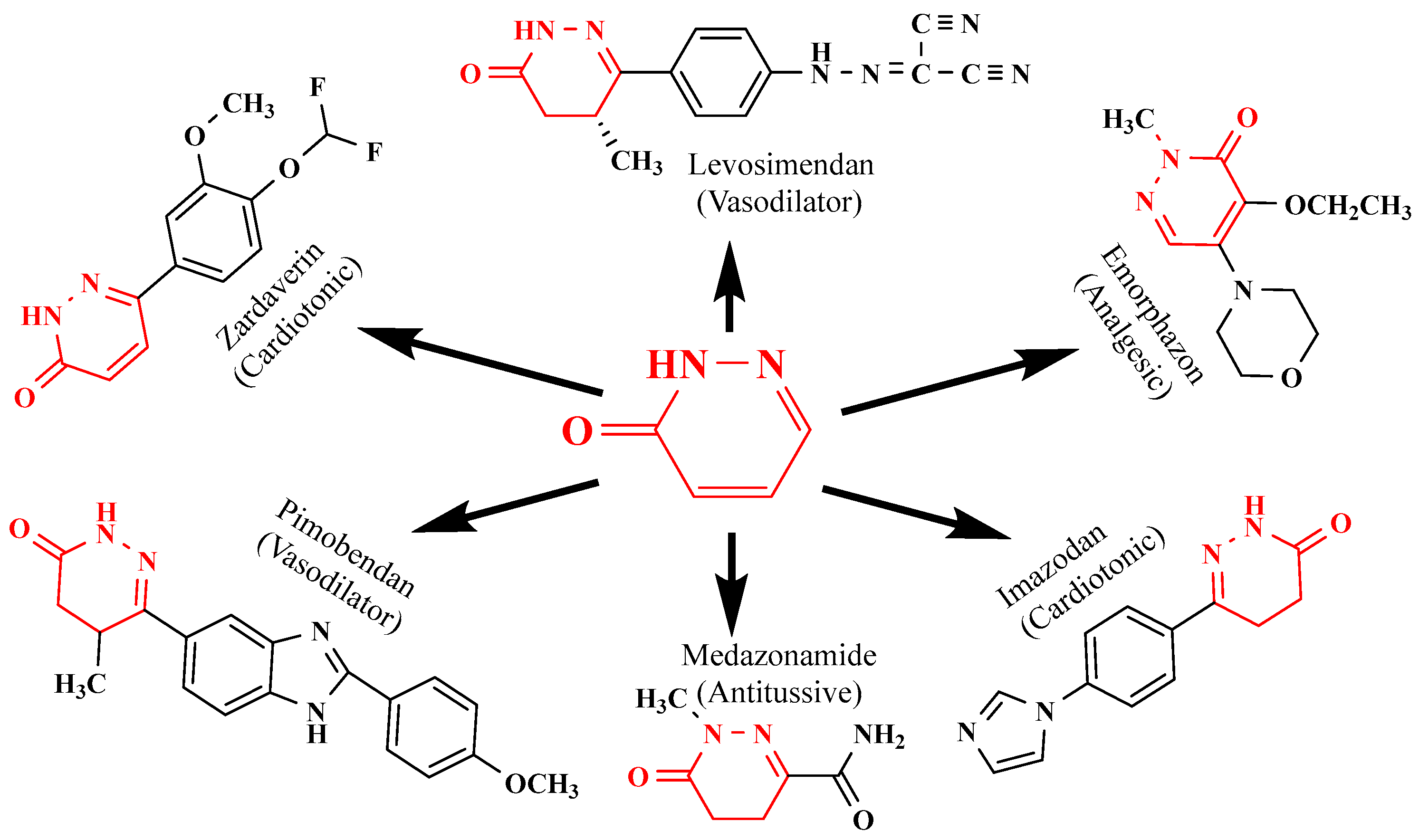
The pharmacological properties of pyridazinone analogs include anti-inflammatory, anticancer, antibacterial, anticonvulsant, analgesic, antioxidant, antihypertensive, antisecretory, and antiulcer effects [13,14,15,16,17,18,19,20,21]. They include lipoxygenase inhibitors, cholinesterase inhibitors, adenosine receptor antagonists, serotonin receptor antagonists, phosphodiesterase inhibitors, glutamate transporter activators, dipeptidyl peptidase inhibitors, cyclooxygenase (COX) inhibitors, and vasodilators [22,23,24,25,26,27,28,29,30]. Additionally, a few commercially available drugs, including pimobendan, levosimendan (a cardiotonic medication), emorphozan, and zardaverine (a non-steroidal anti-inflammatory drug) as analgesics and anti-inflammatory agents, are based on pyridazine rings (Figure 3).
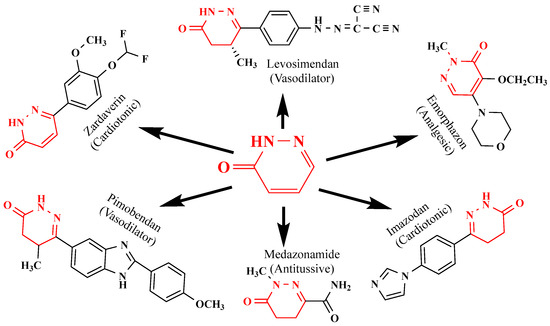
Figure 3.
Structure of pyridazinone derivatives.
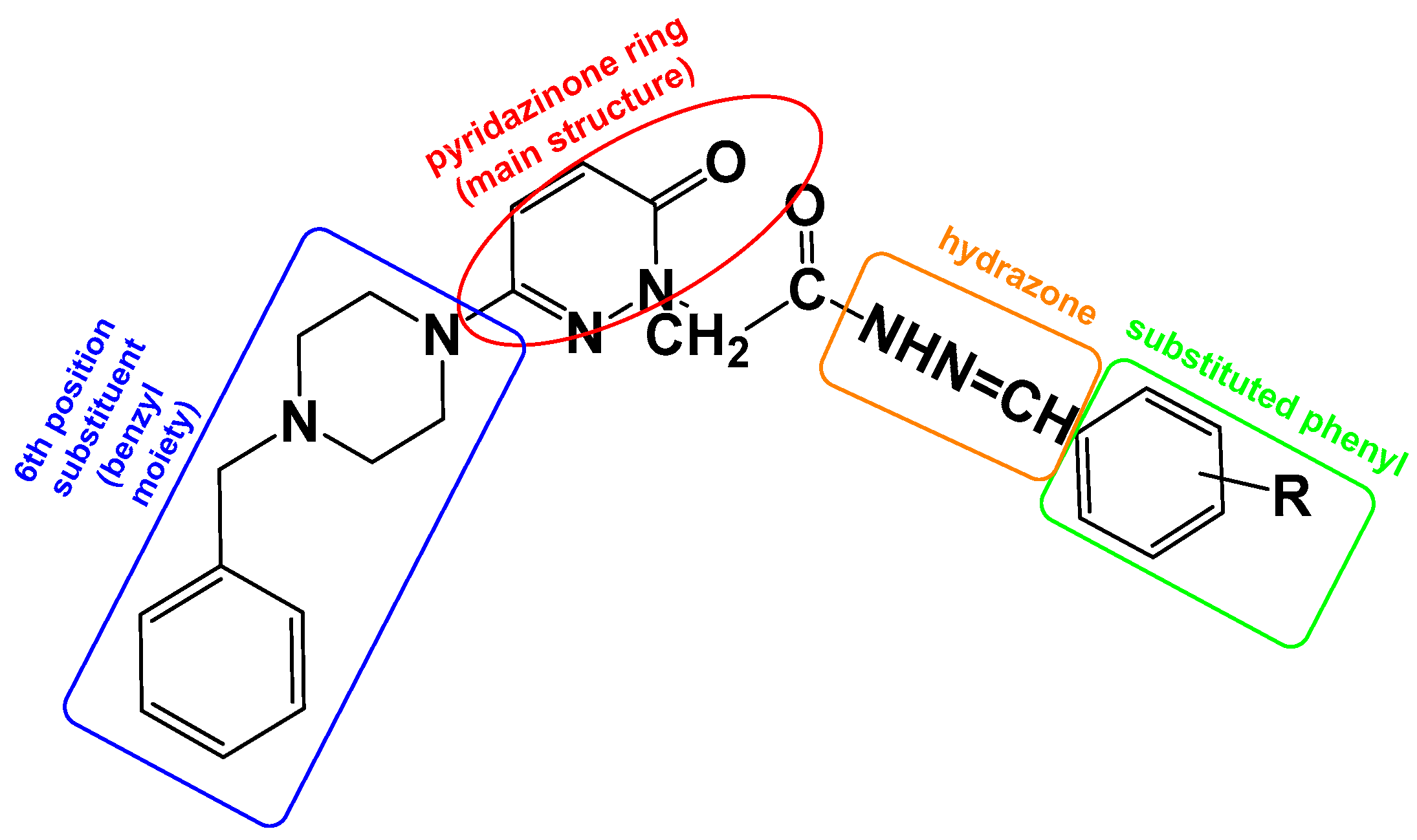
To increase the biological activity of pyridazinone, new pharmacological molecules known as benzylpiperidinopyridazine derivatives were synthesized. The resulting compounds were selective MAO-B inhibitors, and the activity results were corroborated by the molecular modeling scores (Figure 4).
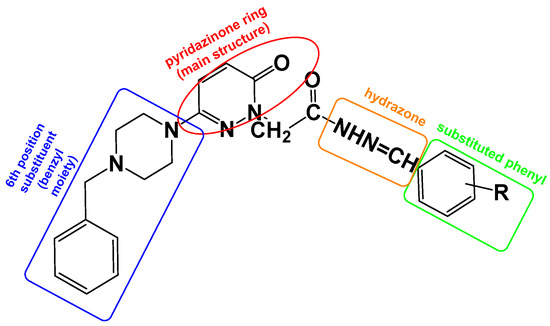
Figure 4.
The design targets to be applied on the molecule within the scope of this study. The pyridazinone ring is protected as the main structure (red), containing the benzyl moiety at the 6th position of pyridazinone (blue), and hydrazone part (orange) and substituted phenyl moiety (green).
2. Results
2.1. Chemistry
This study synthesized and investigated the in vitro enzyme inhibitor activities of 23 compounds that act as selective MAO-B inhibitors, which were obtained from 6-substituted-3(2H)-pyridazinone-2-acetyl-2-(substituted benzalhydrazone) [10,11]. Besides unsubstituted benzaldehyde, benzaldehyde derivatives with disubstituted methylamine, methyl, bromo, fluoro, methyl, and methoxy substituents at various positions were used in synthesizing benzhydrazone derivatives, and the corresponding contributions of these groups to the activity were investigated. The structures of the obtained compounds were confirmed using spectrum data from 1H-NMR, 13C-NMR, and TOF-MS. Their melting temperatures were determined, and the findings are shown in the Supplementary File and Table 1.

Table 1.
Structures, yields, and chemical–physical data of titled compounds.
2.2. Inhibition Studies
2.2.1. The Inhibitory Activities of MAO-A and MAO-B by the Compounds
Twenty-four pyridazinobenzylpiperidine derivatives against MAO-A and MAO-B were evaluated. These pyridazinobenzylpiperidine derivatives were synthesized with two large structures: one residue was attached to the phenyl ring, and two or more residues were attached to the phenyl ring (Figure 4). Seventeen of the twenty-four compounds showed higher MAO-B inhibition than MAO-A except seven compounds (Table 2). Compound S5 showed potent MAO-B inhibition with an IC50 value of 0.203 μM, followed by S16 (IC50 = 0.979 μM). In contrast, compound S15 showed the highest MAO-A inhibition with an IC50 value of 3.691 μM, followed by S5 (IC50 = 3.857 μM) (Table 2). The selectivity index (SI) of MAO-B over MAO-A was the highest for compound S5 (19.04) and the lowest for compound S9 at 0.62 (Table 2).
Table 2.
The inhibition of MAO-A and MAO-B by the S series a.
