A ng/L Level LC-MS Method Using Membrane SPE as Sampling Technology: Determination of Nine Halobenzoquinones in Potable Water


Abstract
1. Introduction
2. Results and Discussions
2.1. Optimization Conditions
2.1.1. Optimization of MS Conditions
2.1.2. Optimization of Chromatographic Conditions
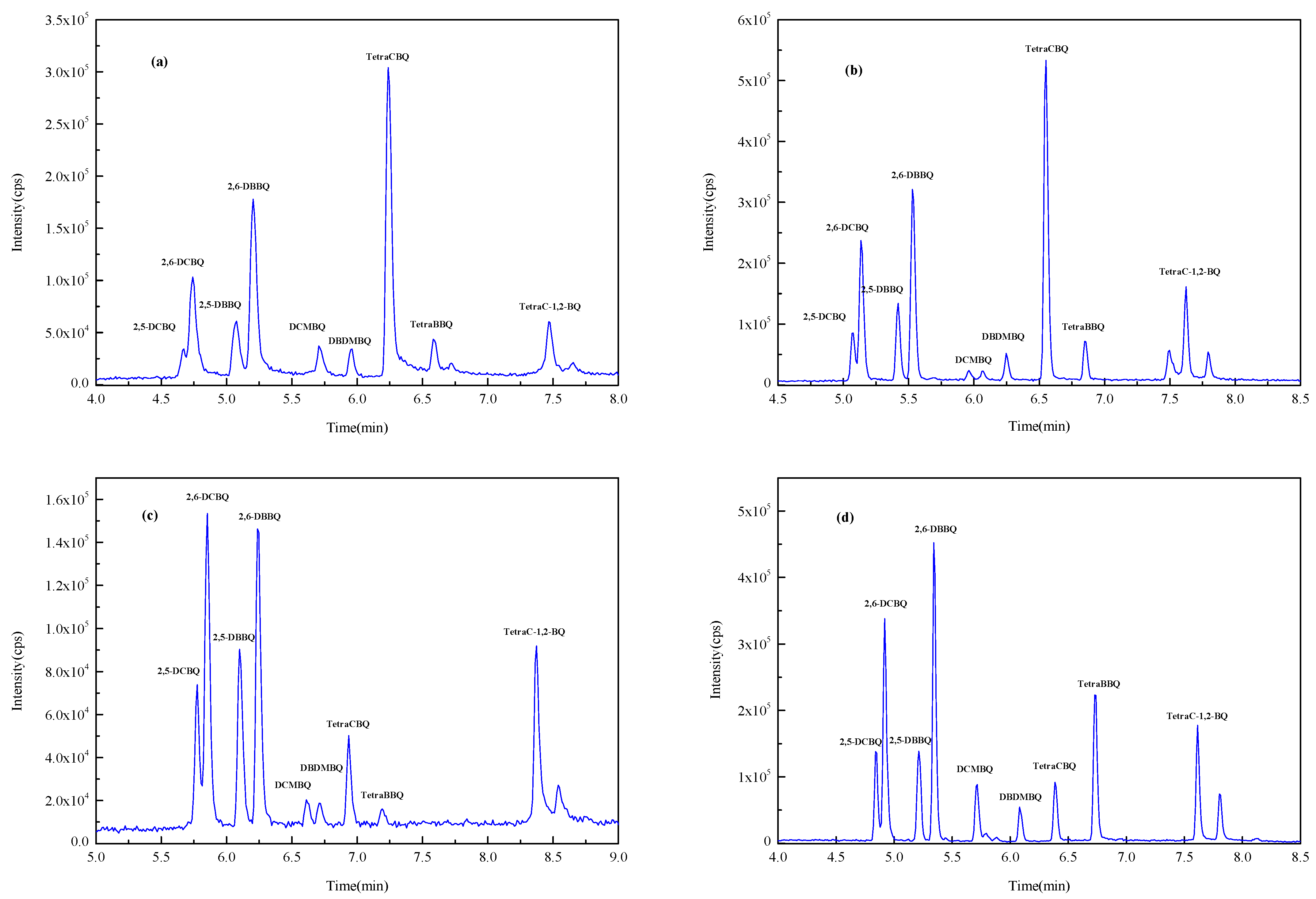
2.2. Selection of SPE Membrane
2.3. Selection of Eluent
2.4. Method Validation
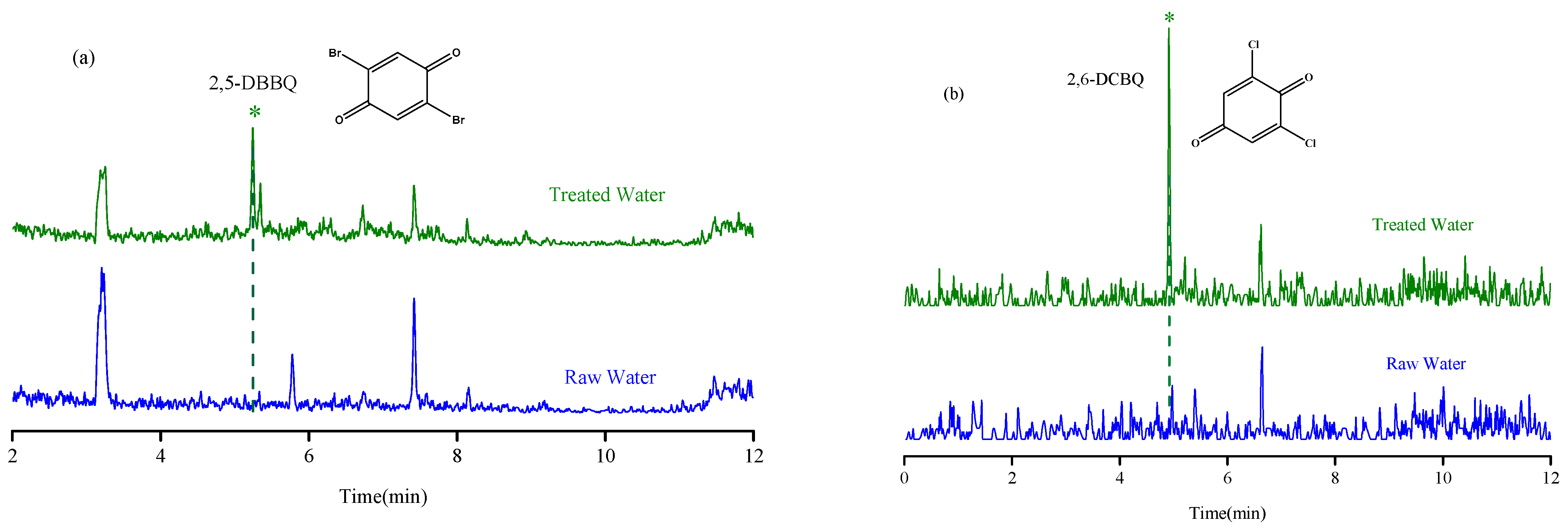
2.5. Practical Application
3. Materials and Methods
3.1. Materials and Reagents
3.2. Instruments and Equipment
3.3. LC-MS Analysis
3.4. Preparation of a Standard Working Curve
3.5. Sample Pretreatment
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Boyce, D.S.; Sproul, O.J.; Buck, C.E. The effect of bentonite clay on ozone disinfection of bacteria and viruses in water. Water Res. 1981, 15, 759–767. [Google Scholar] [CrossRef]
- Wolfe, R.L. Ultraviolet disinfection of potable water. Environ. Sci. Technol. 1990, 24, 768–773. [Google Scholar] [CrossRef]
- Nieuwenhuijsen, M.J.; Toledano, M.B.; Eaton, N.E.; Fawell, J.; Elliott, P. Chlorination disinfection byproducts in water and their association with adverse reproductive outcomes: A review. Occup. Environ. Med. 2000, 57, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Haag, W.R.; Hoigne, J. Ozonation of bromide-containing waters: Kinetics of formation of hypobromous acid and bromate. Environ. Sci. Technol. 1983, 17, 261–267. [Google Scholar] [CrossRef]
- Agus, E.; Voutchkov, N.; Sedlak, D.L. Disinfection by-products and their potential impact on the quality of water produced by desalination systems: A literature review. Desalination 2009, 237, 214–237. [Google Scholar] [CrossRef]
- Richardson, S.D.; Thruston, A.D.; Caughran, T.V.; Chen, P.H.; Collette, T.W.; Floyd, T.L.; Schenck, K.M.; Lykins, B.W.; Sun, G.-r.; Majetich, G. Identification of New Drinking Water Disinfection Byproducts Formed in the Presence of Bromide. Environ. Sci. Technol. 1999, 33, 3378–3383. [Google Scholar] [CrossRef]
