2.1. Characteristics of the Datasets
Our goal was to explore a variety of environments within proteins that represent the structural motifs found in soluble and membrane-bound proteins. In earlier work, we suggested that dividing the 2π × 2π Ramachandran plot for protein backbone angles into sixty-four π/4 × π/4 “chess squares” was an appropriate methodology for determining and cataloguing backbone dependence for sidechain conformational differences, but more importantly sidechain interaction preferences [
34]. We later implemented [
36,
37] a chess square parsing scheme for larger residues to filter sidechain conformations into 60 ± 30°, 180 ± 30°, and 300 ± 30° χ
1 bins for ASN, ASP, CYS, HIS, ILE, LEU, MET, PHE, SER, THR, TRP, and TYR, and these were further and similarly subdivided into (9) χ
1/χ
2 bins for ARG, GLN, GLU, and LYS. PRO residues were parsed into two bins centered around χ
1 = +30° and –30°, while ALA, GLY, and VAL were not parsed. Our nomenclature is that chess squares are named
a1–
h8, from lower left to upper right on the Ramachandran plot, and are always written in bold italic font. The parses are named (for all residues except PRO)
.60,
.180, and
.300, and are appended to the chess square to identify a bin (for ARG, GLN, GLU, and LYS, these names are
.60.60,
.60.180, etc.). PRO parses are named as
.30m and
.30p [
36]. The -S-S- bridged cysteine (CYX) was treated as described earlier [
38].
Later, as mentioned above, we added a second collection of proteins to our studies that focused on those membrane-bound by abstracting a portion of the MemProtMD database [
40] (
http://memprotmd.bioch.ox.ac.uk (accessed on 22 May 2023)) and used the filters thus provided to build three subsets of residues: (1) extracellular, i.e., in the soluble domain, that we termed the mS dataset; (2) intracellular and lipid-facing, termed the mL dataset; and (3) intracellular and core-facing, termed the mC dataset. An alternative interaction calculation method, wherein interactions between the residues and lipids are ignored, was termed the mN dataset.
Table 1 summarizes the data in terms of the numbers of residues in each dataset. The soluble dataset is the largest, encompassing nearly 750,000 residues with occurrence frequencies generally in line with the accepted norm. Altogether, there are about half as many residues in the membrane-bound protein datasets, but these are split unevenly: the mS dataset contains 54% of the residues, the mC dataset contains only 7% of the residues and the mL dataset contains the remaining 39%. In agreement with the expectation that the mS dataset comprises residues much like those found in soluble proteins, the mS occurrence frequencies are in reasonable agreement with those found in the soluble dataset. However, the mC dataset shows some significant and interesting differences (compared to mS): (1) It is much less likely to find an ARG, ASP, GLU, LYS, or PRO as a core-facing residue. This is interesting because these, other than PRO, are most of the residues associated with some sort of regulatory function (PRO is probably just ill-suited for the structural constraints of the core); (2) The most prominent increase in occurrence in the mC dataset is exhibited by PHE, and to a lesser extent by TYR, suggesting a necessary π-stacking interaction role here; and (3) There are no CYX residues and only a small number of CYS residues. Even more dramatic, but largely expected, are the differences between relative populations in the mL dataset compared to mS. The hydrophobic residues, ALA, ILE, LEU, PRO, and VAL, along with PHE, are now 57% of the residues found as lipid-facing (vs. 40% in the mS dataset). In the mL dataset, the four formally-charged residues ARG, ASP, GLU, and LYS now comprise just 7% (vs. 25% in the mS dataset).
2.2. Three-Dimensional Interaction Maps and Map Clustering
To catalogue the array of interactions for each residue’s sidechain—that shape its conformation and role in protein structure at all scales—we calculated three-dimensional maps that enumerated the interactions between that residue and its environment, i.e., the rest of the protein and cofactors, such as water. Hydrophobic, hydrophobic-polar, favorable polar such as hydrogen bonding and acid–base, and unfavorable polar such as acid–acid and base–base interactions were scored such that their strengths are mapped in space [
34,
35]. These maps were binned by the residue type, chess square, and, as appropriate (vide supra), χ
1 and χ
2 parses so that their sidechains were aligned [
37].