2.2.2. Reversibility Studies
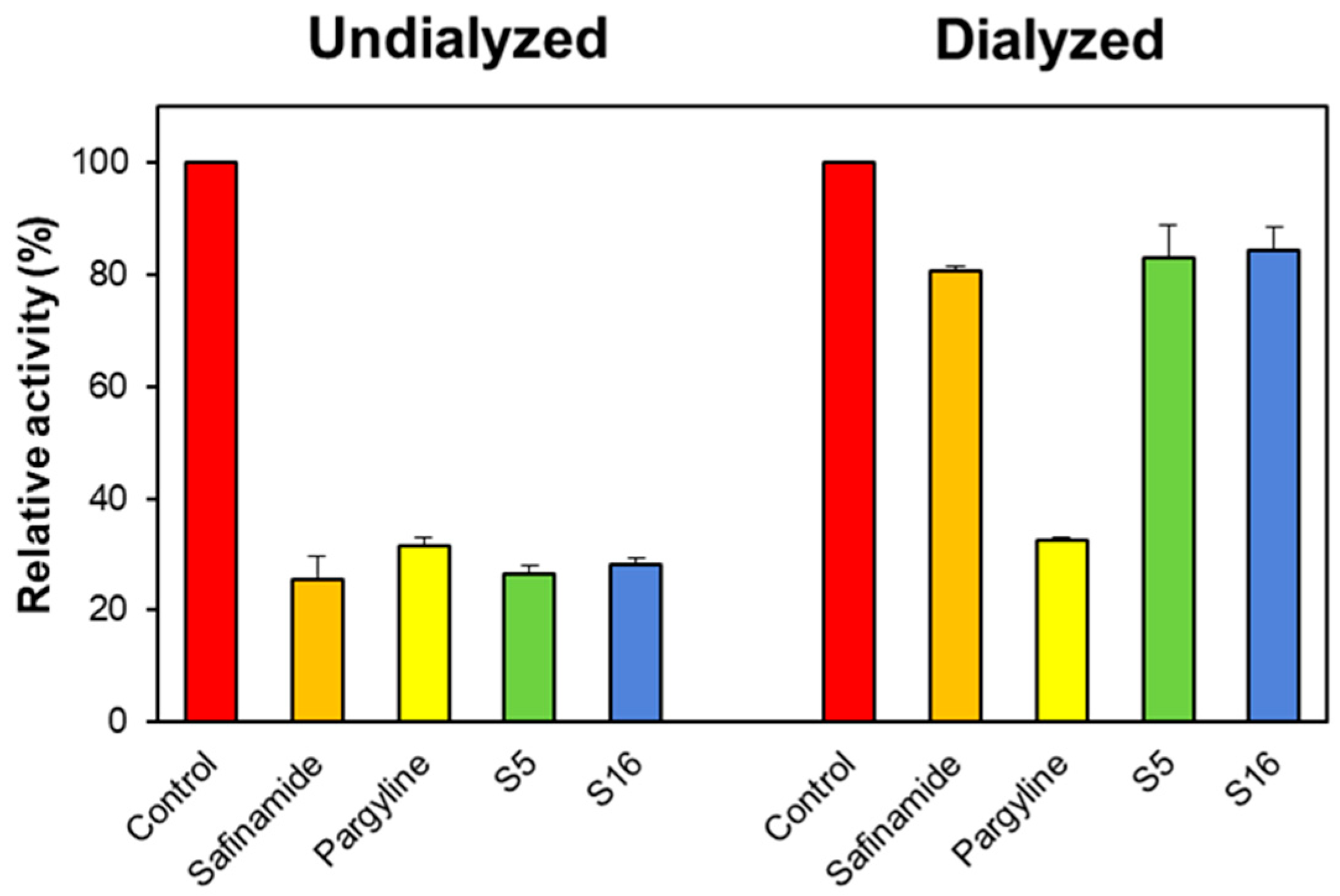
In the reversibility study, the concentrations of S5 and S16 used were approximately 2.0 times their IC50 values (0.4 and 2.0 μM for MAO-B, respectively). The residual activities before and after recovery are represented by AU and AD. Compounds S5 and S16 were recovered from 27.53% to 79.07% and from 29.04% to 81.33%, respectively (Figure 5). The recovery values of S5 and S16 were similar to safinamide (ranging from 28.34% to 81.19%), whereas they differed from pargyline (ranging from 32.66% to 32.93%). These results indicate that S5 and S16 are reversible MAO-B inhibitors.
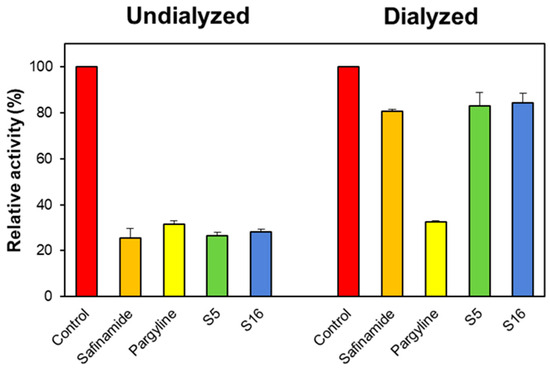
Figure 5.
The recovery of MAO-B inhibition by S5 and S16 using dialysis experiments. The concentrations of S5 and S16 used were approximately 2.0 times their IC50 values (0.4 and 2.0 μM). After 30 min of pre-incubation, the mixtures were dialyzed for 6 h with two buffer changes.
2.2.3. Enzyme and Inhibition Kinetics
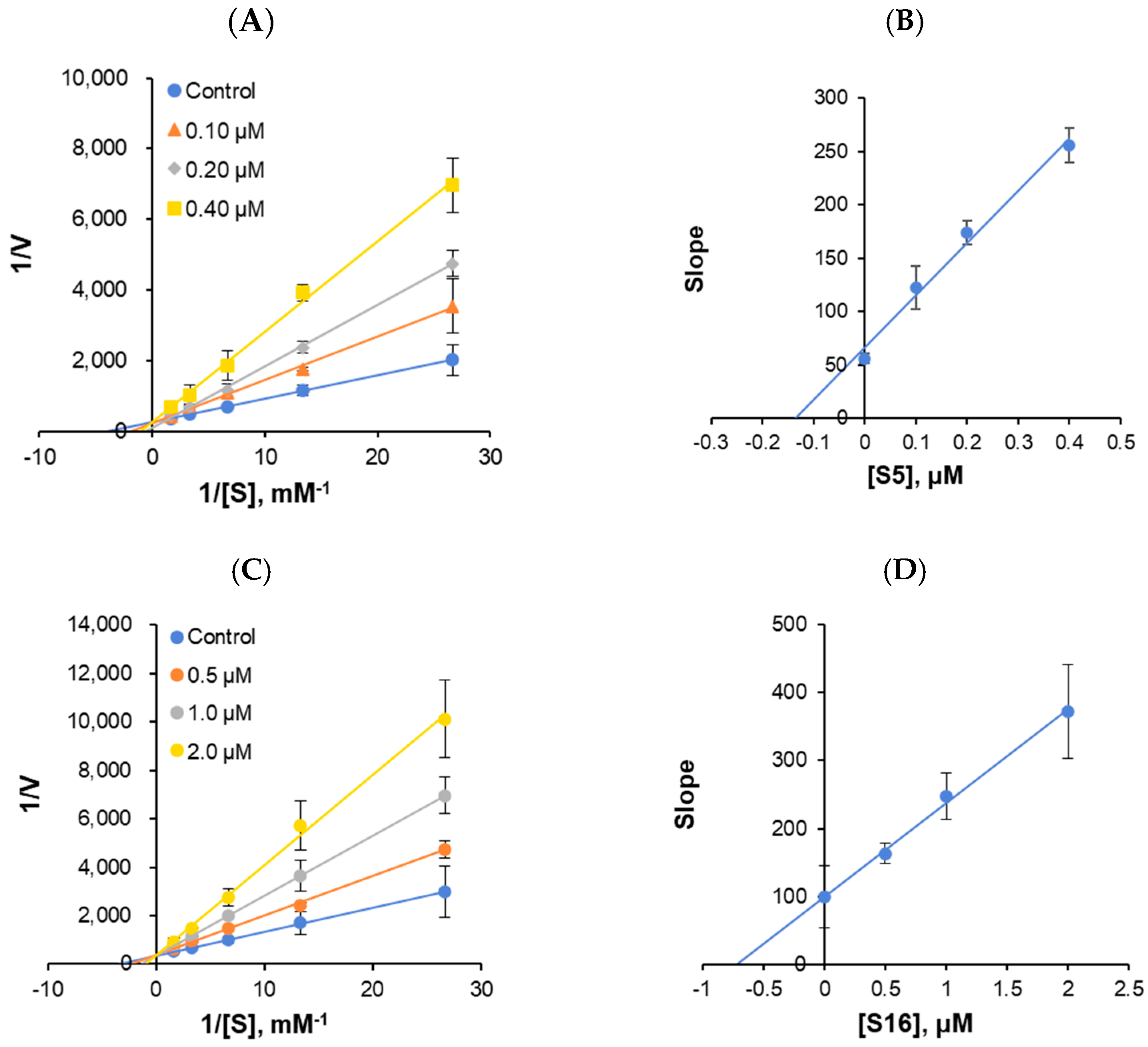
The enzyme kinetics of MAO-B and the types of inhibition were analyzed at five concentrations of benzylamine as a substrate and three inhibitor concentrations. In the LB plots, S5 and S16 appeared to be competitive MAO-B inhibitors (Figure 6A,C). Secondary plots showed that the Ki value was 0.155 ± 0.050 and 0.721 ± 0.074 μM, respectively (Figure 6B,D), suggesting that S5 and S16 are competitive MAO-B inhibitors.
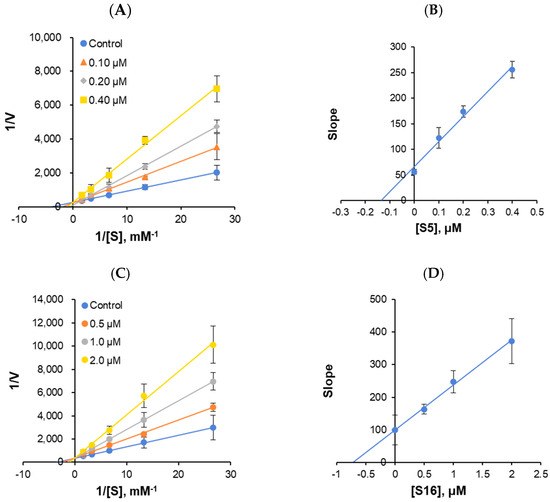
Figure 6.
Lineweaver–Burk (LB) plots for MAO-B inhibition by S5 and S16 (A,C), and their secondary plots (B,D) of the slopes versus inhibitor concentrations. The experiments were analyzed using five substrate concentrations and three inhibitor concentrations.
2.3. Parallel Artificial Membrane Permeability Assay (PAMPA) for Blood–Brain Barrier (BBB) Permeation Study
The results obtained from the PAMPA indicated significant levels of permeation and CNS bioavailability for compounds S5 and S16. S5 showed a Pe value surpassing 4.0 × 10−6 cm·s−1, while S16 came close to this threshold, suggesting higher rates of BBB penetration at 5.75 × 10−6 cm·s−1 and 3.78 × 10−6 cm·s−1, respectively (Table 3).
Table 3.
Blood–brain barrier assay of key compounds using PAMPA method.
The effective delivery of CNS medications relies on successful brain penetration. To evaluate the brain permeability of all the derivatives, PAMPA-BBB was used in this study. The extent of penetration was determined using a specific formula and considering the effective permeability of the substance (Log Pe). Compounds classified as potentially permeating (CNS+) exhibited a Pe value greater than 4.0 × 10−6 cm·s−1, while those categorized as likely non-BBB permeating (CNS−) had a Pe value less than 2.0 × 10−6 cm·s−1.
