
Effect of Electrolysis Conditions on Electrodeposition of Cobalt–Tin Alloys, Their Structure, and Wettability by Liquids


Abstract
1. Introduction
2. Results and Discussion
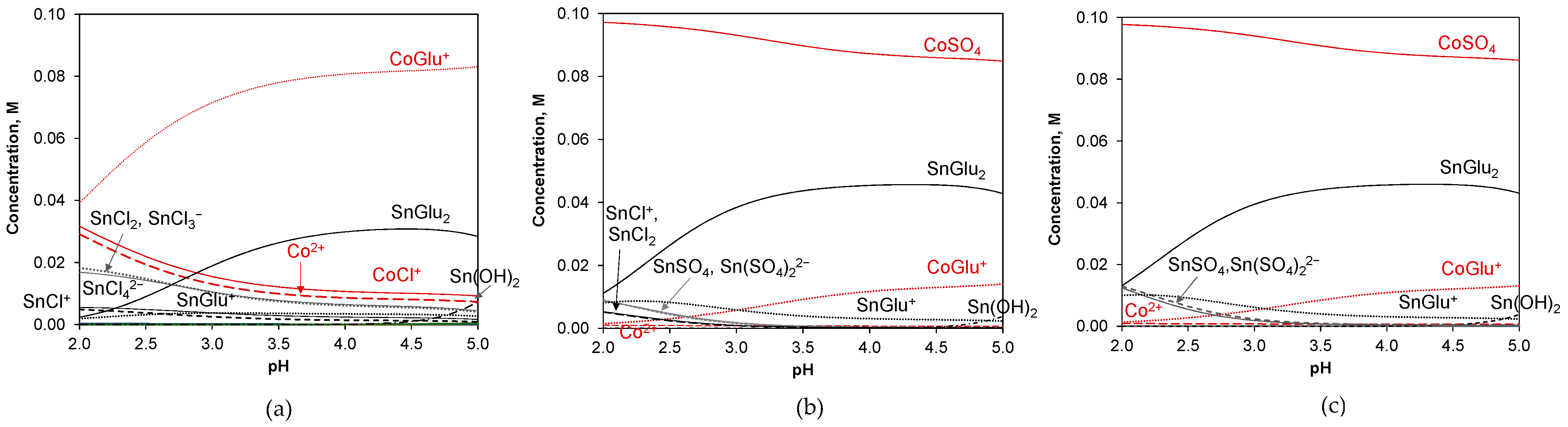
2.1. Bath Speciation
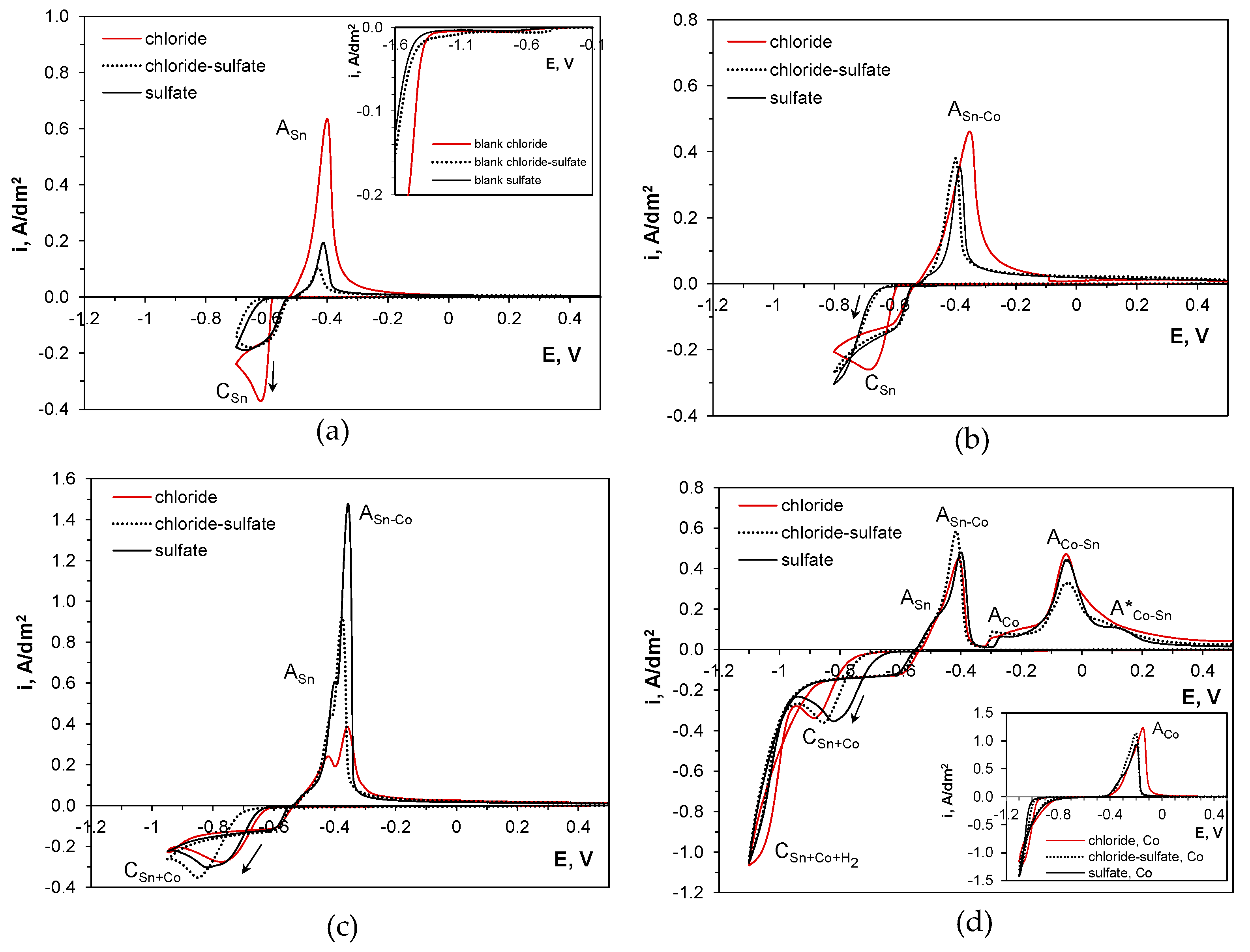
2.2. Cyclic Voltammetry
2.3. Thermodynamic Analysis
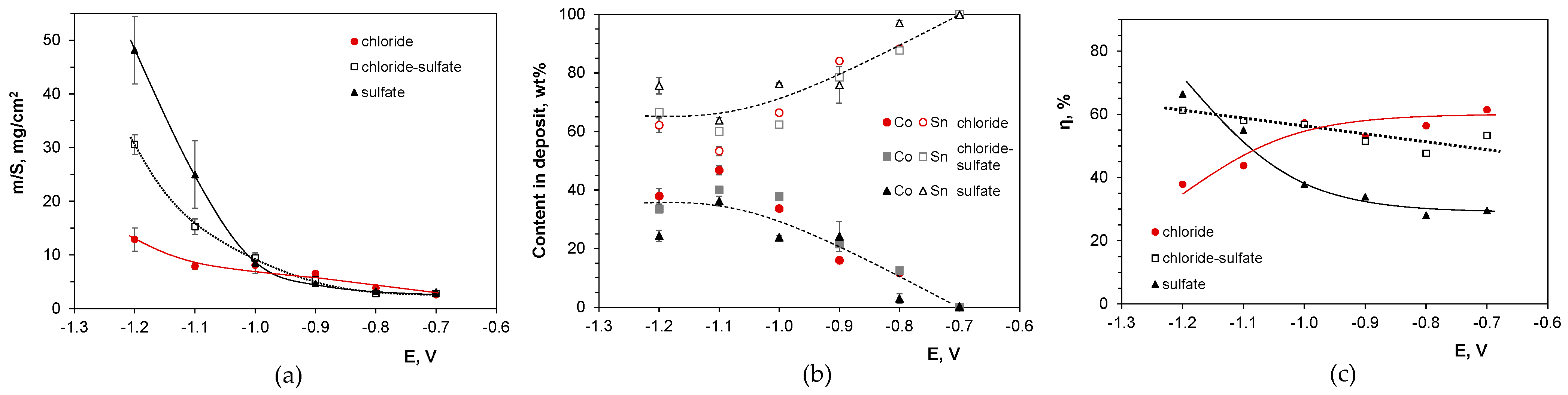
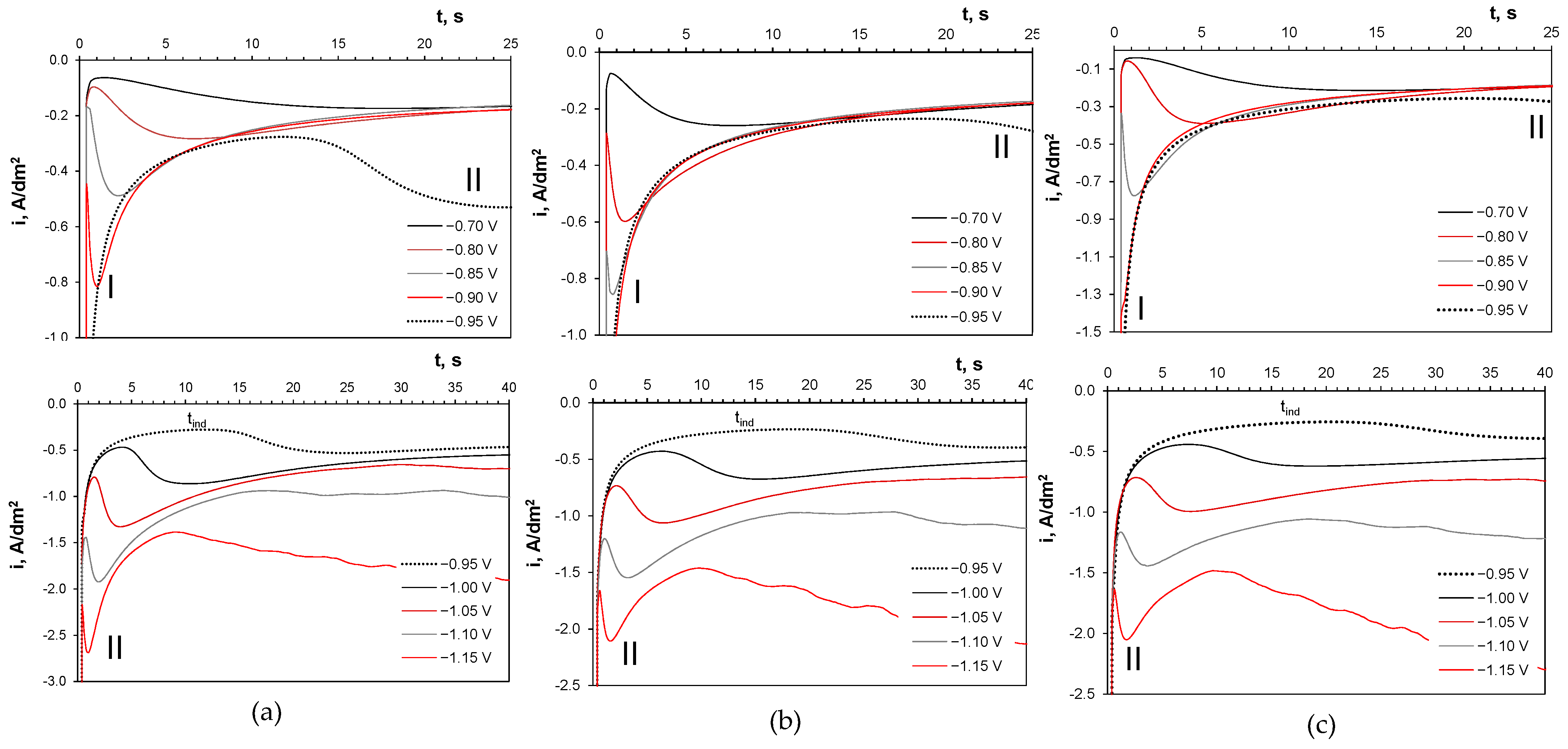
2.4. Potentiostatic Deposition
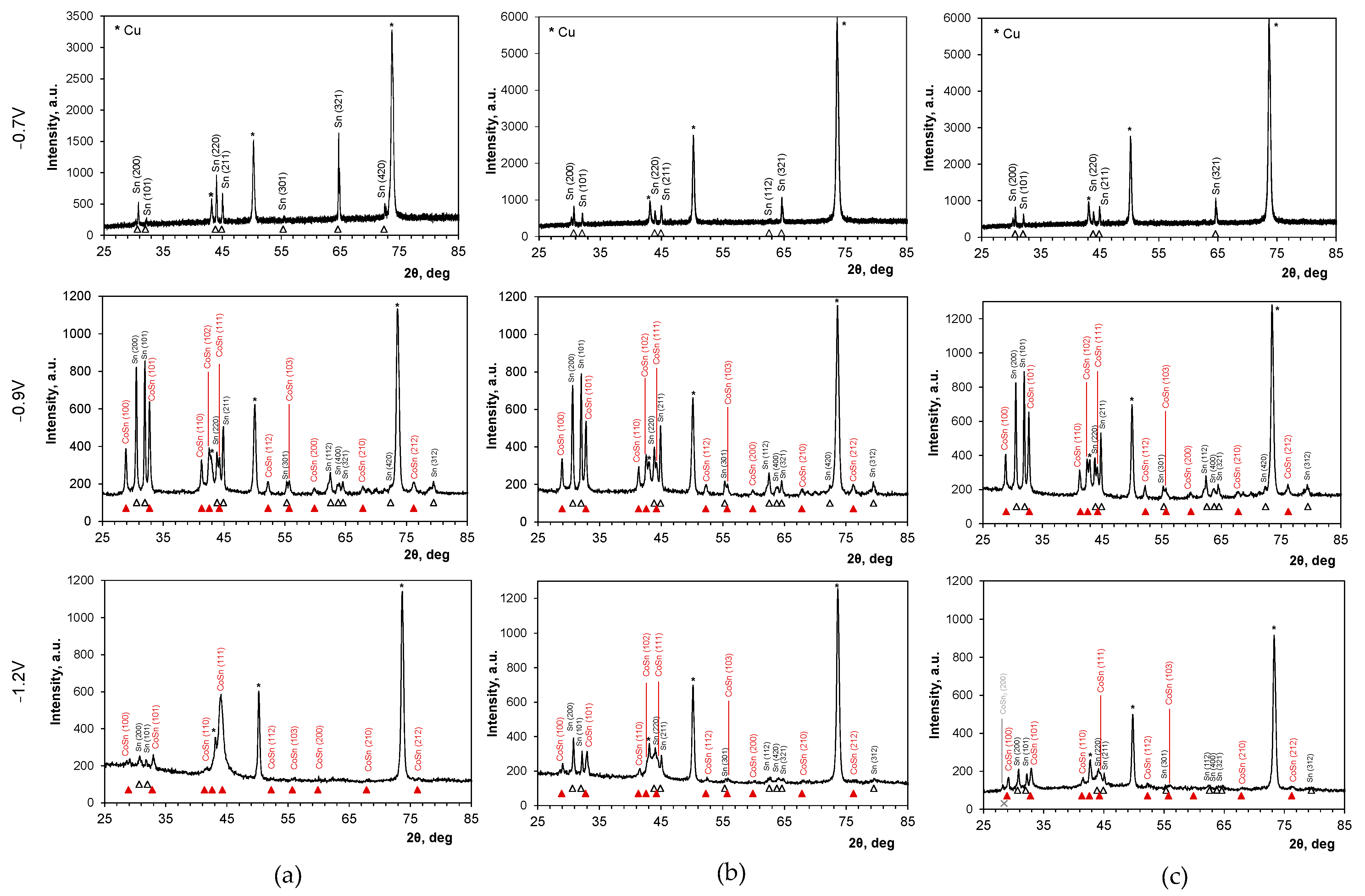
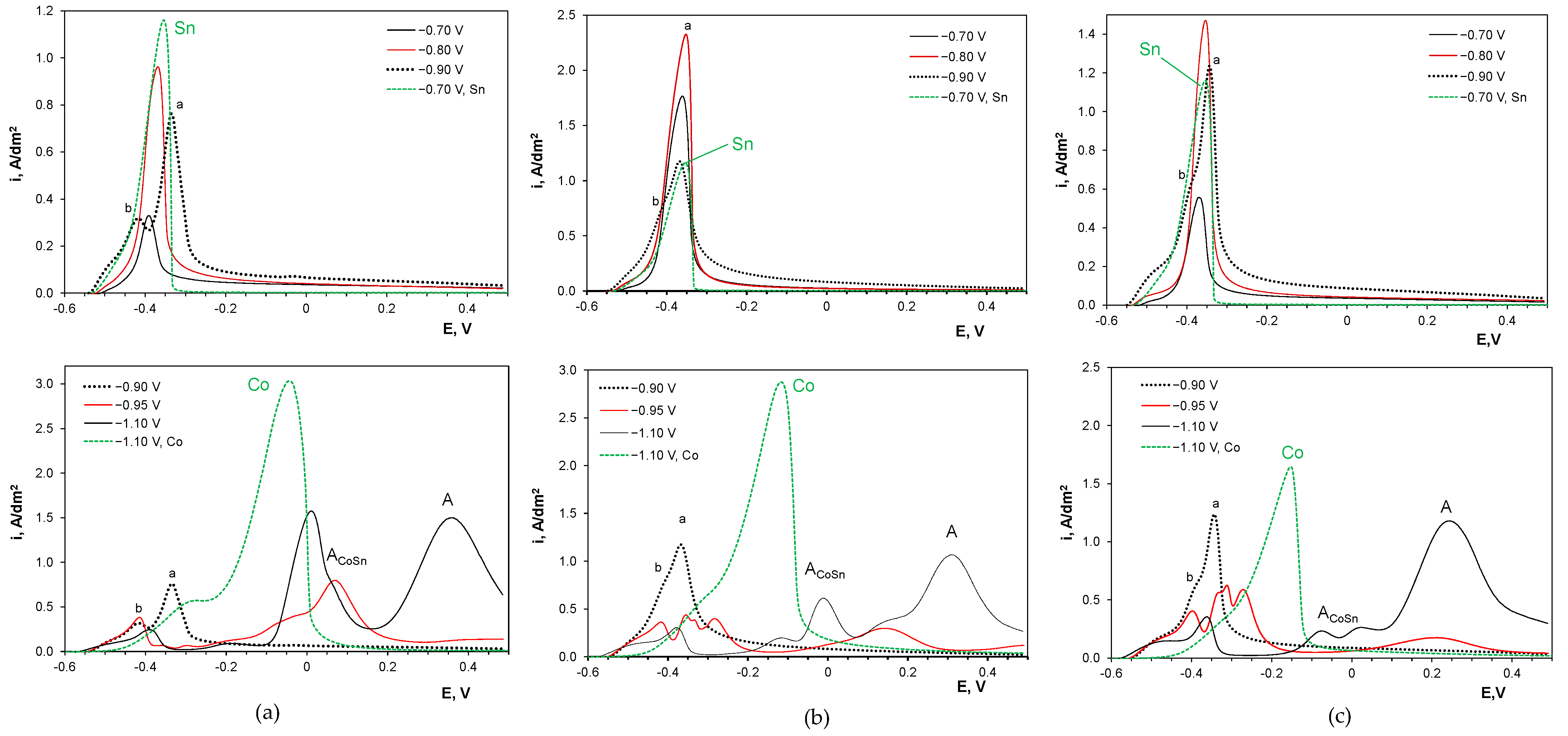
2.5. Anodic Sweep Linear Voltammetry
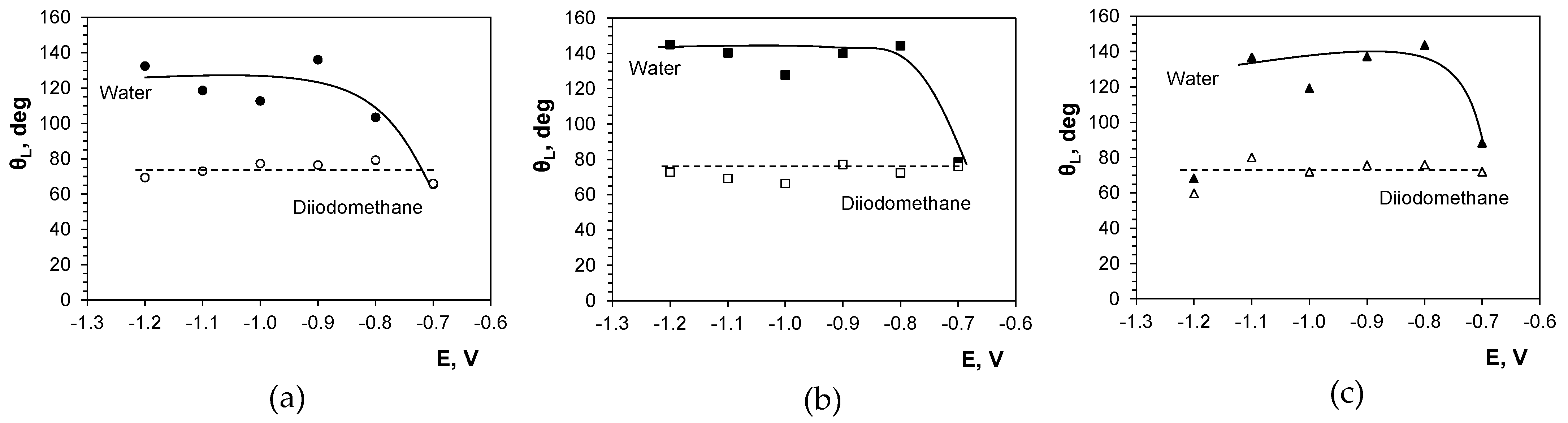
2.6. Surface Wettability
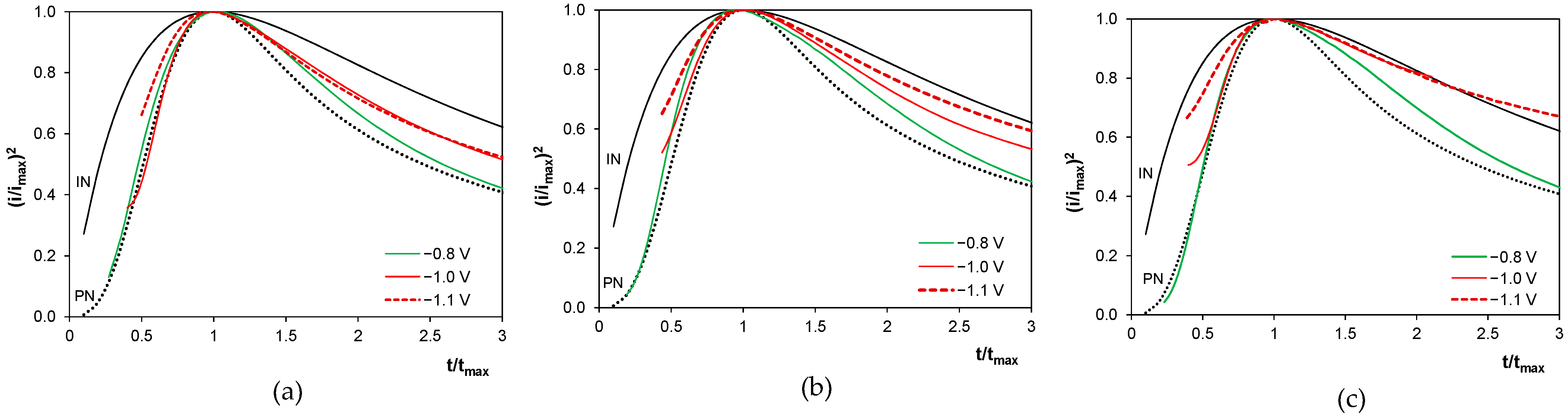
2.7. Nucleation and Growth of Deposits
3. Materials and Methods
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brenner, A. Electrodeposition of Alloys; Academic Press: New York, NY, USA, 1963; Volume II, pp. 339–343. [Google Scholar]
- Abd El Rehim, S.S.; Refaey, S.A.; Schwitzgebelf, G.; Taha, F.; Saleh, M.B. Electrodeposition of Sn-Co alloys from gluconate baths. J. Appl. Electrochem. 1996, 26, 413–418. [Google Scholar] [CrossRef]
- Sujatha, M.; Sabitha, R.; Pushpavanam, M. Electrodeposited cobalt-tin alloy from a neutral gluconate bath. Trans. IMF 2000, 78, 49–52. [Google Scholar] [CrossRef]
- Gomez, E.; Guaus, E.; Torrent, J.; Alcobe, X.; Valles, E. Cobalt-tin electrodeposition from sulfate-gluconate baths. J. Appl. Electrochem. 2001, 31, 349–354. [Google Scholar] [CrossRef]
- Vinokurov, E.G. Prognostication of the composition of a solution for electrodeposition of Sn–Co alloy and determination of its color characteristics. Russ. J. Appl. Chem. 2010, 83, 258–262. [Google Scholar] [CrossRef]
- Medvedev, G.I.; Makrushin, N.A. Electrodeposition of tin–cobalt alloy from a sulfate electrolyte with organic additives. Russ. J. Appl. Chem. 2012, 85, 52–56. [Google Scholar] [CrossRef]
- Valkova, T.; Krastev, I. Influence of glycine on the electrochemical deposition of Sn-Co alloy from gluconate electrolyte. Bulg. Chem. Comm. 2016, 48B, 78–84. [Google Scholar]
- Refaey, S.A.M. Corrosion of Sn–Co alloy in alkaline media and the effect of Cl− and Br− ions. Appl. Surf. Sci. 1999, 147, 67–76. [Google Scholar] [CrossRef]