- Zhai, H.; Zhang, X. Formation and Decomposition of New and Unknown Polar Brominated Disinfection Byproducts during Chlorination. Environ. Sci. Technol. 2011, 45, 2194–2201. [Google Scholar] [CrossRef] [PubMed]
- Cantor, K.P.; Villanueva, C.M.; Silverman, D.T.; Figueroa, J.D.; Real, F.X.; Garcia-Closas, M.; Malats, N. Polymorphisms in GSTT1, GSTZ1, and CYP2E1, Disinfection By-products, and Risk of Bladder Cancer in Spain. Environ. Health Perspect. 2010, 118, 1545–1550. [Google Scholar] [CrossRef]
- Li, X.-F.; Mitch, W.A. Drinking Water Disinfection Byproducts (DBPs) and Human Health Effects: Multidisciplinary Challenges and Opportunities. Environ. Sci. Technol. 2018, 52, 1681–1689. [Google Scholar] [CrossRef]
- Villanueva, C.M.; Cantor, K.P.; Grimalt, J.O.; Malats, N.; Silverman, D.; Tardon, A.; Garcia-Closas, R.; Serra, C.; Carrato, A.; Castaño-Vinyals, G.; et al. Bladder Cancer and Exposure to Water Disinfection By-Products through Ingestion, Bathing, Showering, and Swimming in Pools. Am. J. Epidemiol. 2006, 165, 148–156. [Google Scholar] [CrossRef]
- Kosaka, K.; Nakai, T.; Hishida, Y.; Asami, M.; Ohkubo, K.; Akiba, M. Formation of 2,6-dichloro-1,4-benzoquinone from aromatic compounds after chlorination. Water Res. 2017, 110, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Wang, X.; Jiang, Y.; Kong, D.; Pan, X.; Ma, J.; Liu, Y. Chloro- and bromo-benzoquinone formation and transformation mechanisms in a drinking water-distribution system. J. Hazard. Mater. 2024, 461, 132692. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Luo, J.; Wei, W.; Song, M.; Shi, W.; Li, A.; Zhou, Q.; Pan, Y. Comparative cytotoxicity analyses of disinfection byproducts in drinking water using dimensionless parameter scaling method: Effect of halogen substitution type and number. Water Res. 2023, 240, 120087. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Li, X.; Gong, T.; Tian, G.; Guo, S.; Huo, C.; Wan, J.; Liu, R. New mechanistic insights into halogen-dependent cytotoxic pattern of monohaloacetamide disinfection byproducts. J. Hazard. Mater. 2024, 465, 133132. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Li, J.; Moe, B.; McGuigan, C.F.; Shen, S.; Li, X.-F. Cytotoxicity and Oxidative Damage Induced by Halobenzoquinones to T24 Bladder Cancer Cells. Environ. Sci. Technol. 2013, 47, 2823–2830. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Moe, B.; Vemula, S.; Wang, W.; Li, X.-F. Emerging Disinfection Byproducts, Halobenzoquinones: Effects of Isomeric Structure and Halogen Substitution on Cytotoxicity, Formation of Reactive Oxygen Species, and Genotoxicity. Environ. Sci. Technol. 2016, 50, 6744–6752. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Long, K.; Sha, Y.; Lu, D.; Xia, Y.; Mo, Y.; Yang, Q.; Zheng, W.; Yang, M.; Wei, X. Occurrence and toxicity of halobenzoquinones as drinking water disinfection byproducts. Sci. Total Environ. 2021, 770, 145277. [Google Scholar] [CrossRef] [PubMed]
- Lou, J.; Lu, H.; Wang, W.; He, S.; Zhu, L. Quantitative identification of halo-methyl-benzoquinones as disinfection byproducts in drinking water using a pseudo-targeted LC-MS/MS method. Water Res. 2022, 218, 118466. [Google Scholar] [CrossRef]
- Yu, S.; Yan, Y.; Zhai, H.; Gu, X.; Liu, Y. Determination of dihalobenzoquinones in water using gas chromatography coupled with an electronic capture detector. Chemosphere 2019, 215, 57–61. [Google Scholar] [CrossRef]
- Xue, W.; Li, N.; Zhang, Z.; Li, G. Dummy template based molecularly imprinted solid-phase microextraction coating for analysis of trace disinfection by-product of 2,6-dichloro-1,4-benzoquinone using high-performance liquid chromatography. Talanta 2022, 239, 123065. [Google Scholar] [CrossRef]