2.4. Molecular Docking
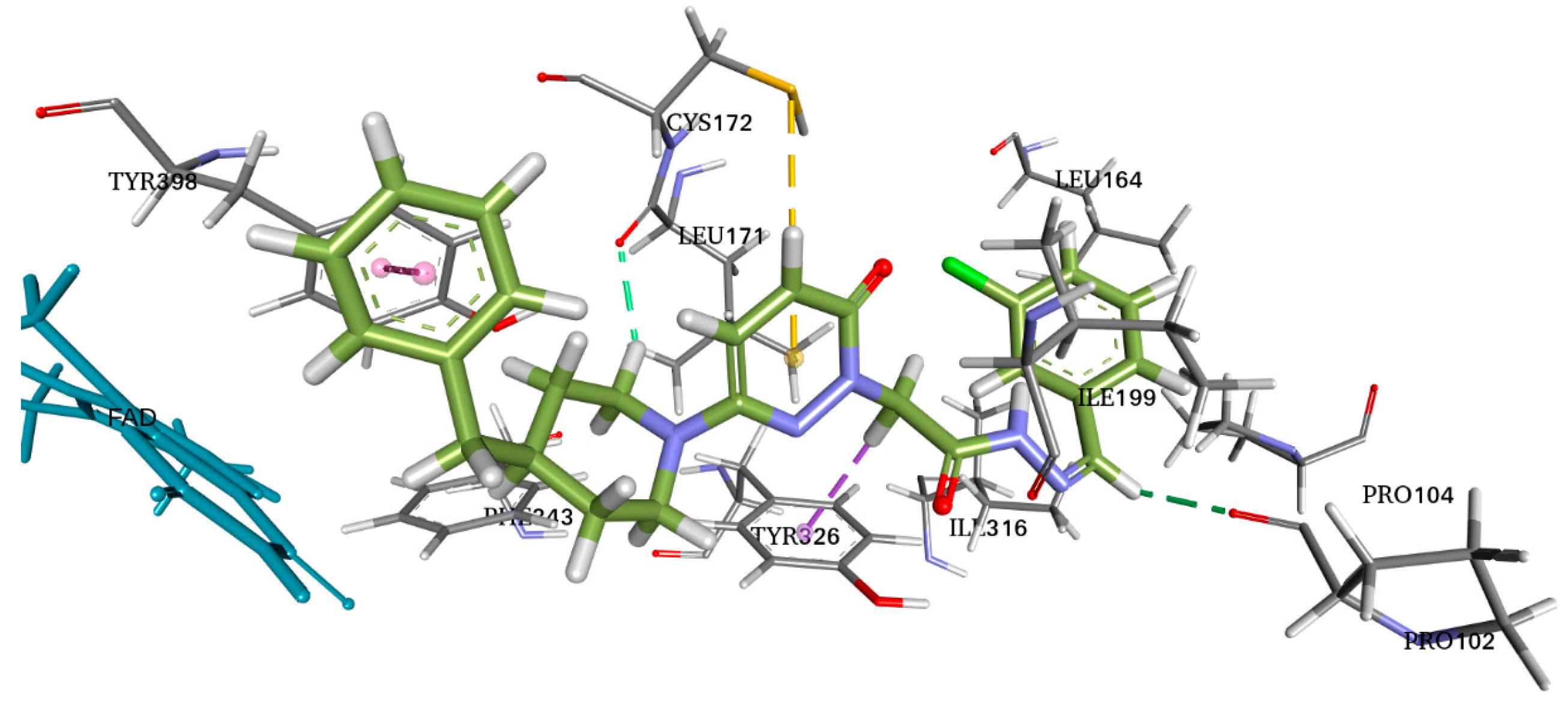
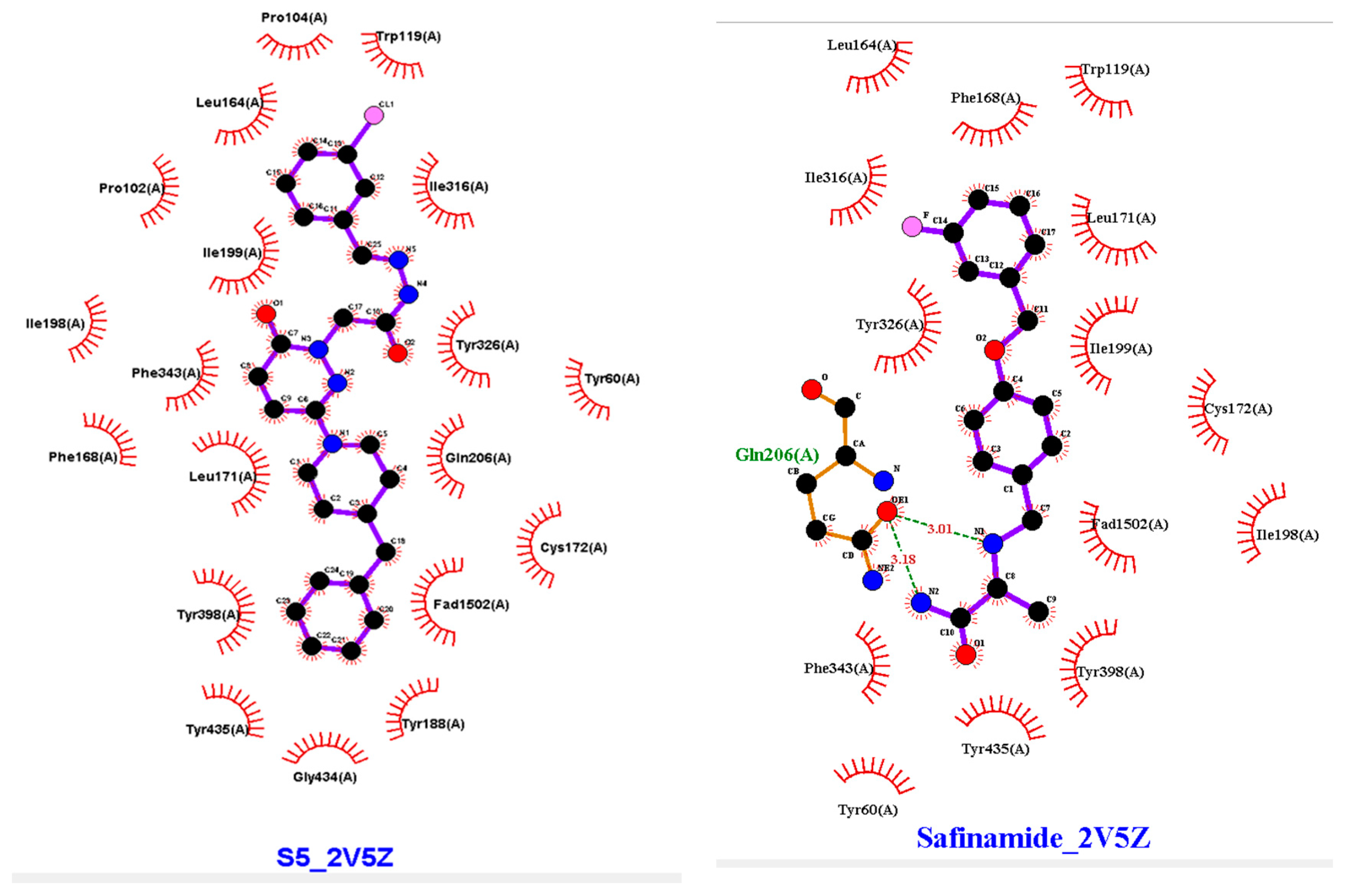
Molecular docking allows for the prediction of the molecular affinity at the binding site, important interactions, and experimental binding mechanisms. With this knowledge of the molecular interactions between the physical and chemical processes, new derivatives can be developed. Therefore, we studied the molecular docking of S5 and 2V5Z (Figure 7).
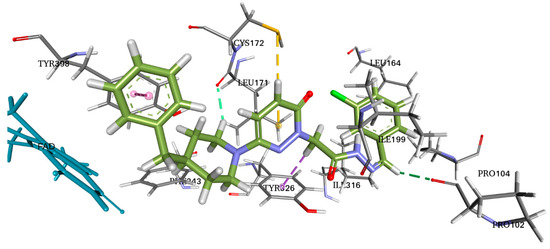
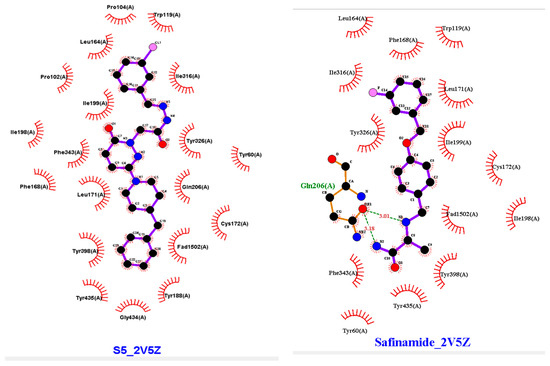
Figure 7.
Two-dimensional (2D) and three-dimensional (3D) interactions of S5 and safinamide with 2V5Z using LigPlot and Biovia Discovery Studio, respectively.
3. Discussion
3.1. Chemistry
The initial step in the synthesis was a benzylpiperidine substitution at position six. The pathway begins with the commercially acquired 3,6-dichloropyridazine and proceeds in five steps to obtain the final products. Mass spectral data demonstrated that the substitution was performed unilaterally and that the first-stage product was obtained with a yield of 52%. The lactam carbonyl present in the pyridazinone ring at 1660 cm−1 in the IR spectral data was used to confirm the results of the hydrolysis of the pyridazine ring in an acidic environment, which was the following step. The SN2 reaction attacks the bromine-bonded carbon of ethyl bromoacetate, producing an acetate derivative. This attack was caused by the unshared electrons of the nitrogen atom in the second position of the pyridazinone ring. Given the ability of bromine to retain electrons, the second nitrogen of the pyridazinone ring attacks the electron-poor carbon atom in the basic environment produced by the presence of potassium carbonate. The ester derivative and hydrazine hydrate react to produce the acetohydrazide derivative, which was produced in the following steps. As the alcohol is stripped from the molecule, the hydrazine hydrate nucleophile hits the carbonyl carbon, initially causing an addition reaction, followed by an elimination reaction by the formation of a double bond between carbon and oxygen.
The final stage involved the reaction of acetohydrazide with both substituted and unsubstituted benzaldehyde derivatives to produce the final compounds. Similar to the previous synthesis step, the next step involves an elimination reaction in which water is released from the structure and a carbon–nitrogen double bond is formed. The addition reaction starts with the nucleophilic attack of the free electrons of the hydrazine nitrogen on the carbon of the benzaldehyde carbonyl. In derivatives from benzaldehyde derivatives with electron-withdrawing substituents on the ring, the resulting compounds were produced in yields as high as 97.37%.
3.2. The Inhibitory Activities of MAO-A and MAO-B by the Compounds
The IC50 value of S5 for MAO-B was lower than a pyridazinone containing dithiocarbamyl moiety 12b1 (IC50 = 6.71 μM) [31], a 4-phenethyl-1-propargylpiperidine 15 (IC50 = 4.3 μM) [32], and a pyridazine–coumarin derivative 9b (IC50 = 0.75 μM) [33] but higher than a pyridazinone derivative TR16 (IC50 = 0.17 μM) [11], a pyridazinone containing the (2-fluorophenyl) piperazine moiety T6 (IC50 = 0.039 μM) [10], and a pyridazinone derivative 4b (IC50 = 0.022 μM) [16]. In addition, the IC50 value of S15 for MAO-A was lower than pyridazinones containing dithiocarbamyl moiety (IC50 >100 μM) [31], pyridazine–coumarin derivatives (IC50 >100 μM) [33], a 4-phenethyl-1-propargylpiperidine 9 (IC50 = 15.5 μM) [32], and a pyridazinone derivative TR8 (IC50 = 12.02 μM) [11] but higher than a pyridazinone containing the (2-fluorophenyl) piperazine moiety T6 (IC50 = 1.57 μM) [10] and a pyridazinone derivative 4f (IC50 = 1.05 μM) [16]. Regarding the structure–activity relationship (SAR), compound S5 (3-Cl of phenyl ring) had 4.82–53.94 times higher MAO-B inhibition than other derivatives. When compared according to the position of the residue, MAO-B inhibition by 2-CN at the phenyl ring (S16) was higher than that by other derivatives with substituents of -CN (S16) > -Br (S3) > -OCH3 (S19) at the 2-position. In addition, the 3-Cl of the phenyl ring (S5) showed the highest MAO-B inhibition compared to the other derivatives, with substituents of -Cl (S5) > -OCH3 (S11) > -F (S7) > -CN (S17) > -CH3 (S10) > -Br (S4) at the 3-position. However, compounds with residues at the 4-position or two or more residues showed weak MAO-B inhibition (Figure 4, Table 2). These results suggested that compounds S5 and S16 are potent selective MAO-B inhibitors.
3.3. Molecular Docking
AutoDock Vina software was used for docking. Vina uses PDBQT molecular structure file types as both input and output. Vina created up to 30 configurations for each docking run, ranking them according to the largest energy difference (kcal/mol) between the optimal and lowest binding modes. The optimal docking scores for S5 and safinamide in affinity were −10.4 and −11.0 kcal/mol, respectively. The S5 and safinamide and S5 bound to the same binding pocket with almost similar amino acid residues. In the case of a native ligand, the major interactions were hydrophobic and bipartite hydrogen bonds between the amide side chain and Gln206 residue. The binding study results indicated that a Pi–sulfur bond holds the pyridazine ring of S5 to Cys172. Pro102 and Leu171 form a typical hydrogen bond with S5. Tyr398 and Tyr326 residues utilize pi–pi stacking to interact with the ligand (Figure 7). As noted above, 5S suppressed MAO-B expression in vitro. Furthermore, we observed that hydrogen bonding and hydrophobic interactions stabilized the 2V5Z and S5 complexes.