- Zhang, J.L.; Gu, C.D.; Fashu, S.; Tong, Y.Y.; Huang, M.L.; Wang, X.L.; Tua, J.P. Enhanced corrosion resistance of Co-Sn alloy coating with a self-organized layered structure electrodeposited from deep eutectic solvent. J. Electrochem. Soc. 2015, 162, D1–D8. [Google Scholar] [CrossRef]
- Pereira, N.M.; Sousa, C.T.; Pereira, C.M.; Araujo, J.P.; Silva, A.F. Enhanced properties of Co-Sn coatings electrodeposited from choline chloride-based deep eutectic solvents. Cryst. Growth Des. 2017, 17, 5208–5215. [Google Scholar] [CrossRef]
- Cho, S.K.; Han, H.S.; Lee, C.K.; Ahn, C.I.; Park, J.I. Cobalt-tin plating in a pyrophosphate bath. Mat. Sci. Forum. 2003, 439, 57–61. [Google Scholar] [CrossRef]
- Georgiou, E.P.; Buijnsters, J.G.; Wang, H.; Drees, D.; Basak, A.K.; Celis, J.P. Nanostructured gradient Co-Sn electrodeposits as alternative to Sn connector contacts. Surf. Coat. Technol. 2015, 271, 148–155. [Google Scholar] [CrossRef]
- Vitina, I.; Belmane, V.; Krumina, A.; Rubene, V. Changes in the phase composition and structure of electrodeposited Sn-Co alloys in Sn-Co/Sn layer systems upon heating. Surf. Coat. Technol. 2011, 205, 2893–2898. [Google Scholar] [CrossRef]
- Hy, R.Z.; Liu, H.; Zeng, M.Q.; Liu, J.W.; Zhu, M. Progress on Sn-based thin film anode materials for lithium-ion batteries. Chin. Sci. Bull. 2012, 57, 4119–4130. [Google Scholar]
- Fan, X.-Y.; Yang, F.-Z.; Sun, S.-G. Fabrication and properties of macroporous tin–cobalt alloy film electrodes for lithium-ion batteries. J. Power Sources 2007, 170, 450–455. [Google Scholar]
- Fan, X.Y.; Ke, F.S.; Wei, G.Z.; Huang, L.; Sun, S.G. Sn-Co alloy anode using porous Cu as current collector for lithium ion battery. J. Alloys Compd. 2009, 476, 70–73. [Google Scholar] [CrossRef]
- Ui, K.; Kikuchi, S.; Jimba, Y.; Kumagai, N. Preparation of Co–Sn alloy film as negative electrode for lithium secondary batteries by pulse electrodeposition method. J. Power Sources 2011, 196, 3916–3920. [Google Scholar] [CrossRef]