- Zhao, Y.; Qin, F.; Boyd, J.M.; Anichina, J.; Li, X.-F. Characterization and Determination of Chloro- and Bromo-Benzoquinones as New Chlorination Disinfection Byproducts in Drinking Water. Anal. Chem. 2010, 82, 4599–4605. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Wang, W.; Qian, Y.; Boyd, J.M.; Zhao, Y.; Li, X.-F. Ultra Pressure Liquid Chromatography–Negative Electrospray Ionization Mass Spectrometry Determination of Twelve Halobenzoquinones at ng/L Levels in Drinking Water. Anal. Chem. 2013, 85, 4520–4529. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Qian, Y.; Li, J.; Moe, B.; Huang, R.; Zhang, H.; Hrudey, S.E.; Li, X.-F. Analytical and Toxicity Characterization of Halo-hydroxyl-benzoquinones as Stable Halobenzoquinone Disinfection Byproducts in Treated Water. Anal. Chem. 2014, 86, 4982–4988. [Google Scholar] [CrossRef] [PubMed]
- Cuthbertson, A.A.; Bach, C.; Richardson, S.D.; Dauchy, X. A novel automated method for the quantification of ten halobenzoquinones in drinking water using online solid-phase extraction coupled with liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2020, 1612, 460642. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, L.; Lu, J.; Chen, Y.; Zhang, L. Determination of 13 halobenzoquinone disinfection by-products in drinking water using solid phase extraction-ultra performance liquid chromatography-triple quadrupole mass spectrometry. Chin. J. Chromatogr. 2023, 41, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Han, Q.; Xia, J.; Xia, L.; Ding, M.; Tang, J. Graphene-based solid-phase extraction disk for fast separation and preconcentration of trace polycyclic aromatic hydrocarbons from environmental water samples. J. Separartion Sci. 2013, 36, 1834–1842. [Google Scholar] [CrossRef] [PubMed]
- Česen, M.; Heath, E. Disk-based solid phase extraction for the determination of diclofenac and steroidal estrogens E1, E2 and EE2 listed in the WFD watch list by GC–MS. Sci. Total Environ. 2017, 590–591, 832–837. [Google Scholar] [CrossRef]