4. Materials and Methods
4.1. Chemistry
Scheme 1 shows the preparation of compounds S1–S24. All chemicals were obtained from commercial suppliers. Chloroform–methanol (90:10) was used as the mobile phase in Merck Kieselgel 60 F254 aluminum plates (Darmstadt, Germany), and the reactions were monitored using thin-layer chromatography. The spots were identified under 254 nm UV light. Melting points (mp) were measured without correction using a Thomas–Hoover capillary melting point device (Fredericksburg, VA, USA). An Avonce 600 UltrashieldTM (Bruker, Rheinstetten, Germany) NMR spectrometer was used to record 1H- and 13C-NMR (300 MHz) spectra. Tetramethylsilane was used as an internal reference, and the compounds were dissolved in dimethyl sulfoxide (DMSO-d6) for NMR spectroscopy. The chemical shifts are represented as δ (ppm) values. The terms “singlet”, “doublet”, “triplet”, “quartet”, “multiplet”, and “doublet of doublet” were used to identify the splitting patterns. Using positive ion (ESI+) and negative ion (ESI−) electrospray ionization procedures, the HRMS spectra of the synthesized compounds were acquired from their solutions in methanol using the Waters LCT Premier XE UPLC/MS TOFF system (Milford, MA, USA) and MassLynx 4.1 software.
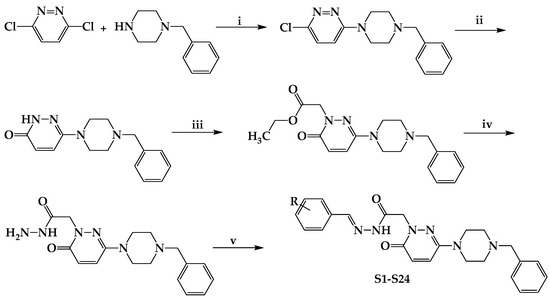
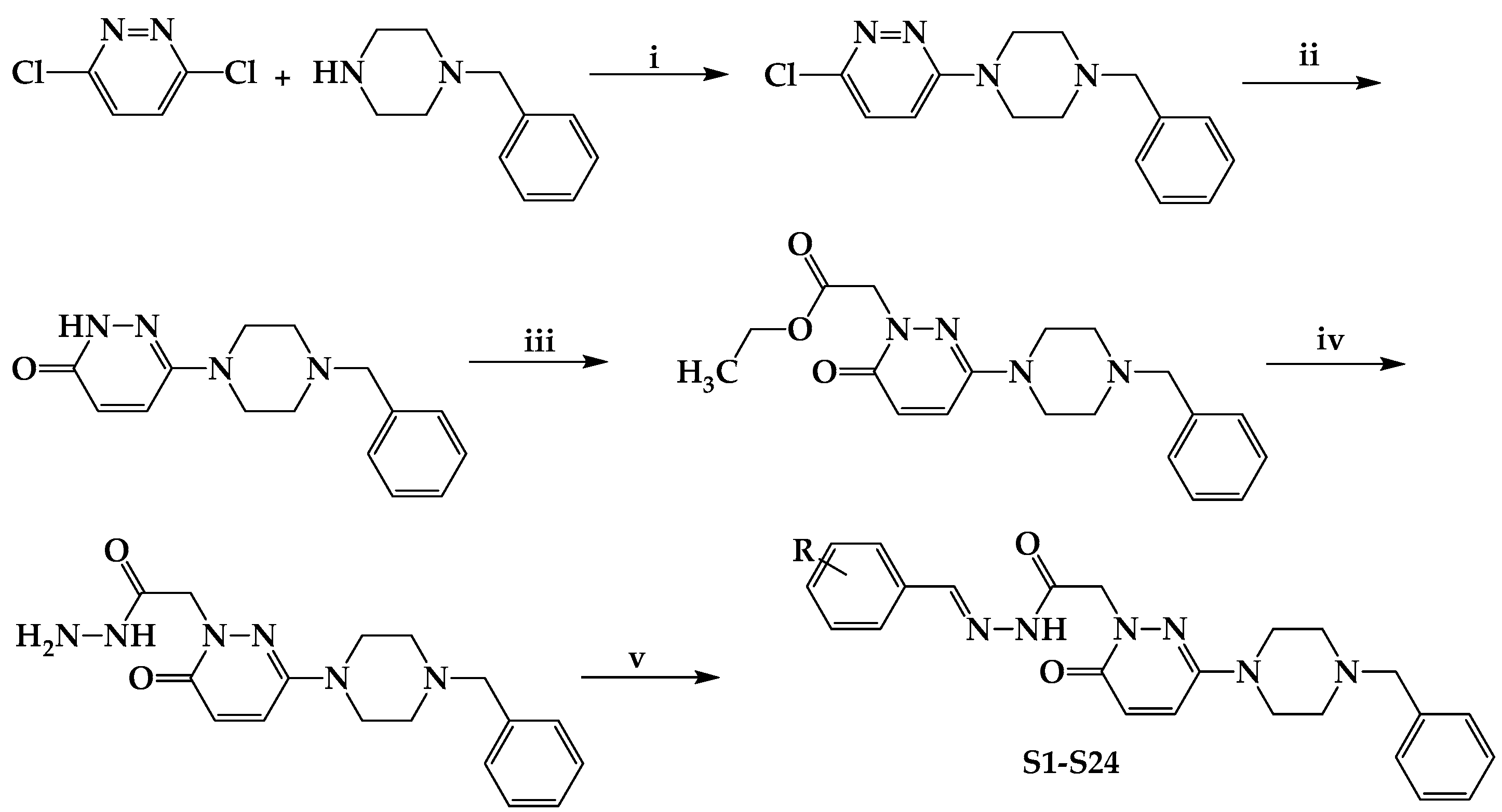
Scheme 1.
Synthesis and molecular structures of S1–S24. (i) EtOH, reflux for 6 h; (ii) AcOH, reflux for 6 h; (iii) BrCH2COOCH2CH3, K2CO3, acetone, reflux for 24 h; (iv) H2NNH2.H2O, MeOH, stirred for 3 h at rt; (v) suitable benzaldehyde; EtOH, reflux for 6 h. R; H, F, Br, Cl, CN, CH3, C2H5, OCH3, and N(CH3)2.
4.1.1. Experimental Procedure for Synthesis
According to the literature procedure, 0.01 mol of 3,6-dichloropyridazine and 0.01 mol of 4-benzylpiperidine were mixed in 15 mL of ethanol under reflux and heated for 6 h to synthesize 3-chloro-6-(4-benzylpiperidine-1-yl)pyridazine. The reaction medium was added to ice water, and the filtered precipitate was refined by crystallizing ethanol. A solution of 0.05 mol of 3-chloro-6-(4-benzylpiperidine-1-yl)pyridazine in 30 mL glacial acetic acid was refluxed for 6 h to obtain 6-(4-benzylpiperidine-1-yl)-3(2H)-pyridazinone. Reduced pressure was used to remove the acetic acid, and the residue was dissolved in water and extracted using chloroform. After drying over sodium sulfate, the organic phase was evaporated at lower pressure. The residue was separated from ethanol using recrystallization. A total of 0.01 mol 6-(4-benzylpiperidine-1-yl)-3(2H)-pyridazinone, 0.02 mol ethyl bromoacetate, and 0.02 mol potassium carbonate in 40 mL of acetone were refluxed overnight to obtain ethyl 6-(4-benzylpiperidine-1-yl)-3(2H)-pyridazinone-2-ylacetate. After cooling, the solvent was evaporated, the organic salts were filtered, and the residue was refined using recrystallization from n-hexane to obtain the esters. To create 6-(4-benzylpiperidine-1-yl)-3(2H)-pyridazinone -2-ylacetohydrazide, 0.01 mol of ethyl 6-(4-benzylpiperidine-1-yl)-3(2H)-pyridazinone-2-yl acetate in 25 mL of methanol was combined with hydrazine hydrate (99%, 3 mL), and the mixture was stirred constantly for 3 h at room temperature. After filtering the precipitate, the ethanol was dried, cleaned with water, and recrystallized [15].
4.1.2. Experimental Protocol for Producing the Chemicals Listed in the Title (S1–S24)
In 15 mL of ethanol, 0.01 mol of 6-(4-(4-benzylpiperidine-1-yl)-3(2H)-pyridazinone-2-yl acetohydrazide and 0.01 mol of substituted/nonsubstituted benzaldehyde were mixed and refluxed for 6 h. The mixture was then placed in frozen water to stop the reaction. The precipitate was filtered, dried, and crystallized in methanol–water (Scheme 1).
Eight of the compounds (S1, S2, S6, S8, S11, S12, S13, and S14) were created earlier, and their analgesic and anti-inflammatory properties were assessed before being published [15]. All the compounds’ spectrum data matched their given structures, as indicated below:
- N′-2-bromobenzylidene-2-(3-(4-benzylpiperidin-1-yl)-6-oxopyridazin-1(6H)-yl)acetohydrazide (S3)
S3 was obtained as a white solid by following a standard protocol with 6-(4-benzylpiperidine-1-yl)-3(2H)-pyridazinone-2-ylacetohydrazide and 2-bromobenzaldehyde. (m.p. 188–189, yield 66.39%). 1H-NMR (CDCl3, 300 MHz) TM 1.24–1.37 (2H, m, piperidine protons), 1.69–1.74 (3H, m, piperidine protons), 2.57 (d, 2H, benzyl CH2), 2.64–2.78 (4H piperidine protons), 5.27 (2H, s, CH2-CO-), 6.90–7.35 (m, 9H, phenyl protons), 7.57–7.60 (1H, d, pyridazinone H5), 7.95–7.98 (1H, d, pyridazinone H4), 8.18 (1H, s, N=CH), 9.44 (1H, s, CO-NH-N). 13C-NMR (CDCl3, 300 MHz), δ 31.37 (1C; CH2), 31.47 (1C; CH2), 37.82 (1C; CH), 43.04 (1C; CH2), 47.05 (2C; CH2-N), 53.58 (1C; N-CH2-C=O), 111.15 (1C; -CN), 125.97 (1C; N=CH), 127.04 (1C; pyridazinone C5), 128.27 (1C; pyridazinone C4), 130.18, 130.63, 130.74, 132.79, 132.88, 133.44 (6C; phenyl carbons), 139.72 (1C; 2-bromophenyl C6), 140.22 (1C; 2-bromophenyl C5) 143.68 (1C; 2-bromophenyl C4), 149.59 (1C; 2-bromophenyl C1), 150.07 (1C; 2-bromophenyl C3), 158.81 (1C; pyridazinone C6), 159.00 (1C; 2-bromophenyl C2), 164.54 (1C; -C=O) and 168.93 (1C; pyridazinone C3). C25H26BrN5O2 MS (ESI+) (Calc.: 508.1348; Found: 508.1352; 510.1351) (M+; 100.0%).
- N′-3-bromobenzylidene-2-(3-(4-benzylpiperidin-1-yl)-6-oxopyridazin-1(6H)-yl)acetohydrazide (S4)
S4 was obtained as a white solid by following standard protocol with 6-(4-benzylpiperidine-1-yl)-3(2H)-pyridazinone-2-ylacetohydrazide and 3-bromobenzaldehyde (m.p. 190–191, yield 69.67%). 1H-NMR (CDCl3, 300 MHz) TM 1.24–1.37 (2H, m, piperidine protons), 1.69–1.74 (2H, m, piperidine protons), 2.57 (d, 2H, benzyl CH2), 2.68–2.73 (1H, m, piperidine proton), 3.77–3.89 (4H piperidine protons), 5.27 (2H, s, CH2-CO-), 6.90–7.37 (m, 9H, phenyl protons), 7.56–7.60 (1H, d, pyridazinone H5), 7.95–7.98 (1H, d, pyridazinone H4), 8.18 (1H, s, N=CH), 9.44 (1H, s, CO-NH-N). 13C-NMR (CDCl3, 300 MHz), δ 31.36 (1C; CH2), 31.47 (1C; CH2), 37.80 (1C; CH), 43.03 (1C; CH2), 47.07 (2C; CH2-N), 53.46 (1C; N-CH2-C=O), 113.16 (1C; -CN), 126.01 (1C; N=CH), 128.29 (1C; pyridazinone C5), 129.63 (1C; pyridazinone C4), 130.38, 130.48, 130.66, 130.98, 131.10, 131.44 (6C; phenyl carbons), 134.98 (1C; 3-bromophenyl C6), 142.00 (1C; 3-bromophenyl C5), 146.18 (1C; 3-bromophenyl C4), 149.57 (1C; 3-bromophenyl C1), 150.14 (1C; 3-bromophenyl C2), 158.69 (1C; pyridazinone C6), 159.00 (1C; 3-bromophenyl C3), 164.38 (1C; -C=O) and 169.08 (1C; pyridazinone C3). C25H26BrN5O2 MS (ESI+) (Calc.: 508.4200; Found: 508.42) (M+; 100.0%).