- Gnanamuthu, R.M.; Jo, Y.N.; Lee, C.W. Brush electroplated CoSn2 alloy film for application in lithium-ion batteries. J. Alloys Compd. 2013, 564, 95–99. [Google Scholar] [CrossRef]
- Groult, H.; El Ghallali, H.; Barhoun, A.; Briot, E.; Perrigaud, L.; Hernandorena, S.; Lantelme, F. Preparation of Co–Sn alloys by electroreduction of Co(II) and Sn(II) in molten LiCl–KCl. Electrochim. Acta 2010, 55, 1926–1932. [Google Scholar] [CrossRef]
- Liu, Y.; Lu, H.; Kou, X. Electrodeposited Ni-Co-Sn alloy as a highly efficient electrocatalyst for water splitting. Int. J. Hydrogen Energy 2019, 44, 8099–80108. [Google Scholar] [CrossRef]
- Chandran, A.M.; Karumuthil, S.C.; Singh, A.K.; Prasad, B.L.V. Electrodeposited Co-Mn-Sn multicomponent alloy as an efficient electrocatalyst for hydrogen evolution reaction. Int. J. Hydrogen Energy 2024, 49, 658–667. [Google Scholar]
- Survila, A.; Mockus, Z.; Kanapeckaite, S.; Stalnionis, G. Kinetics of Sn(II) reduction in acid sulphate solutions containing gluconic acid. J. Electroanal. Chem. 2012, 667, 59–65. [Google Scholar] [CrossRef]
- Mockus, Z.; Norkus, E.; Vaitkus, R.; Kalinauskas, P.; Grinciene, G.; Tamasauskaite-Tamasiunaite, L. Inhibition of Sn(II) oxidation by air oxygen in acidic gluconate-containing solutions. J. Electrochem. Soc. 2020, 167, 122503. [Google Scholar] [CrossRef]
- Guaus, E.; Torrent-Burgues, J. Tin–zinc electrodeposition from sulphate–gluconate baths. J. Electroanal. Chem. 2003, 549, 25–36. [Google Scholar] [CrossRef]
- Rudnik, E.; Chowaniec, G. Effect of organic additives on electrodeposition of tin from acid sulfate solution. Metall. Foundry Eng. 2018, 44, 41–52. [Google Scholar] [CrossRef]
- Rudnik, E. Effect of anions on the electrodeposition of tin from acidic gluconate baths. Ionics 2013, 19, 1047–1059. [Google Scholar] [CrossRef]
- Rudnik, E.; Włoch, G. Studies on the electrodeposition of tin from acidic chloride-gluconate solutions. Appl. Surf. Sci. 2013, 265, 839–849. [Google Scholar] [CrossRef]
- Rudnik, E. The influence of sulfate ions on the electrodeposition of Ni-Sn alloys from acidic chloride-gluconate baths. J. Electroanal. Chem. 2014, 726, 97–106. [Google Scholar] [CrossRef]