- Günter, A.; Balsaa, P.; Werres, F.; Schmidt, T.C. Influence of the drying step within disk-based solid-phase extraction both on the recovery and the limit of quantification of organochlorine pesticides in surface waters including suspended particulate matter. J. Chromatogr. A 2016, 1450, 1–8. [Google Scholar] [CrossRef]
- Li, L.; Xu, K.; Huang, Z.; Xu, X.; Iqbal, J.; Zhao, L.; Du, Y. Rapid determination of trace Cu2+ by an in-syringe membrane SPE and membrane solid-phase spectral technique. Anal. Methods 2021, 13, 4691–4698. [Google Scholar] [CrossRef]
- Miaomiao, Z.; Xin, Z.; Yunxia, W.; Juan, W. Determination of total flavonoid components in Ginkgo biloba L. Tea by UPLC. Northwest Pharm. J. 2017, 32, 28–31. [Google Scholar]
- Liu, Z.; Wu, X.; Si, Z.; Kong, D.; Yang, D.; Zhou, F.; Wang, Z. Simultaneous determination of nine constituents by validated UFLC-MS/MS in the plasma of cough variant asthma rats and its application to pharmacokinetic study after oral administration of Huanglong cough oral liquid. J. Pharm. Biomed. Anal. 2021, 193, 113726. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Zhou, H.; Wu, X.; Song, C.; Zheng, J.; Lei, M.; Mu, P.; Wu, P. Simultaneous determination of the residues of anesthetics and sedatives in fish using LC-QLIT-MS/MS combined with DSPE. Food Chem. 2023, 403, 134407. [Google Scholar] [CrossRef] [PubMed]
- Mutzner, L.; Vermeirssen, E.L.M.; Mangold, S.; Maurer, M.; Scheidegger, A.; Singer, H.; Booij, K.; Ort, C. Passive samplers to quantify micropollutants in sewer overflows: Accumulation behaviour and field validation for short pollution events. Water Res. 2019, 160, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Becker, B.; Kochleus, C.; Spira, D.; Möhlenkamp, C.; Bachtin, J.; Meinecke, S.; Vermeirssen, E.L.M. Passive sampler phases for pesticides: Evaluation of AttractSPE™ SDB-RPS and HLB versus Empore™ SDB-RPS. Environ. Sci. Pollut. Res. 2021, 28, 11697–11707. [Google Scholar] [CrossRef] [PubMed]