- N′-3-chlorobenzylidene-2-(3-(4-benzylpiperidin-1-yl)-6-oxopyridazin-1(6H)-yl)acetohydrazide (S5)
S5 was obtained as a white solid by following standard protocol with 6-(4-benzylpiperidine-1-yl)-3(2H)-pyridazinone-2-ylacetohydrazide and 3-chlorobenzaldehyde (m.p. 137–138, yield 87.29%). 1H-NMR (CDCl3, 300 MHz) TM 1.2–1.32 (4H, m, piperidin protons), 2.56 (d, 2H, benzyl CH2), 2.69–2.73 (1H, m, piperidin proton), 3.74–3.85 (4H piperidin protons), 5.25 (2H, s, CH2-CO-), 7.17–7.35 (m, 9H, phenyl protons, pyridazinone H5), 7.5 (1H, d, pyridazinone H4), 7.78 (1H, s, 3-chlorophenyl H2), 7.64 (1H, s, N=CH), 10.05 (1H, s, CO-NH-N). 13C-NMR (CDCl3, 300 MHz), δ 31.36 (1C; CH2), 31.46 (1C; CH2), 37.80 (1C; CH), 43.04 (1C; CH2), 47.08 (2C; CH2-N), 53.34 (1C; N-CH2-C=O), 125.60 (1C; N=CH), 128.31 (1C; pyridazinone C5), 129.12 (1C; pyridazinone C4), 129.79, 129.97, 130.11, 130.29, 130.42, 130.70 (6C; phenyl carbons), 140.18 (1C; 3-chlorophenyl C6), 143.03 (1C; 3-chloroyphenyl C5) 147.31 (1C; 3-chlorophenyl C2), 149.51 (1C; 3-chlorophenyl C1), 150.08 (1C; 3-chlorophenyl C4), 158.72 (1C; pyridazinone C6), 159.01 (1C; 3-chlorophenyl C3), 164.29 (1C; -C=O) and 169.07 (1C; pyridazinone C3). C25H26ClN5O2 MS (ESI+) (Calc.: 464.1353; Found: 464.1353) (M+; 100.0%).
- N′-3-fluorobenzylidene-2-(3-(4-benzylpiperidin-1-yl)-6-oxopyridazin-1(6H)-yl)acetohydrazide (S7)
S7 was obtained as a white solid by following standard protocol with 6-(4-benzylpiperidine-1-yl)-3(2H)-pyridazinone-2-ylacetohydrazide and 3-fluorobenzaldehyde (m.p. 197–198, yield 92.33%). 1H-NMR (CDCl3, 300 MHz) TM 1.23–1.32 (3H, m, piperidin protons), 2.55–2.57 (d, 2H, benzyl CH2), 2.69–2.74 (2H, m, piperidin proton), 3.74–3.86 (4H piperidin protons), 5.25 (2H, s, CH2-CO-), 7.14–7.39 (m, 11H, phenyl protons, pyridazinone H4,5), 7.78 (1H, s, N=CH), 9.87 (1H, s, CO-NH-N). 13C-NMR (CDCl3, 300 MHz), δ 31.36 (1C; CH2), 31.47 (1C; CH2), 37.81 (1C; CH), 43.04 (1C; CH2), 47.08 (2C; CH2-N), 53.32 (1C; N-CH2-C=O), 113.20 (1C; N=CH), 117.25 (1C; pyridazinone C5), 128.28 (1C; pyridazinone C4), 129.12, 130.13, 130.29, 130.34, 130.43, 130.72 (6C; phenyl carbons), 140.19 (1C; 3-fluorophenyl C6), 143.11 (1C; 3-fluoroyphenyl C5), 147.56 (1C; 3-fluorophenyl C2), 149.49 (1C; 3-fluorophenyl C1), 150.08 (1C; 3-fluorophenyl C4), 158.70 (1C; pyridazinone C6), 163.81 (1C; 3-fluorophenyl C3), 164.30 (1C; -C=O) and 168.97 (1C; pyridazinone C3). C25H26FN5O2 MS (ESI+) (Calc.: 448.2149; Found: 448.2147) (M+; 100.0%).
- N′-2-methylbenzylidene-2-(3-(4-benzylpiperidin-1-yl)-6-oxopyridazin-1(6H)-yl)acetohydrazide (S9)
S9 was obtained as a white solid by following standard protocol with 6-(4-benzylpiperidine-1-yl)-3(2H)-pyridazinone-2-ylacetohydrazide and 2-methylbenzaldehyde (m.p. 222–223, yield 43.56%). 1H-NMR (CDCl3, 300 MHz) TM 1.26–1.74 (5H, m, piperidin protons), 2.44 (s, 3H, CH3), 2.71 (d, 2H, benzyl CH2), 3.76–3.85 (4H piperidin protons), 4.82 (2H, s, CH2-CO-), 6.88–7.72 (m, 9H, phenyl protons), 7.73 (1H, d, pyridazinone H5), 7.94 (1H, d, pyridazinone H4), 8.32 (1H, s, N=CH), 10.59 (1H, s, CO-NH-N). 13C-NMR (CDCl3, 300 MHz), δ 20.16 (1C; CH3), 31.37 (1C; CH2), 31.49 (1C; CH2), 37.82 (1C; CH), 43.05 (1C; CH2), 47.10 (2C; CH2-N), 53.33 (1C; N-CH2-C=O), 125.98 (1C; N=CH), 126.25 (1C; pyridazinone C5), 127.49 (1C; pyridazinone C4), 129.97, 130.20, 130.48, 130.69, 130.75, 131.11 (6C; phenyl carbons), 131.62, 137.10, 140.21, 143.40, 147.35, 149.42 (6C; 2-methylphenyl carbons), 150.04 (1C; pyridazinone C6), 158.71 (1C; -C=O) and 168.54 (1C; pyridazinone C3). C26H29N5O2 MS (ESI+) (Calc.: 444.2400; Found: 444.2409) (M+; 100.0%).
- N′-3-methylbenzylidene-2-(3-(4-benzylpiperidin-1-yl)-6-oxopyridazin-1(6H)-yl)acetohydrazide (S10)
S10 was obtained as a white solid by following standard protocol with 6-(4-benzylpiperidine-1-yl)-3(2H)-pyridazinone-2-ylacetohydrazide and 3-methylbenzaldehyde (m.p. 106–107, yield 86.98%). 1H-NMR (CDCl3, 300 MHz) TM 1.23–1.32 (2H, m, piperidin protons), 2.35 (s, 3H, CH3), 2.55 (2H, d, benzyl CH2), 2.69–2.73 (3H piperidin protons), 3.74–3.86 (4H piperidin protons), 5.27 (2H, s, CH2-CO-), 7.21–7.30 (m, 9H, phenyl protons, pyridazinone H5), 7.44 (1H, s, 3-methylphenyl C2), 7.47 (1H, d, pyridazinone H4), 7.74 (1H, s, N=CH), 9.57 (1H, s, CO-NH-N). 13C-NMR (CDCl3, 300 MHz), δ 21.34 (1C; CH3), 31.36 (1C; CH2), 31.48 (1C; CH2), 37.81 (1C; CH), 43.05 (1C; CH2), 47.10 (2C; CH2-N), 53.28 (1C; N-CH2-C=O), 124.67 (1C; N=CH), 125.41 (1C; pyridazinone C5), 127.64 (1C; pyridazinone C4), 128.27, 128.62, 129.12, 130.47, 130.74, 131.12 (6C; phenyl carbons), 131.35, 138.45, 144.61, 149.12, 149.45, 150.03, (6C; 3-methylphenyl carbons), 159.06 (1C; pyridazinone C6), 164.20 (1C; -C=O) and 168.80 (1C; pyridazinone C3). C26H29N5O2 MS (ESI+) (Calc.: 444.2400; Found: 444.2419) (M+; 100.0%).
- N′-4-ethylbenzylidene-2-(3-(4-benzylpiperidin-1-yl)-6-oxopyridazin-1(6H)-yl)acetohydrazide (S15)
S15 was obtained as a white solid by following standard protocol with 6-(4-benzylpiperidine-1-yl)-3(2H)-pyridazinone-2-ylacetohydrazide and 4-ethylbenzaldehyde (m.p. 225–226, yield 78.62%). 1H-NMR (CDCl3, 300 MHz) TM 1.24–1.29 (3H, t, CH3), 1.28–1.32 (2H, q, -CH2-), 1.71–1.72 (3H, m, piperidin proton), 2.58 (d, 2H, benzyl CH2), 2.66–2.74 (2H, m, piperidin proton), 3.78–3.89 (4H piperidin protons), 5.26 (2H, s, CH2-CO-), 7.14–7.30 (m, 9H, phenyl protons), 7.75–7.78 (1H, d, pyridazinone H5), 7.82–7.85 (1H, d, pyridazinone H4), 8.12 (1H, s, N=CH), 10.96 (1H, s, CO-NH-N). 13C-NMR (CDCl3, 300 MHz), δ 15.35 (1C; CH3), 28.85 (1C; CH2), 31.36 (1C; CH2), 31.49 (1C; CH2), 37.82 (1C; CH), 43.05 (1C; CH2), 47.10 (2C; CH2-N), 53.24 (1C; N-CH2-C=O), 125.97 (1C; N=CH), 126.28 (1C; pyridazinone C5), 127.32 (1C; pyridazinone C4), 127.87, 128.14, 128.31, 129.12, 130.76, 131.01 (6C; phenyl carbons), 144.36, 146.93, 149.07, 149.71, 150.02, 158.70 (6C; 4-methylphenyl carbons), 159.07 (1C; pyridazinone C6), 164.15 (1C; -C=O) and 168.61 (1C; pyridazinone C3). C27H31N5O2 MS (ESI+) (Calc.: 458.2556; Found: 458.2556) (M+; 100.0%).
- N′-2-cyanobenzylidene-2-(3-(4-benzylpiperidin-1-yl)-6-oxopyridazin-1(6H)-yl)acetohydrazide (S16)
S16 was obtained as a white solid by following standard protocol with 6-(4-benzylpiperidine-1-yl)-3(2H)-pyridazinone-2-ylacetohydrazide and 2-cyanobenzaldehyde (m.p. 189–190, yield 66.85%). 1H-NMR (CDCl3, 300 MHz) TM 1.21–1.29 (2H, m, piperidin protons), 1.67–1.75 (2H, m, piperidin protons), 2.56 (d, 2H, benzyl CH2), 2.69 (1H, m, piperidin proton), 3.76–3.80 (4H piperidin protons), 5.25 (2H, s, CH2-CO-), 7.18–7.38 (m, 9H, phenyl protons), 7.87–7.89 (1H, d, pyridazinone H5), 8.06 (1H, d, pyridazinone H4), 8.43 (1H, s, N=CH), 10.50 (1H, s, CO-NH-N). 13C-NMR (CDCl3, 300 MHz), δ 31.37 (1C; CH2), 31.47 (1C; CH2), 37.82 (1C; CH), 43.04 (1C; CH2), 47.05 (2C; CH2-N), 53.58 (1C; N-CH2-C=O), 111.15 (1C; -CN), 125.97 (1C; N=CH), 127.04 (1C; pyridazinone C5), 128.27 (1C; pyridazinone C4), 130.18, 130.63, 130.74, 132.79, 132.88, 133.44 (6C; phenyl carbons), 139.72 (1C; 2-cyanophenyl C6), 140.22 (1C; 2-cyanophenyl C5) 143.68 (1C; 2-cyanophenyl C4), 149.59 (1C; 2-cyanophenyl C1), 150.07 (1C; 2-cyanophenyl C3), 158.81 (1C; pyridazinone C6), 159.00 (1C; 2-cyanophenyl C2), 164.54 (1C; -C=O) and 168.93 (1C; pyridazinone C3). C26H26N6O2 MS (ESI+) (Calc.: 455.2195; Found: 455.2191) (M+; 100.0%).