- Kuzmann, E.; Felner, I.; Sziraki, L.; Stichleutner, S.; Homonnay, Z.; El-Sharif, M.; Chisholm, C.U. Magnetic anisotropy and microstructure in electrodeposited quaternary Sn-Fe-Ni-Co alloys with amorphous character. Materials 2022, 15, 3015. [Google Scholar] [CrossRef]
- Tripathi, D.; Ray, P.; Singh, A.V.; Kishore, V.; Singh, S.L. Durability of slippery liquid-infused surfaces: Challenges and advances. Coatings 2023, 13, 1095. [Google Scholar] [CrossRef]
- Kaden, N.; Schlimbach, R.; Garcia, A.R.; Droder, K. A systematic literature analysis on electrolyte filling and wetting in lithium-ion battery production. Batteries 2023, 9, 164. [Google Scholar] [CrossRef]
- Rudnik, E.; Dashbold, N. Electrolytic recovery of tin from solder alloy in acid chloride solutions. Rudy Met. Rec. 2015, 60, 414–420. (In Polish) [Google Scholar]
- The IUPAC Stability Constants Database; Academic Software and IUMAC, 1992–2000. Available online: https://www.acadsoft.co.uk (accessed on 21 June 2024).
- Ashton, F.; Pickering, W.F. Cobalt(II) gluconate complexes. Aust. J. Chem. 1970, 23, 1367–1373. [Google Scholar] [CrossRef]
- Pettine, M.; Millero, F.J.; Macchi, G. Hydrolysis of tin(II) in aqueous solutions. Anal. Chem. 1981, 53, 1039–1043. [Google Scholar] [CrossRef]
- Müller, B.; Seward, T.M. Spectrophotometric determination of the stability of tin (II) chloride complexes in aqueous solution up to 300 °C. Geochim. Cosmochim. Acta 2001, 65, 4187–4199. [Google Scholar] [CrossRef]
- Kochkodan, V.; Darwish, N.B.; Hilal, N. The chemistry of boron in water. In Boron Separation Processes; Kabay, N., Bryjak, M., Hilal, N., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 35–63. [Google Scholar]
- Smith, P.J. (Ed.) Chemistry of Tin; Springer Science+Buisness Media: Dordrecht, The Netherlands, 1998. [Google Scholar]
- Soto, A.B.; Arce, E.M.; Palomar-Pardavé, M.; Gonzalez, I. Electrochemical nucleation of cobalt on glassy carbon electrode from ammonium chloride solutions. Electrochim. Acta 1996, 41, 2647–2655. [Google Scholar] [CrossRef]
- Grujicic, D.; Pesic, B. Electrochemical and AFM study of cobalt nucleation mechanisms on glassy carbon from ammonium sulfate solutions. Electrochim. Acta 2004, 49, 4719–4732. [Google Scholar] [CrossRef]