- Shaw, M.; Eaglesham, G.; Mueller, J.F. Uptake and release of polar compounds in SDB-RPS Empore™ disks; implications for their use as passive samplers. Chemosphere 2009, 75, 1–7. [Google Scholar] [CrossRef]

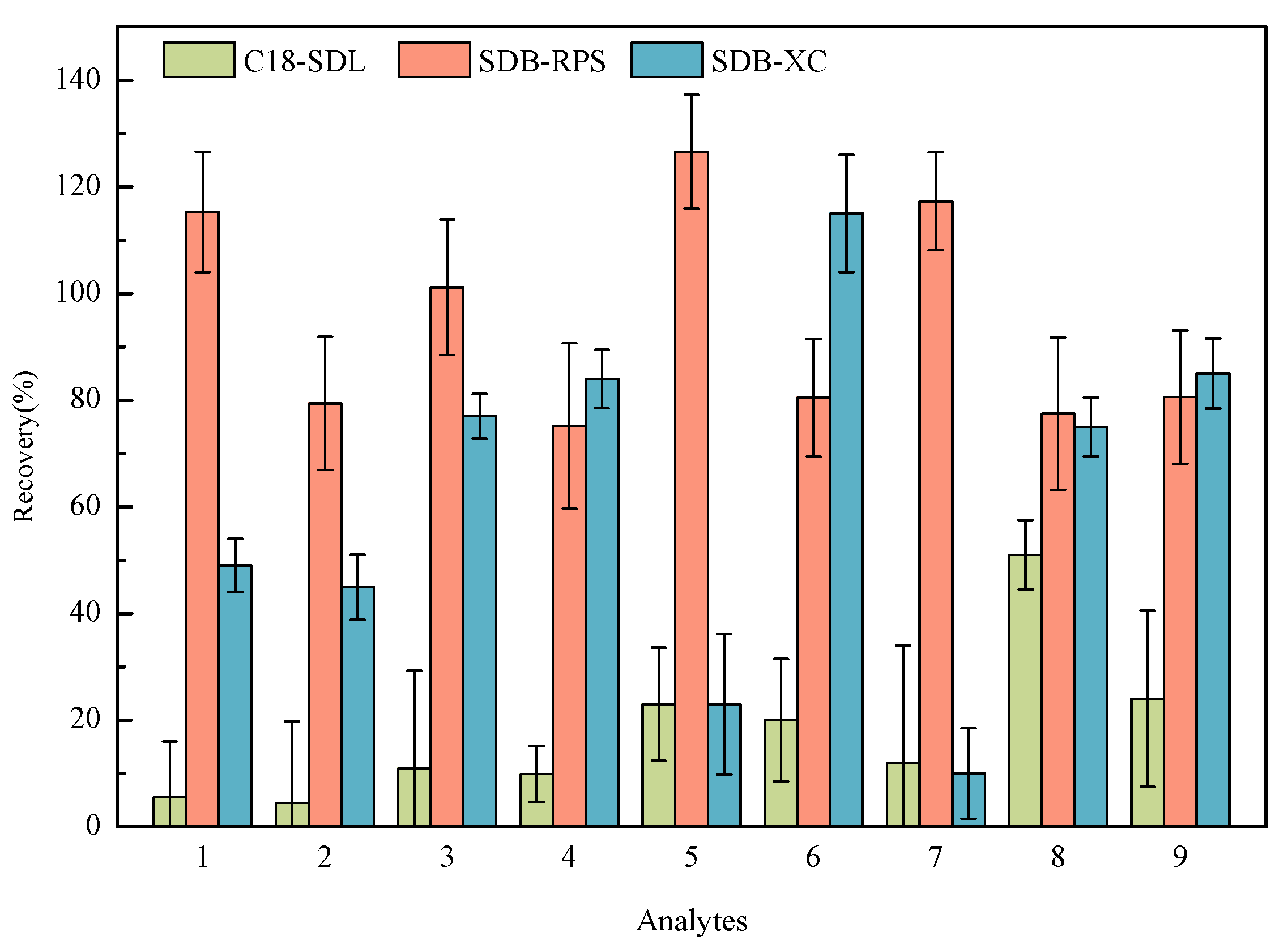


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | ESI (−/+) | Retention Time (min) | Q1 (m/z) | Q3 (m/z) | DP (V) | CE (eV) |
|---|---|---|---|---|---|---|
| 2,5-DCBQ | − | 4.86 | 176.9 | 112.9 */34.9 | −90 | −23/−30 |
| 2,5-DBBQ | − | 5.22 | 266.8 | 78.8 */80.9 | −105 | −50/−50 |
| 2,6-DCBQ | − | 4.93 | 176.9 | 112.9 */34.9 | −90 | −23/−30 |
| 2,6-DBBQ | − | 5.36 | 266.8 | 78.8 */80.9 | −105 | −50/−50 |
| TetraCBQ | − | 6.39 | 245.0 | 34.9 */208.8 | −124 | −44/−20 |
| TetraBBQ | − | 6.74 | 424.8 | 78.9 */81.0 | −110 | −70/−70 |
| TetraC-1,2-BQ | − | 7.62 | 245.0 | 34.9 */208.8 | −124 | −44/−20 |
| DBDMBQ | − | 6.09 | 309.0 | 214.8 */142.8 | −130 | −21/−34 |
| DCMBQ | − | 5.72 | 191.0 | 35.0 */127.0 | −50 | −35/−22 |
| Analyte | Linear Range (μg/L) | Linear | R2 | Recovery, % (RSD, %) | LOD (ng/L) | LOQ (ng/L) | ||
|---|---|---|---|---|---|---|---|---|
| Spiked at 10 ng/L | Spiked at 50 ng/L | Spiked at 500 ng/L | ||||||
| 2,5-DCBQ | 4~1000 | 2980.143x − 7053.38 | 0.9994 | 109.3 (12.9) | 115.3 (11.3) | 102.3 (9.0) | 0.5 | 0.15 |
| 2,5-DBBQ | 4~1000 | 4369.877x − 5346.38 | 0.9987 | 86.0 (8.4) | 79.4 (12.5) | 94.1 (7.7) | 0.5 | 1.5 |
| 2,6-DCBQ | 4~1000 | 6618.669x − 7598.79 | 0.9989 | 123.3 (6.9) | 101.2 (12.7) | 109.5 (10.5) | 0.1 | 0.3 |
| 2,6-DBBQ | 4~1000 | 14,314.21x − 25,460.2 | 0.9993 | 84.0 (10.8) | 75.2 (15.5) | 96.3 (9.2) | 0.2 | 0.6 |
| TetraCBQ | 4~1000 | 3656.618x − 3000.86 | 0.9989 | 112.5 (11.9) | 126.6 (10.7) | 105.3 (8.8) | 0.7 | 2.1 |
| TetraBBQ | 4~1000 | 6012.050x − 5526.22 | 0.9987 | 87.0 (6.8) | 80.5 (11.0) | 107.6 (9.2) | 0.3 | 0.9 |
| TetraC-1,2-BQ | 4~1000 | 1173.196x − 7213.38 | 0.9963 | 108.5 (14.7) | 117.3 (9.2) | 105.6 (11.5) | 0.5 | 1.5 |
| DBDMBQ | 4~1000 | 337.1917x + 1170.10 | 0.9974 | 82.3 (7.8) | 77.5 (14.3) | 97.2 (12.1) | 0.3 | 0.9 |