- N′-3-cyanobenzylidene-2-(3-(4-benzylpiperidin-1-yl)-6-oxopyridazin-1(6H)-yl)acetohydrazide (S17)
S17 was obtained as a white solid by following standard protocol with 6-(4-benzylpiperidine-1-yl)-3(2H)-pyridazinone-2-ylacetohydrazide and 3-cyanobenzaldehyde (m.p. 128–129, yield 86.49%). 1H-NMR (CDCl3, 300 MHz) TM 1.25–1.33 (4H, m, piperidin protons), 2.55 (d, 2H, benzyl CH2), 2.69 (1H, m, piperidin proton), 3.76–3.85 (4H piperidin protons), 5.25 (2H, s, CH2-CO-), 6.85–6.91 (m, 9H, phenyl protons), 7.21 (1H, s, 3-cyanophenyl H2), 7.28 (1H, d, pyridazinone H5), 7.69 (1H, d, pyridazinone H4), 7.98 (1H, s, N=CH), 10.67 (1H, s, CO-NH-N). 13C-NMR (CDCl3, 300 MHz), δ 31.36 (1C; CH2), 31.47 (1C; CH2), 37.80 (1C; CH), 43.03 (1C; CH2), 47.07 (2C; CH2-N), 53.46 (1C; N-CH2-C=O), 113.16 (1C; -CN), 126.01 (1C; N=CH), 128.29 (1C; pyridazinone C5), 129.63 (1C; pyridazinone C4), 130.38, 130.48, 130.66, 130.98, 131.10, 131.44 (6C; phenyl carbons), 134.98 (1C; 3-cyanophenyl C6), 142.00 (1C; 3-cyanophenyl C5), 146.18 (1C; 3-cyanophenyl C4), 149.57 (1C; 3-cyanophenyl C1), 150.14 (1C; 3-cyanophenyl C2), 158.69 (1C; pyridazinone C6), 159.00 (1C; 3-cyanophenyl C3), 164.38 (1C; -C=O) and 169.08 (1C; pyridazinone C3). C26H26N6O2 MS (ESI+) (Calc.: 455.2195; Found: 455.2200) (M+; 100.0%).
- N′-4-cyanobenzylidene-2-(3-(4-benzylpiperidin-1-yl)-6-oxopyridazin-1(6H)-yl)acetohydrazide (S18)
S18 was obtained as a white solid by following standard protocol with 6-(4-benzylpiperidine-1-yl)-3(2H)-pyridazinone-2-ylacetohydrazide and 4-cyanobenzaldehyde (m.p. 129–130, yield 69.47%). 1H-NMR (CDCl3, 300 MHz) TM 1.25–1.33 (2H, m, piperidin protons), 1.62–1.72 (2H, m, piperidin protons), 2.57 (d, 2H, benzyl CH2), 2.66–2.75 (1H, m, piperidin proton), 3.77–3.87 (4H piperidin protons), 5.28 (2H, s, CH2-CO-), 7.15–7.32 (m, 9H, phenyl protons), 7.73 (1H, d, pyridazinone H5), 8.01 (1H, d, pyridazinone H4), 9.74 (1H, s, N=CH), 10.73 (1H, s, CO-NH-N). 13C-NMR (CDCl3, 300 MHz), δ 31.36 (1C; CH2), 31.47 (1C; CH2), 37.82 (1C; CH), 43.03 (1C; CH2), 47.06 (2C; CH2-N), 53.23 (1C; N-CH2-C=O), 104.88 (1C; -CN), 126.05 (1C; N=CH), 128.28 (1C; pyridazinone C5), 129.11 (1C; pyridazinone C4), 130.43, 130.71, 140.04, 140.15, 144.38 (6C; phenyl carbons), 149.05 (1C; 4-cyanophenyl C6), 149.48 (1C; 4-cyanophenyl C2), 150.04 (1C; 4-cyanophenyl C1), 153.37 (1C; 4-cyanophenyl C3), 153.49 (1C; 4-cyanophenyl C5), 158.67 (1C; pyridazinone C6), 159.04 (1C; 4-cyanophenyl C4), 164.17 (1C; -C=O) and 168.74 (1C; pyridazinone C3). C26H26N6O2 MS (ESI+) (Calc.: 455.2195; Found: 455.2202) (M+; 100.0%).
- N′-2-methoxybenzylidene-2-(3-(4-benzylpiperidin-1-yl)-6-oxopyridazin-1(6H)-yl)acetohydrazide (S19)
S19 was obtained as a white solid by following standard protocol with 6-(4-benzylpiperidine-1-yl)-3(2H)-pyridazinone-2-ylacetohydrazide and 2-methoxybenzaldehyde (m.p. 206–207, yield 92.37%). 1H-NMR (CDCl3, 300 MHz) TM 1.23–1.33 (2H, m, piperidin protons), 1.65–1.75 (2H, m, piperidin protons), 2.56 (d, 2H, benzyl CH2), 2.64–2.74 (1H, m, piperidin proton), 3.74–3.79 (4H piperidin protons), 3.86 (3H, s, OCH3), 5.25 (2H, s, CH2-CO-), 7.12–7.30 (m, 9H, phenyl protons), 7.47–7.49 (1H, d, pyridazinone H5), 7.65 (1H, d, pyridazinone H4), 8.36 (1H, s, N=CH), 10.53 (1H, s, CO-NH-N). 13C-NMR (CDCl3, 300 MHz), δ 31.35 (1C; CH2), 31.49 (1C; CH2), 37.83 (1C; CH), 43.05 (1C; CH2), 47.11 (2C; CH2-N), 53.26 (1C; N-CH2-C=O), 55.56 (1C; OCH3), 120.80 (1C; N=CH), 125.97 (1C; pyridazinone C5), 126.49 (1C; pyridazinone C4), 127.18, 128.27, 128.31, 129.12, 130.54, 130.76 (6C; phenyl carbons), 131.58 (1C; 2-methoxyphenyl C6), 131.78 (1C; 2-methoxyphenyl C5) 140.06 (1C; 2-methoxyphenyl C4), 144.78 (1C; 2-methoxyphenyl C1), 149.99 (1C; 2-methoxyphenyl C3), 158.14 (1C; pyridazinone C6), 159.07 (1C; 2-methoxyphenyl C2), 164.14 (1C; -C=O) and 168.27 (1C; pyridazinone C3). C26H29N5O3 MS (ESI+) (Calc.: 460.2349; Found: 460.2352) (M+; 100.0%).
- N′-3,5-dimethoxybenzylidene-2-(3-(4-benzylpiperidin-1-yl)-6-oxopyridazin-1(6H)-yl)acetohydrazide (S20)
S20 was obtained as a white solid by following standard protocol with 6-(4-benzylpiperidine-1-yl)-3(2H)-pyridazinone-2-ylacetohydrazide and 3,5-dimethoxybenzaldehyde (m.p. 208–209, yield 97.37%). 1H-NMR (CDCl3, 300 MHz) TM 1.25–1.32 (4H, m, piperidin protons), 2.56 (d, 2H, benzyl CH2), 2.64–2.72 (1H, m, piperidin proton), 3.75–3.76 (2H, m, piperidin protons), 3.78 (2H, m, piperidin protons), 3.84 (3H, s, OCH3), 3.88 (3H, s, OCH3), 5.22 (2H, s, CH2-CO-), 7.18–7.34 (m, 9H, phenyl protons, pyridazinone H4,5), 8.03 (1H, s, 3,5-dimethoxyphenyl H4), 8.52 (1H, s, N=CH), 10.36 (1H, s, CO-NH-N). 13C-NMR (CDCl3, 300 MHz), δ 31.35 (1C; CH2), 31.47 (1C; CH2), 37.80 (1C; CH), 43.04 (1C; CH2), 47.07 (2C; CH2-N), 53.21 (1C; N-CH2-C=O), 55.38 (1C; OCH3), 55.47 (1C; OCH3), 104.56 (1C; N=CH), 125.98 (1C; pyridazinone C5), 126.04 (1C; pyridazinone C4), 126.31, 126.57, 128.27, 129.12, 130.73, 135.41 (6C; phenyl carbons), 140.20 (1C; 3,5-dimethoxyphenyl C6), 141.56 (1C; 3,5-dimethoxyphenyl C2), 144.28 (1C; 3,5-dimethoxyphenyl C1), 149.06 (1C; 3,5-dimethoxyphenyl C4), 149.44 (1C; pyridazinone C6), 158.67 (1C; 3,5-dimethoxyphenyl C5), 160.99 (1C; 3,5-dimethoxyphenyl C3), 164.25 (1C; -C=O) and 168.76 (1C; pyridazinone C3). C27H31N5O4 MS (ESI+) (Calc.: 490.2454; Found: 490.2461) (M+; 100.0%).
- N′-2,4,6-trimethoxybenzylidene-2-(3-(4-benzylpiperidin-1-yl)-6-oxopyridazin-1(6H)-yl)acetohydrazide (S21)
S21 was obtained as a white solid by following standard protocol with 6-(4-benzylpiperidine-1-yl)-3(2H)-pyridazinone-2-ylacetohydrazide and 2,4,6-trimethoxybenzaldehyde (m.p. 160–161, yield 54.81%). 1H-NMR (CDCl3, 300 MHz) TM 1.20–1.32 (2H, m, piperidin protons), 1.65–1.75 (2H, m, piperidin protons), 2.56 (d, 2H, benzyl CH2), 2.66–2.67 (1H, m, piperidin proton), 3.80 (4H piperidin protons), 3.82 (3H, s, OCH3), 3.84 (6H, s, OCH3), 5.23 (2H, s, CH2-CO-), 7.17–7.30 (m, 10H, phenyl protons, pyridazinone H4,5), 8.02 (1H, s, 2,4,6-trimethoxyphenyl H2), 8.33 (1H, s, N=CH), 10.29 (1H, s, CO-NH-N). 13C-NMR (CDCl3, 300 MHz), δ 31.36 (1C; CH2), 31.47 (1C; CH2), 37.79 (1C; CH), 43.03 (1C; CH2), 47.06 (2C; CH2-N), 53.23 (1C; N-CH2-C=O), 56.11 (1C; OCH3), 56.21 (1C; OCH3), 56.26 (1C; OCH3), 104.88 (1C; N=CH), 126.00 (1C; pyridazinone C5), 126.05 (1C; pyridazinone C4), 126.32, 128.28, 129.04, 129.11, 130.43, 130.71 (6C; phenyl carbons), 140.15 (1C; 2,4,6-trimethoxyphenyl C5), 144.38 (1C; 2,4,6-trimethoxyphenyl C1), 149.48 (1C; 2,4,6-trimethoxyphenyl C3), 150.04 (1C; pyridazinone C6), 153.37 (1C; 2,4,6-trimethoxyphenyl C2), 153.49 (1C; 2,4,6-trimethoxyphenyl C4), 159.04 (1C; 2,4,6-trimethoxyphenyl C6), 164.17 (1C; -C=O) and 168.74 (1C; pyridazinone C3). C28H33N5O5 MS (ESI+) (Calc.: 520.2560; Found: 520.2563) (M+; 100.0%).
- N′-2,3-dimethoxybenzylidene-2-(3-(4-benzylpiperidin-1-yl)-6-oxopyridazin-1(6H)-yl)acetohydrazide (S22)
S22 was obtained as a white solid by following standard protocol with 6-(4-benzylpiperidine-1-yl)-3(2H)-pyridazinone-2-ylacetohydrazide and 2,3-dimethoxybenzaldehyde (m.p. 212–213, yield 76.95%). 1H-NMR (CDCl3, 300 MHz) TM 1.24–1.31 (4H, m, piperidin protons), 1.68–1.72 (1H, m, piperidin protons), 2.55–2.71 (4H, benzyl CH2, piperidin proton), 3.75–3.77 (3H, m, piperidin protons), 3.86 (3H, s, OCH3), 3.89 (3H, s, OCH3), 5.25 (2H, s, CH2-CO-), 6.88–7.48 (m, 10H, phenyl protons, pyridazinone H4,5), 8.09 (1H, s, N=CH), 9.06 (1H, s, CO-NH-N). 13C-NMR (CDCl3, 300 MHz), δ 31.35 (1C; CH2), 31.48 (1C; CH2), 37.81 (1C; CH), 43.04 (1C; CH2), 47.09 (2C; CH2-N), 53.27 (1C; N-CH2-C=O), 55.79 (1C; OCH3), 55.84 (1C; OCH3), 113.86 (1C; N=CH), 124.13 (1C; pyridazinone C5), 124.30 (1C; pyridazinone C4), 126.28, 126.53, 127.25, 128.26, 129.11, 130.73 (6C; phenyl carbons), 140.05 (1C; 2,3-dimethoxyphenyl C6), 144.49 (1C; 2,3-dimethoxyphenyl C5), 148.55 (1C; 2,3-dimethoxyphenyl C1), 149.40 (1C; 2,3-dimethoxyphenyl C4), 152.83 (1C; pyridazinone C6), 158.69 (1C; 2,3-dimethoxyphenyl C2), 159.03 (1C; 2,3-dimethoxyphenyl C3), 164.23 (1C; -C=O) and 168.40 (1C; pyridazinone C3). C27H31N5O4 MS (ESI+) (Calc.: 490.2454; Found: 490.2457) (M+; 100.0%).