- Łętowski, F.; Niemiec, J. Diagrams of electrochemical equilibria E-pH at 25 °C. Part IV. Co-H2O-NH3-H2SO4 system. Rocz. Chem. 1969, 43, 281–290. (In Polish) [Google Scholar]
- Rudnik, E.; Dashbold, N. Effect of Cl− and SO42− ions on electrodeposition of cobalt from acidic gluconate solutions. Russ. J. Electrochem. 2019, 55, 1305–1319. [Google Scholar] [CrossRef]
- Tripkovic, D.V.; Strmcnik, D.; van der Vliet, D.; Stamenkovic, V.; Markovic, N.M. The role of anions in surface electrochemistry. Farad. Discuss. 2008, 140, 25–40. [Google Scholar] [CrossRef]
- Laszlo, P. Electrochemical Methods of Nanostructure Preparation; Springer: Berlin/Heidelberg, Germany, 2021. [Google Scholar]
- Matsushima, J.T.; Trivinho-Strixino, F.; Pereira, E.C. Investigation of cobalt deposition using the electrochemical quartz crystal microbalance. Electrochim. Acta 2006, 51, 1960–1966. [Google Scholar] [CrossRef]
- Kröger, F.A. Cathodic deposition and characterization of metallic or semiconducting binary alloys or compounds. J. Electrochem. Soc. 1978, 125, 2028–2034. [Google Scholar] [CrossRef]
- Liu, L.; Andersson, C.; Liu, J. Thermodynamic assessment of the Sn-Co lead free solder system. J. Electr. Mater. 2004, 33, 935–939. [Google Scholar] [CrossRef]
- Ishida, K.; Nishizawa, T. The Co-Sn (cobalt-tin) system. J. Phase Equilib. 1991, 12, 88–93. [Google Scholar] [CrossRef]
- Tamura, N.; Kato, Y.; Mikami, A.; Kamino, M.; Matsuta, S.; Fujitani, S. Study on Sn-Co alloy anodes for lithium secondary batteries. I. Amorphous system. J. Electrochem. Soc. 2006, 153, A1626–A1632. [Google Scholar] [CrossRef]
- Gul, H.; Uysal, M.; Cetinkaya, T.; Guler, M.O.; Alp, A.; Akbut, H. Preparation of Sn-Co alloy electrode for lithium ion batteries by pulse electrodeposition. Int. J. Hydrogen Energy 2014, 39, 21414–21419. [Google Scholar] [CrossRef]
- Jaen, J.; Varsanyi, M.L.; Czako-Nagy, I.; Buzas, A.; Vertes, A.; Kiss, L. Structural studies of electrodeposited cobalt-tin alloys. Electrochim. Acta 1984, 29, 119–1122. [Google Scholar] [CrossRef]