Next, our pairwise similarity metric [
34] was applied to each map pair in its bin, and these metrics were set into a two-dimensional matrix in preparation for clustering. We used the k-means unified gap method [
42] based on the testing of a numerous other methods [
34], and manually set the maximum number of clusters based on our early experiences to maximize the likelihood that the residue types and/or chess squares we wished to compare would have corresponding cluster counts [
39].
Table 1 indicates the number of clusters found for each residue type in each dataset. These vary over a wide range and are dependent on the number of residues present (weakly populated datasets will have fewer), the number of parses (ALA has one, while LYS has nine), and the structural complexity that in turn dictates the maximum allowed (ALA-4, ARG-18, ASN-12, ASP-12, CYS-6, CYX-12, GLN-12, GLU-12, GLY-9, HIS-12, ILE-9, LEU-9, LYS-18, MET-18, PHE-12, PRO-6, SER-6, THR-6, TRP-12, TYR-12, and VAL-9). It was our hypothesis that these cluster-derived and weighted-average map sets represent a significant reduction in data compared to the original structural data (at least 50-fold in the soluble dataset), and that we have sampled enough that it is unlikely that many new motifs would appear with larger protein datasets. For classification, each cluster is identified by the most representative residue member (in an ordinal list), which is called the exemplar, and is the residue (map) closest to the cluster’s centroid. Our nomenclature is that cluster names are written in bold font. Thus, in our approach, one member within the set of average maps in the specified backbone conformation very likely captures the constellation of interactions between any residue and its environment. In other words, this is the “hydropathic valence” of the residue type in its secondary structure (chess square). To summarize, these maps are conserved motifs that are information-rich backbone-dependent rotamer and interaction libraries.
Our earlier articles [
34,
35,
36,
37,
38,
39] provide examples of cluster-derived hydropathic interaction maps like those described above. We will show a handful of targeted cases below.
2.3. Solvent-Accessible and Lipid-Accessible Surface Areas
To understand further the role each residue plays in structure, we calculated the solvent-accessible surface area (SASAs) for all residue sidechains using the GETAREA algorithm and server [
41]. The SASA is a metric representing the degree of exposure/buriedness of a residue’s sidechain and at its basic level reveals the residue’s position within the protein, i.e., surface or not. It becomes a more powerful structure-understanding tool when the identity of the residue is considered: high solvent exposure in a hydrophobic residue has an entirely different meaning than high solvent exposure in a polar residue. This is particularly relevant in considering membrane-bound proteins because lipid-facing residues are generally hydrophobic rather than polar but have large calculated SASAs because the membrane bilayer is not per se protein and thus not recognized by GETAREA. For this reason, we implemented an adjustment, which we call lipid-accessible surface-area (LASA), where accessible surface area is categorized as SASA or LASA based on the ratios of score sums involving atoms in the DPPC molecules to the total score.
Table 2 sets out the average solvent-accessible and lipid-accessible surface areas by residue type for the sidechains of the four datasets. Also listed are the reference random coil SASAs for Gly-X-Gly tripeptides [
41]. Notes: (1) As GLY does not have a sidechain, its actual SASA is 0.0. We did all of our calculations on its full structure and the random coil value listed is that value; (2) Bridging cysteine (CYX) is not recognized by GETAREA, and our calculations actually consider each half of CYX to be –CB-SG-SG’-CB’. The random coil number presented in
Table 2 is arbitrarily 150% of the CYS random coil SASA. These two values will only be used as normalization factors in the calculations below.