- N′-3,4,5-trimethoxybenzylidene-2-(3-(4-benzylpiperidin-1-yl)-6-oxopyridazin-1(6H)-yl)acetohydrazide (S23)
S23 was obtained as a white solid by following standard protocol with 6-(4-benzylpiperidine-1-yl)-3(2H)-pyridazinone-2-ylacetohydrazide and 3,4,5-trimethoxybenzaldehyde (m.p. 146–147, yield 86.16%). 1H-NMR (CDCl3, 300 MHz) TM 1.27–1.71 (4H, m, piperidin protons), 2.55–2.69 (4H, benzyl CH2, piperidin proton), 3.78–3.81 (3H piperidin protons), 3.85 (9H, s, OCH3), 5.28 (2H, s, CH2-CO-), 6.89–7.32 (m, 10H, phenyl protons, pyridazinone H4,5), 7.30 (1H, s, 3,4,5-trimethoxyphenyl H2), 8.03 (1H, s, N=CH), 10.36 (1H, s, CO-NH-N). 13C-NMR (CDCl3, 300 MHz), δ 31.36 (1C; CH2), 31.47 (1C; CH2), 37.79 (1C; CH), 43.03 (1C; CH2), 47.06 (2C; CH2-N), 53.23 (1C; N-CH2-C=O), 56.11 (1C; OCH3), 56.21 (1C; OCH3), 56.26 (1C; OCH3), 104.88 (1C; N=CH), 126.00 (1C; pyridazinone C5), 126.05 (1C; pyridazinone C4), 126.32, 128.28, 129.04, 129.11, 130.43, 130.71 (6C; phenyl carbons), 140.15 (1C; 3,4,5-trimethoxyphenyl C2), 144.38 (1C; 3,4,5-trimethoxyphenyl C1), 149.48 (1C; 3,4,5-trimethoxyphenyl C6), 150.04 (1C; pyridazinone C6), 153.37 (1C; 3,4,5-trimethoxyphenyl C3), 153.49 (1C; 3,4,5-trimethoxyphenyl C4), 159.04 (1C; 3,4,5-trimethoxyphenyl C5), 164.17 (1C; -C=O) and 168.74 (1C; pyridazinone C3). C28H33N5O5 MS (ESI+) (Calc.: 520.2560; Found: 520.2558) (M+; 100.0%).
- N′-2,4-dimethoxybenzylidene-2-(3-(4-benzylpiperidin-1-yl)-6-oxopyridazin-1(6H)-yl)acetohydrazide (S24)
S24 was obtained as a white solid by following standard protocol with 6-(4-benzylpiperidine-1-yl)-3(2H)-pyridazinone-2-ylacetohydrazide and 2,4-dimethoxybenzaldehyde (m.p. 174–175, yield 61.26%). 1H-NMR (CDCl3, 300 MHz) TM 1.28–1.30 (4H, m, piperidin protons), 2.68 (d, 4H, benzyl CH2, piperidin proton), 3.76–3.80 (3H, m, piperidin protons), 3.88 (3H, s, OCH3), 3.90 (3H, s, OCH3), 4.81 (2H, s, CH2-CO-), 6.39–7.99 (m, 9H, phenyl protons, pyridazinone H4,5), 8.05 (1H, s, 2,4-dimethoxyphenyl H4), 8.33 (1H, s, N=CH), 10.29 (1H, s, CO-NH-N). 13C-NMR (CDCl3, 300 MHz), δ 31.35 (1C; CH2), 31.47 (1C; CH2), 37.80 (1C; CH), 43.04 (1C; CH2), 47.07 (2C; CH2-N), 53.21 (1C; N-CH2-C=O), 55.38 (1C; OCH3), 55.47 (1C; OCH3), 104.56 (1C; N=CH), 125.98 (1C; pyridazinone C5), 126.04 (1C; pyridazinone C4), 126.31, 126.57, 128.27, 129.12, 130.73, 135.41 (6C; phenyl carbons), 140.20 (1C; 2,4-dimethoxyphenyl C6), 141.56 (1C; 2,4-dimethoxyphenyl C3), 144.28 (1C; 2,4-dimethoxyphenyl C1), 149.06 (1C; 2,4-dimethoxyphenyl C5), 149.44 (1C; pyridazinone C6), 158.67 (1C; 2,4-dimethoxyphenyl C2), 160.99 (1C; 2,4-dimethoxyphenyl C4), 164.25 (1C; -C=O) and 168.76 (1C; pyridazinone C3). C27H31N5O4 MS (ESI+) (Calc.: 490.2454; Found: 490.2448) (M+; 100.0%).
4.2. Enzyme Inhibition Studies
4.2.1. Enzyme Assays and Kinetics
The inhibition of hMAO-A and hMAO-B (recombinant; Sigma-Aldrich, St. Louis, MO, USA) was evaluated using 0.06 mM kynuramine and 0.3 mM benzylamine, respectively [34]. The absorbance of the MAO-A and MAO-B reactions was measured using the continuous method at 250 nm and 316 nm, respectively, for 45 min [34] with slight modifications. In the kinetic study, a Lineweaver–Burk (LB) plot was generated using five substrate concentrations, and the Km value of benzylamine for MAO-B was calculated as 0.29 mM.
4.2.2. An Inhibition Study of MAO-A and MAO-B by the Compounds
In the primary screening, 24 compounds were evaluated for their inhibition of MAO-A or MAO-B at 10 µM concentration. The half-inhibitory concentrations (IC50) of the compounds were calculated using GraphPad Prism software 5 [35]. The compounds with IC50 values of 40 µM or higher were indicated as >40 µM. The selectivity index (SI) was calculated as the IC50 of MAO-A/IC50 of MAO-B. The inhibition of compounds was compared with reference inhibitors such as toloxatone and clorgyline (reversible and irreversible inhibitors, respectively) for MAO-A and safinamide and pargyline (reversible and irreversible inhibitors, respectively) for MAO-B inhibitors [35]. For the inhibition kinetics, lead compounds were used at three concentrations, approximately 0.5, 1.5, and 2.0 times the IC50 value [34], and the inhibition constant (Ki) was determined using the secondary plot of their slopes in the LB plots.
4.2.3. Reversibility Studies
The reversibility of the lead compounds to MAO-B was evaluated using a dialysis tube (D-Tube Dialyzer Maxi, MWCO 6–8 kDa, Sigma-Aldrich, St. Louis, MO, USA). Their reversibilities were analyzed by the values of undialyzed (AU) and dialyzed (AD) activities, measuring approximately 2.0 times the IC50 value after pre-incubation for 30 min [34]. The reversibility patterns of the lead compounds were determined by comparing AU and AD with those reported in the literature [34].
4.3. Blood–Brain Barrier Permeability Study
In early drug studies, a parallel artificial membrane permeation approach was employed to predict the passive transcellular permeability of a drug through the blood–brain barrier (BBB). This involved setting up a sandwich-like structure in the PAMPA using a microtiter plate with 96 wells and a Millipore filter plate with 96 wells (IPVH, 125 m thick filter, 0.45 m pore), which were then submerged in 0.1 mL of n-dodecane. Drug samples were initially prepared as stock solutions in DMSO at 10 mm doses and stored at 0 °C. Before being introduced into a 96-well filter plate, these stock solutions were further diluted at pH 7.4 to achieve final sample concentrations of 0.01, 0.1, 0.5, and 1 mM, with the DMSO content limited to 1% (v/v). The resulting diluted solutions were then transferred to the donor wells (270 µL each), with 200 µL of pH 7.4 buffering agent added to the acceptor well. To create the “sandwich”, the donor plate (with the analyte at the bottom, an aqueous recipient on top, and an artificial lipid barrier in the middle) was accurately positioned over the acceptor filter plate. The drug material diffused from the donor well into the acceptor well through the lipid membrane, with the “sandwich” structure remaining intact throughout the process. UV spectroscopy was used to measure the drug concentration in the donor, recipient, and reference wells, and the extent of permeation was assessed using a specific expression [36].
4.4. Molecular Docking
The molecular docking software AutoDock Vina and the PDBQT molecular structure file format were used [37]. Consequently, AutoDockTools-1.5.6, from the MGLTools-1.5.6 package, was used to create ligands and receptors in the PDBQT file format. ChemDraw 23 was used to draw the S5 chemical structure in pdf format. Furthermore, Chembio3D pro was utilized for optimization, for improved molecular confirmation, using energy minimization with a 0.01 RMS gradient. Three-dimensional ligands were corrected by substituting hydrogen atoms in the water molecules. The crystal structure of 2V5Z was obtained from the Protein Data Bank. Following the extraction of the co-crystallized ligands, ions, and water molecules, a 40 × 40 × 40 grid box with a grid spacing of 0.375, X = 51.901, Y = 156.468, and Z = 28.561 was formed. The receptor file [38] was saved in the PDBQT file format. Vina was run on a Windows 10 PC with a Core i5 processor. A standard configuration file was established to specify grid box coordinates, exhaustiveness, computer usage, and output data for the docking experiment. The search was set to eight intensities to cover as much of the ground as possible. The docking experiments were initiated using a command line.
The docking experiment produced a PDBQT file with 20 different docked ligand positions on the receptor. The ideal position was selected by considering the docking score, number of H-bonds, and visual inspection of each docking position. The PDB format was used to store the selected receptor and ligand positions. The resulting PDB file was examined for ligand–receptor interactions using LigPlot + version 2.2 [39,40] and Biovia Discovery Studio 2021.
5. Conclusions
Twenty-four derivatives of pyridazinobenzylpiperidine were prepared, and their ability to inhibit monoamine oxidase (MAO) was assessed. The compound displayed greater MAO-B than MAO-A inhibition. Compound S15 demonstrated the most potent MAO-A inhibition, whereas compounds S5 and S16 demonstrated significant competitive and reversible MAO-B inhibition. Overall, if we look into the selectivity index, we can conclude that most of the compounds showed non-specific MAO inhibition. Additionally, BBB penetration was demonstrated by compounds S5 and S16, and molecular docking experiments indicated that compound S5 stabilized the protein–ligand complex. Based on these findings, lead compounds may be used as medications to treat neurological conditions.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/molecules29133097/s1, Figures S1–S16: Structures of sixteen compounds comprising 1H-NMR spectrum, 13C-NMR spectrum, and ESI-MS spectrum.