- Eckold, P.; Niewa, R.; Hügel, W. Texture of electrodeposited tin layers and its influence on their corrosion behavior. Microel. Reliab. 2014, 54, 2578–2585. [Google Scholar] [CrossRef]
- Eckold, P.; Sellers, M.S.; Niewa, R.; Hügel, W. The surface energies of β-Sn—A new concept for corrosion and whisker mitigation. Microel. Reliab. 2015, 55, 2799–2807. [Google Scholar] [CrossRef]
- Garret, C.S.; Massalski, T.B. Structure of Metals; Pergamon Press: Oxford, UK, 1980. [Google Scholar]
- Nasirpouri, F. Electrodeposition of Nanostructured Materials; Springer: Cham, Switzerland, 2017. [Google Scholar]
- Casella, I.G.; Contursi, M. Cobalt oxide electrodeposition on various electrode substrates from alkaline medium containing Co–gluconate complexes: A comparative voltammetric study. J. Solid State Electrochem. 2012, 66, 3739–3764. [Google Scholar] [CrossRef]
- Honciuc, A. Chemistry of Functional Materials, Surfaces and Interfaces; Elsevier Science: Amsterdam, The Netherlands, 2021. [Google Scholar]
- Jańczuk, B.; Białopiotrowicz, T. Surface free-energy components of liquids and low energy solids and contact angles. J. Coll. Interf. Sci. 1989, 127, 189–201. [Google Scholar] [CrossRef]
- Wenzel, R.N. Resistance of solid surfaces to wetting by water. Ind. Eng. Chem. 1936, 28, 988–994. [Google Scholar] [CrossRef]
- Cassie, A.B.D.; Baxter, S. Wettability of porous surfaces. Trans. Farad. Soc. 1944, 40, 546–551. [Google Scholar] [CrossRef]
- Marmur, A. Wetting on hydrophobic rough surfaces: To be heterogeneous or not to be? Langmuir 2003, 19, 8343–8348. [Google Scholar] [CrossRef]
- Sharifker, B.R.; Hills, G. Theoretical and experimental studies of multiple nucleation. Electrochim. Acta 1983, 28, 879–889. [Google Scholar] [CrossRef]
- Rudnik, E. Effect of pH-dependent bath speciation on cobalt electrodeposition from sulfate-gluconate solutions. Trans. Nonferr. Met. Soc. China 2024. accepted. [Google Scholar]
- Alderighi, L.; Gans, P.; Ienco, A.; Peters, D.; Sabatini, A.; Vacca, A. Hyperquad simulation and speciation (HySS): A utility program for the investigation of equilibria involving soluble and partially soluble species. Coord. Chem. Rev. 1999, 184, 311–318. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction | Equilibrium Constant (Logarithmic Values) |