| DCMBQ | 4~1000 | 3991.976x − 6143.96 | 0.9974 | 73.5 (9.6) | 80.6 (12.5) | 91.9 (8.3) | 0.1 | 0.3 |
| Quantitative Technique | Purification Method | Extraction Solvent/Column /Membrane | Sample Loading Time (min) | Analytes | Linear Range | LOD | Detected Concentration (ng/L) | Ref. |
|---|---|---|---|---|---|---|---|---|
| GC-ECD | LLE a | Methyl-tert-butyl ether (MtBE) | - | 2,6-DCBQ and 2,6-DBBQ | - | 0.8–0.9 ng/L | 2.4–20.5 ng/L | [19] |
| LC-MS | SPE b | Waters Oasis HLB cartridges (6 mL, 200 mg) | 62.5 | 4 HBQs | 1–100 ng/L | 0.3–2.0 ng/L | 0.5–165 ng/L | [21] |
| LC-MS | 12 HBQs | 1–20 ng/L | 0.2–6.6 ng/L | 2.5–21.3 ng/L | [22] | |||
| LC-MS | 4 HBQs and 4 OH-HBQs | - | 0.03–0.8 ng/L | 0.3–20.3 ng/L | [23] | |||
| LC-MS | MSPE c | SDB-RPS membrane | 10 | 9 HBQs | 4–1000 ng/L | 0.1–0.7 ng/L | 2.0–5.1 ng/L | This work |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Q.; Zhou, H.; Wu, X.; Jiang, J.; Zhan, B.; Wu, P. A ng/L Level LC-MS Method Using Membrane SPE as Sampling Technology: Determination of Nine Halobenzoquinones in Potable Water. Molecules 2024, 29, 2856. https://doi.org/10.3390/molecules29122856
Huang Q, Zhou H, Wu X, Jiang J, Zhan B, Wu P. A ng/L Level LC-MS Method Using Membrane SPE as Sampling Technology: Determination of Nine Halobenzoquinones in Potable Water. Molecules. 2024; 29(12):2856. https://doi.org/10.3390/molecules29122856
Chicago/Turabian StyleHuang, Qin, Hua Zhou, Xianglun Wu, Jiaqi Jiang, Bingdong Zhan, and Pinggu Wu. 2024. "A ng/L Level LC-MS Method Using Membrane SPE as Sampling Technology: Determination of Nine Halobenzoquinones in Potable Water" Molecules 29, no. 12: 2856. https://doi.org/10.3390/molecules29122856
APA StyleHuang, Q., Zhou, H., Wu, X., Jiang, J., Zhan, B., & Wu, P. (2024). A ng/L Level LC-MS Method Using Membrane SPE as Sampling Technology: Determination of Nine Halobenzoquinones in Potable Water. Molecules, 29(12), 2856. https://doi.org/10.3390/molecules29122856