Author Contributions
Conceptualization, Z.Ö., H.K. and B.M.; methodology, Z.Ö., H.K. and B.M.; software, Z.Ö., H.K., S.K. (Sunil Kumar) and B.M.; validation, Z.Ö., H.K., J.M.O., and B.M.; formal analysis, Y.N.Z., S.K. (Semanur Kılıç), M.A., J.M.O., and B.M.; investigation, Z.Ö., H.K. and B.M.; resources, Z.Ö., M.A., A.B.Ö. and H.K.; data curation, H.K., S.K. (Sunil Kumar), and B.M.; writing—original draft preparation, Z.Ö. and H.K.; writing—review and editing, Z.Ö., B.M. and H.K.; visualization, Y.N.Z., J.M.O. and S.K. (Sunil Kumar); supervision, Z.Ö., H.K. and B.M.; project administration, H.K. and Z.Ö.; funding acquisition, H.K. and Z.Ö. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the Research Foundation of İnönü University (TYL-2023-3186), Inonu University, Malatya, Türkiye.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article and Supplementary Materials.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Bohnen, N.I.; Yarnall, A.J.; Weil, R.S.; Moro, E.; Moehle, M.S.; Borghammer, P.; Bedard, M.A.; Albin, R.G. Cholinergic system changes in Parkinson’s disease: Emerging therapeutic approaches. Lancet Neurol. 2022, 21, 381–392. [Google Scholar] [CrossRef]
- Chew, Z.X.; Lim, C.L.; Ng, K.Y.; Chye, S.M.; Ling, A.P.; Koh, R.Y. The role of monoamine oxidase B inhibitors in the treatment of Parkinson’s disease-An update. CNS Neurol. Disord. Drug Targets 2023, 22, 329–352. [Google Scholar]
- Fujii, T.; Nagamori, S.; Wiriyasermkul, P.; Zheng, S.; Yago, A.; Shimizu, T.; Tabuchi, Y.; Okumura, T.; Fujii, T.; Takeshima, H.; et al. Parkinson’s disease-associated ATP13A2/PARK9 functions as a lysosomal H+, K+-ATPase. Nature Commun. 2023, 14, 2174. [Google Scholar] [CrossRef]
- Regensburger, M.; Ip, C.W.; Kohl, Z.; Schrader, C.; Urban, P.P.; Kassubek, J.; Jost, W.H. Clinical benefit of MAO-B and COMT inhibition in Parkinson’s disease: Practical considerations. J. Neural Transm. 2023, 130, 847–861. [Google Scholar] [CrossRef]
- Baweja, G.S.; Gupta, S.; Kumar, B.; Patel, P.; Asati, V. Recent updates on structural insights of MAO-B inhibitors: A review on target-based approach. Mol. Divers. 2023, 1–23. [Google Scholar] [CrossRef]
- Lv, Y.; Fan, M.; He, J.; Song, X.; Guo, J.; Gao, B.; Zhang, J.; Zhang, C.; Xie, Y. Discovery of Novel Benzimidazole Derivatives as Selective and Reversible Monoamine Oxidase B Inhibitors for Parkinson’s Disease Treatment. Eur. J. Med. Chem. 2024, 274, 116566. [Google Scholar] [CrossRef]
- Al-Saad, O.M.; Gabr, M.; Darwish, S.S.; Rullo, M.; Pisani, L.; Miniero, D.V.; Liuzzi, G.M.; Kany, A.M.; Hirch, A.K.N.; Abadi, A.H.; et al. Novel 6-hydroxybenzothiazol-2-carboxamides as potent and selective monoamine oxidase B inhibitors endowed with neuroprotective activity. Eur. J. Med. Chem. 2024, 269, 116266. [Google Scholar] [CrossRef]
- Kondeva-Burdina, M.; Mateev, E.; Angelov, B.; Tzankova, V.; Georgieva, M. In Silico Evaluation and In Vitro Determination of Neuroprotective and MAO-B Inhibitory Effects of Pyrrole-Based Hydrazones: A Therapeutic Approach to Parkinson’s Disease. Molecules 2022, 27, 8485. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Oh, J.M.; Abdelgawad, M.A.; Abourehab, M.A.; Tengli, A.K.; Singh, A.K.; Ahmad, I.; Patel, H.; Mathew, B.; Kim, H. Development of isopropyl-tailed chalcones as a new class of selective MAO-B inhibitors for the treatment of Parkinson’s disorder. ACS Omega 2023, 8, 6908–6917. [Google Scholar] [CrossRef] [PubMed]
- Çeçen, M.; Oh, J.M.; Özdemir, Z.; Büyüktuncel, S.E.; Uysal, M.; Abdelgawad, M.A.; Musa, A.; Gambacorta, N.; Nicolotti, O.; Mathew, B.; et al. Design, synthesis, and biological evaluation of pyridazinones containing the (2-fluorophenyl) piperazine moiety as selective MAO-B inhibitors. Molecules 2020, 25, 5371. [Google Scholar] [CrossRef] [PubMed]
- Alagöz, M.A.; Oh, J.M.; Zenni, Y.N.; Özdemir, Z.; Abdelgawad, M.A.; Naguib, I.A.; Ghoneim, M.M.; Gambacorta, N.; Nicolotti, O.; Kim, H.; et al. Development of a novel class of pyridazinone derivatives as selective MAO-B inhibitors. Molecules 2022, 27, 3801. [Google Scholar] [CrossRef]
- Yang, C.; Wang, X.; Gao, C.; Liu, Y.; Ma, Z.; Zang, J.; Wang, H.; Liu, L.; Liu, Y.; Sun, H.; et al. Molecular Mechanism and Structure-activity Relationship of the Inhibition Effect between Monoamine Oxidase and Selegiline Analogues. Curr. Comput.-Aid. Drug 2024, 20, 474–485. [Google Scholar] [CrossRef]
- Hirano, M.; Samukawa, M.; Isono, C.; Kusunoki, S.; Nagai, Y. The effect of rasagiline on swallowing function in Parkinson’s disease. Heliyon 2024, 10, e23407. [Google Scholar] [CrossRef]
- Blair, H.A.; Dhillon, S. Safinamide: A review in Parkinson’s disease. CNS Drugs 2017, 31, 169–176. [Google Scholar] [CrossRef]
- Özdemir, Z.; Gökçe, M.; Karakurt, A. Synthesis and analgesic, anti-inflammatory and antimicrobial evaluation of 6-substituted-3 (2H)-pyridazinone-2-acetyl-2-(substituted benzal) hydrazone derivatives. FABAD J. Pharm. Sci. 2012, 37, 111–122. [Google Scholar]
- Özdemir, Z.; Alagöz, M.A.; Uslu, H.; Karakurt, A.; Erikci, A.; Ucar, G.; Uysal, M. Synthesis, molecular modelling and biological activity of some pyridazinone derivatives as selective human monoamine oxidase-B inhibitors. Pharmacol. Rep. 2020, 72, 692–704. [Google Scholar] [CrossRef]
- Hamed, M.Y.; Aly, A.F.; Abdullah, N.H.; Ismail, M.F. Synthesis, characterization and antifungal evaluation of novel pyridazin-3 (2 H)-one derivatives. Polycycl. Aromat. Comp. 2023, 43, 2356–2375. [Google Scholar] [CrossRef]
- Partap, S.; Akhtar, M.J.; Yar, M.S.; Hassan, M.Z.; Siddiqui, A.A. Pyridazinone hybrids: Design, synthesis and evaluation as potential anticonvulsant agents. Bioorg. Chem. 2018, 77, 74–83. [Google Scholar] [CrossRef]
- Szczukowski, Ł.; Redzicka, A.; Wiatrak, B.; Krzyżak, E.; Marciniak, A.; Gębczak, K.; Gebarowski, T.; Świątek, P. Design, synthesis, biological evaluation and in silico studies of novel pyrrolo [3,4-d] pyridazinone derivatives with promising anti-inflammatory and antioxidant activity. Bioorg. Chem. 2020, 102, 104035. [Google Scholar] [CrossRef]
- Imran, M.; Nayeem, N. Synthesis and antihypertensive activity of some novel pyridazinones. Orient. J. Chem. 2016, 32, 267–274. [Google Scholar] [CrossRef]
- Yamada, T.; Shimamura, H.; Tsukamoto, Y.; Yamaguchi, A.; Ohki, M. Pyridazinones. 3. Synthesis, antisecretory, and antiulcer activities of 2-cyanoguanidine derivatives. J. Med. Chem. 1983, 26, 1144–1149. [Google Scholar] [CrossRef]
- Mikus, J.; Świątek, P.; Przybyła, P.; Krzyżak, E.; Marciniak, A.; Kotynia, A.; Redzicka, A.; Wiatrak, B.; Jawień, P.; Gębarowski, T.; et al. Synthesis, Biological, Spectroscopic and Computational Investigations of Novel N-Acylhydrazone Derivatives of Pyrrolo [3,4-d] pyridazinone as Dual COX/LOX Inhibitors. Molecules 2023, 28, 5479. [Google Scholar] [CrossRef]
- He, Z.X.; Gong, Y.P.; Zhang, X.; Ma, L.Y.; Zhao, W. Pyridazine as a privileged structure: An updated review on anticancer activity of pyridazine containing bioactive molecules. Eur. J. Med. Chem. 2021, 209, 112946. [Google Scholar] [CrossRef]
- Giovannoni, M.P.; Ciciani, G.; Cilibrizzi, A.; Crocetti, L.; Daniele, S.; Mannelli, L.D.C.; Ghelardini, C.; Giacomelli, C.; Guerrini, G.; Martini, C.; et al. Further studies on pyrazolo[1′,5′:1,6] pyrimido[4,5-d] pyridazin-4(3H)-ones as potent and selective human A1 adenosine receptor antagonists. Eur. J. Med. Chem. 2015, 89, 32–41. [Google Scholar] [CrossRef]
- Cao, X.; Chen, Y.; Zhang, Y.; Qiu, Y.; Yu, M.; Xu, X.; Liu, X.; Liu, B.-F.; Zhang, G. Synthesis and biological evaluation of new 6-hydroxypyridazinone benzisoxazoles: Potential multi-receptor-targeting atypical antipsychotics. Eur. J. Med. Chem. 2016, 124, 713–728. [Google Scholar] [CrossRef]
- Abd-Rabo, Z.S.; George, R.F.; Zaafar, D.K.; Gawish, A.Y.; Serry, A.M. Design, synthesis, and biological evaluation of some new 2-phenyl-3,6-pyridazinedione derivatives as PDE-5 inhibitors. Bioorg. Chem. 2024, 145, 107213. [Google Scholar] [CrossRef]
- Xing, X.; Chang, L.C.; Kong, Q.; Colton, C.K.; Lai, L.; Glicksman, M.A.; Lin, C.L.G.; Cuny, G.D. Structure–activity relationship study of pyridazine derivatives as glutamate transporter EAAT2 activators. Bioorg. Med. Chem. Lett. 2011, 21, 5774–5777. [Google Scholar] [CrossRef]
- Asif, M.; Almehmadi, M.M.; Alsaiari, A.A. Diverse pharmacological potential of pyridazine analogs against various diseases. Med. Chem. 2024, 20, 245–267. [Google Scholar]
- Rosa, F.A.; Gonçalves, D.S.; Pianoski, K.E.; da Silva, M.J.; Ames, F.Q.; Aguiar, R.P.; Volpato, H.; Lazarin-Bidóia, D.; Nakamura, C.V.; Bersani-Amado, C.A. Discovery of a new pyrido [2,3-d] pyridazine-2, 8-dione derivative as a potential anti-inflammatory agent through COX-1/COX-2 dual inhibition. RSC Med. Chem. 2024, 15, 1038–1045. [Google Scholar] [CrossRef]
- Allam, H.A.; Kamel, A.A.; El-Daly, M.; George, R.F. Synthesis and vasodilator activity of some pyridazin-3(2H)-one based compounds. Future Med. Chem. 2020, 12, 37–50. [Google Scholar] [CrossRef]
- Besada, P.; Viña, D.; Costas, T.; Costas-Lago, M.C.; Vila, N.; Torres-Terán, I.; Sturlese, M.; Moro, S.; Terán, C. Pyridazinones containing dithiocarbamoyl moieties as a new class of selective MAO-B inhibitors. Bioorg. Chem. 2021, 115, 105203. [Google Scholar] [CrossRef]
- Mazej, T.; Knez, D.; Meden, A.; Gobec, S.; Sova, M. 4-phenethyl-1-propargylpiperidine-derived dual inhibitors of butyrylcholinesterase and monoamine oxidase B. Molecules 2021, 26, 4118. [Google Scholar] [CrossRef]
- Costas-Lago, M.C.; Besada, P.; Rodríguez-Enríquez, F.; Viña, D.; Vilar, S.; Uriarte, E.; Borges, F.; Terán, C. Synthesis and structure-activity relationship study of novel 3-heteroarylcoumarins based on pyridazine scaffold as selective MAO-B inhibitors. Eur. J. Med. Chem. 2017, 139, 1–11. [Google Scholar] [CrossRef]
- Baek, S.C.; Park, M.H.; Ryu, H.W.; Lee, J.P.; Kang, M.-G.; Park, D.; Park, C.M.; Oh, S.-R.; Kim, H. Rhamnocitrin isolated from Prunus padus var. seoulensis: A potent and selective reversible inhibitor of human monoamine oxidase A. Bioorg. Chem. 2019, 83, 317–325. [Google Scholar] [CrossRef]
- El-Damasy, A.K.; Oh, J.M.; Kim, H.J.; Mun, S.-G.; Al-Karmalawy, A.A.; Alnajjar, R.; Choi, Y.-J.; Kim, J.-J.; Nam, G.; Kim, H.; et al. Novel coumarin benzamides as potent and reversible monoamine oxidase-B inhibitors: Design, synthesis, and neuroprotective effects. Bioorg. Chem. 2024, 142, 106939. [Google Scholar] [CrossRef]
- Di, L.; Kerns, E.H.; Fan, K.; McConnell, O.J.; Carter, G.T. High throughput artificial membrane permeability assay for blood-brain barrier. Eur. J. Med. Chem. 2003, 38, 223–232. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Binda, C.; Wang, J.; Pisani, L.; Caccia, C.; Carotti, A.; Salvati, P.; Edmondson, D.E.; Mattevi, A. Structures of human monoamine oxidase B complexes with selective noncovalent inhibitors: Safinamide and coumarin analogs. J. Med. Chem. 2007, 50, 5848–5852. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand–protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Jejurikar, B.L.; Rohane, S.H. Drug designing in discovery studio. Asian J. Res. Chem. 2021, 14, 135–138. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).