|---|---|
| Co2+ + H2O ↔ CoOH+ + H+ | −9.687 |
| Co2+ + 2H2O ↔ Co(OH)2 + 2H+ | −18.794 |
| Co2+ + 3H2O ↔ Co(OH)3− + 3H+ | −31.491 |
| Co2+ + 4H2O ↔ Co(OH)42− + 4H+ | −46.288 |
| 2Co2+ + H2O ↔ Co2OH3+ + H+ | −10.997 |
| Co2+ + SO42− ↔ CoSO4 | 2.36 |
| Co2+ + Cl− ↔ CoCl+ | 0.22 |
| Co2+ + 2Cl− ↔ CoCl2 | −3.95 |
| Co2+ + 3Cl− ↔ CoCl3− | −3.02 |
| Co2+ + 4Cl− ↔ CoCl42− | −9.06 |
| Co2+ + Glu− ↔ CoGlu+ | 2.31 |
| Co2+ + Glu− ↔ CoH−1Glu + H+ | −4.96 |
| Co2+ + Glu− ↔ CoH−2Glu− + 2H+ | −13.29 |
| Co2+ + 3Glu− ↔ CoH−1Glu32− + H+ | −1.27 |
| Co2+ + 3Glu− ↔ CoH−2Glu33− + 2H+ | −9.21 |
| 2Co2+ + 2Glu− ↔ Co2H−3Glu2− + 3H+ | −17.89 |
| Co2+ + NH3 ↔ Co(NH3)2+ | 1.99 |
| Co2+ + 2NH3 ↔ Co(NH3)22+ | 3.50 |
| Co2+ + 3NH3 ↔ Co(NH3)32+ | 4.43 |
| Co2+ + 4NH3 ↔ Co(NH3)42+ | 5.07 |
| Co2+ + 5NH3 ↔ Co(NH3)52+ | 5.13 |
| Co2+ + 6NH3 ↔ Co(NH3)62+ | 4.39 |
| Sn2+ + H2O ↔ SnOH+ + H+ | −4.1 |
| Sn2+ + 2H2O ↔ Sn(OH)2 + 2H+ | −7.8 |
| Sn2+ + 3H2O ↔ Sn(OH)3− + 3H+ | −17.6 |
| Sn2+ + SO42− ↔ SnSO4 | 1.29 |
| Sn2+ + 2SO42− ↔ Sn(SO4)22− | 1.65 |
| Sn2+ + Cl− ↔ SnCl+ | 1.42 |
| Sn2+ + 2Cl− ↔ SnCl2 | 2.18 |
| Sn2+ + 3Cl− ↔ SnCl3− | 2.33 |
| Sn2+ + 4Cl− ↔ SnCl42− | 2.03 |
| Sn2+ + Glu− ↔ SnGlu+ | 3.01 |
| Sn2+ + 2Glu− ↔ SnGlu2 | 5.29 |
| H+ + SO42−↔ HSO4− | 1.12 |
| H+ + Cl− ↔ HCl | −7.0 |
| H+ + Glu− ↔ HGlu | 3.35 |
| H+ + NH3 ↔ NH4+ | 9.22 |
| Phase | ΔGo, kJ·mol−1 | Shift of Quasi-Rest Potential, V | Difference in Quasi-Rest Potentials, V | |
|---|---|---|---|---|
| CoSn | −19.0 | 0.099 | 0.099 | 0.130 |
| CoSn2 | −12.7 | 0.066 | 0.033 | 0.097 |
| α-Co3Sn2 | −16.8 | 0.029 | 0.044 | 0.145 |
| β-Co3Sn2 | −16.5 | 0.029 | 0.043 | 0.144 |
| Deposition Potential, V | Texture Coefficient Tc(hkl) | ||||||
|---|---|---|---|---|---|---|---|
| Sn(200) 30.7° | Sn(101) 32.0° | Sn(220) 43.9° | Sn(321) 64.7° | CoSn (100) 28.9° | CoSn(101) 32.8° | CoSn(110) 41.3° | |
| Chloride bath | |||||||
| −0.7 | 0.1 | 0.1 | 0.9 | 2.9 | nd | nd | nd |
| −0.9 | 1.3 | 1.3 | 1.0 | 0.4 | 0.9 | 1.5 | 0.7 |
| −1.1 | 2.3 | 1.7 | 0.0 | 0.0 | 0.5 | 1.5 | 1.0 |
| −1.2 | 2.2 | 1.8 | 0.0 | 0.0 | 0.7 | 1.7 | 0.6 |
| Chloride–sulfate bath | |||||||
| −0.7 | 0.4 | 0.3 | 0.8 | 2.5 | nd | nd | nd |
| −0.9 | 1.4 | 0.8 | 1.5 | 0.3 | 0.8 | 1.5 | 0.7 |
| −1.1 | 1.2 | 0.8 | 1.6 | 0.4 | 0.9 | 1.4 | 0.6 |
| −1.2 | 0.9 | 0.6 | 2.0 | 0.5 | 0.7 | 1.5 | 0.7 |
| Sulfate bath | |||||||
| −0.7 | 0.4 | 0.3 | 0.8 | 2.5 | nd | nd | nd |
| −0.9 | 0.8 | 1.0 | 1.6 | 0.6 | 0.8 | 1.6 | 0.6 |
| −1.1 | 0.8 | 0.8 | 1.7 | 0.7 | 0.8 | 1.4 | 0.8 |
| −1.2 | 0.8 | 0.8 | 2.0 | 0.4 | 0.8 | 1.3 | 0.9 |
| Deposition Potential, V (Ag/AgCl) | Total Free Surface Energy γS, mJ∙m−2 | ||
|---|---|---|---|
| Chloride | Chloride–Sulfate | Sulfate | |
| −0.8 | 25.8 | 37.3 | 33.3 |
| −0.9 | 30.4 | 31.1 | 31.5 |
| −1.0 | 31.2 | 32.6 | 28.2 |
| −1.1 | 28.4 | 39.0 | 27.3 |
| −1.2 | 35.9 | 37.0 | 26.4 |
| Component | Bath Composition, M | ||
|---|---|---|---|
| Chloride | Chloride–Sulfate | Sulfate | |
| SnCl2 | 0.05 | 0.05 | - |
| SnSO4 | - | - | 0.05 |
| CoCl2 | 0.1 | - | - |
| CoSO4 | - | 0.1 | 0.1 |
| NH4Cl | 0.5 | - | - |
| (NH4)2SO4 | - | 0.5 | 0.5 |
| C6H11O7Na | 0.2 | 0.2 | 0.2 |
| H3BO3 | 0.5 | 0.5 | 0.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rudnik, E.; Włoch, G.; Walkowicz, M. Effect of Electrolysis Conditions on Electrodeposition of Cobalt–Tin Alloys, Their Structure, and Wettability by Liquids. Molecules 2024, 29, 3084. https://doi.org/10.3390/molecules29133084
Rudnik E, Włoch G, Walkowicz M. Effect of Electrolysis Conditions on Electrodeposition of Cobalt–Tin Alloys, Their Structure, and Wettability by Liquids. Molecules. 2024; 29(13):3084. https://doi.org/10.3390/molecules29133084
Chicago/Turabian StyleRudnik, Ewa, Grzegorz Włoch, and Monika Walkowicz. 2024. "Effect of Electrolysis Conditions on Electrodeposition of Cobalt–Tin Alloys, Their Structure, and Wettability by Liquids" Molecules 29, no. 13: 3084. https://doi.org/10.3390/molecules29133084
APA StyleRudnik, E., Włoch, G., & Walkowicz, M. (2024). Effect of Electrolysis Conditions on Electrodeposition of Cobalt–Tin Alloys, Their Structure, and Wettability by Liquids. Molecules, 29(13), 3084. https://doi.org/10.3390/molecules29133084