While most indications are that the structural characteristics and the residues themselves are very similar between the soluble dataset and the mS dataset, there are notable differences in the average SASAs between the two. While we cannot be definitive, it seems likely that this is due to the different ways the structure models within the two datasets were collected and processed. In general, for smaller residues (ALA, CYS, ILE, LEU, SER, and VAL), the SASAs are largely the same, but for most of the larger residues (ARG, GLN, GLU, HIS LYS, TRP, and TYR), the mS dataset residues are less solvent-exposed. Since the latter set was subjected to extensive molecular dynamics, it may be that the larger sidechains were optimized into more compact conformations that may be less native-like.
For the mC and mL datasets, we calculated both residue-average SASAs and LASAs. The mC LASA data account for 4% to 17% (average 9%) of the total accessible surface areas for these residues. This must represent residues whose backbones are in the core region but whose sidechains make contact with lipid molecules. The three residue types with the largest LASAs are somewhat unexpected: HIS, MET, and PHE. Perhaps, the sidechains of aromatic planar residues (TRP but not TYR also has a high LASA) have affinity and access to the lipid. Interestingly, only the MET of the long chain residues reaches the lipids: ARG, GLN, GLU, and LYS have below-average LASAs, likely because they are less hydrophobic.
In the mL dataset, the LASA captures 72% of the accessible surface area, with largely expected trends. The larger hydrophobic residues (including PHE and TRP) show between 84% and 89% LASA, while in contrast, ASN, ASP, GLN, and GLU show between 49% and 60% LASA. ALA and PRO likely have less prominent LASAs, 76% and 77%, respectively, because they are making a larger fraction of their interactions with neighboring residues. In the following section, we will examine the interaction character for each residue type and dataset.
Examining the SASA and LASA metrics on this residue-by-residue basis is a gross oversimplification of the data we have generated.
Supplementary Tables S1–S5 list these data on a cluster-by-cluster basis for: the soluble residue dataset (
Table S1), soluble domain of the membrane dataset (mS,
Table S2), the core-facing residues of the membrane dataset (mC,
Table S3), the lipid-facing residues (lipid interactions on) of the membrane dataset (mL,
Table S4), and the lipid-facing residues (lipid interactions off) of the membrane dataset (mN,
Table S5). For discussion, a very small subset of these data (~0.3%) is provided in
Table 3 and
Table 4, for the soluble proteins, the soluble domain of the membrane proteins (mS), and the core-facing residues of the membrane proteins (mC) (
Table 3) and the mL and mN residue sets (
Table 4). Data for one bin of residue data for five somewhat diverse residues, ALA (
c5), ASP (
c5.300), ILE (
c5.300), PHE (
c5.300), and THR (
c5.300), are listed. (The
c5 chess square is in the α-helix region of the Ramachandran plot.) Note that the ASP maps were calculated at a pH that evenly divides the ASP residues into charged, deprotonated aspartates and neutral protonated aspartic acids using concepts and procedures described earlier [
37].
It is clear from
Table 3 and
Table 4 that the SASA/LASA values are not monotonic from residue to residue, or even within the same residue and chess square. They are, in fact, a distinguishing feature of each cluster. However, we showed earlier that the clustered maps can be numerically compared with similarity metrics and that there can be very similar 3D map motifs generated in different chess squares for the same residue, e.g., for ALA [
35], or between different datasets, e.g., soluble proteins and soluble domains of membrane proteins [
39]. We are not going to repeat that exercise here, but do highlight that ALA
c5 3020 and ALAmS
c5 905 were shown to have similar profiles, and as expected, their SASA values are very nearly the same. Also described earlier is that there is a degree of similarity between the core-facing membrane residue cluster maps (mC) and those of the soluble set [
39], but the significantly smaller sample size of the former (~30-fold) is a confounding factor. The clusters of core-facing residue interaction maps show a similar diversity of SASAs as other cluster sets, but are generally more buried probably due to the confined space available in that environment.
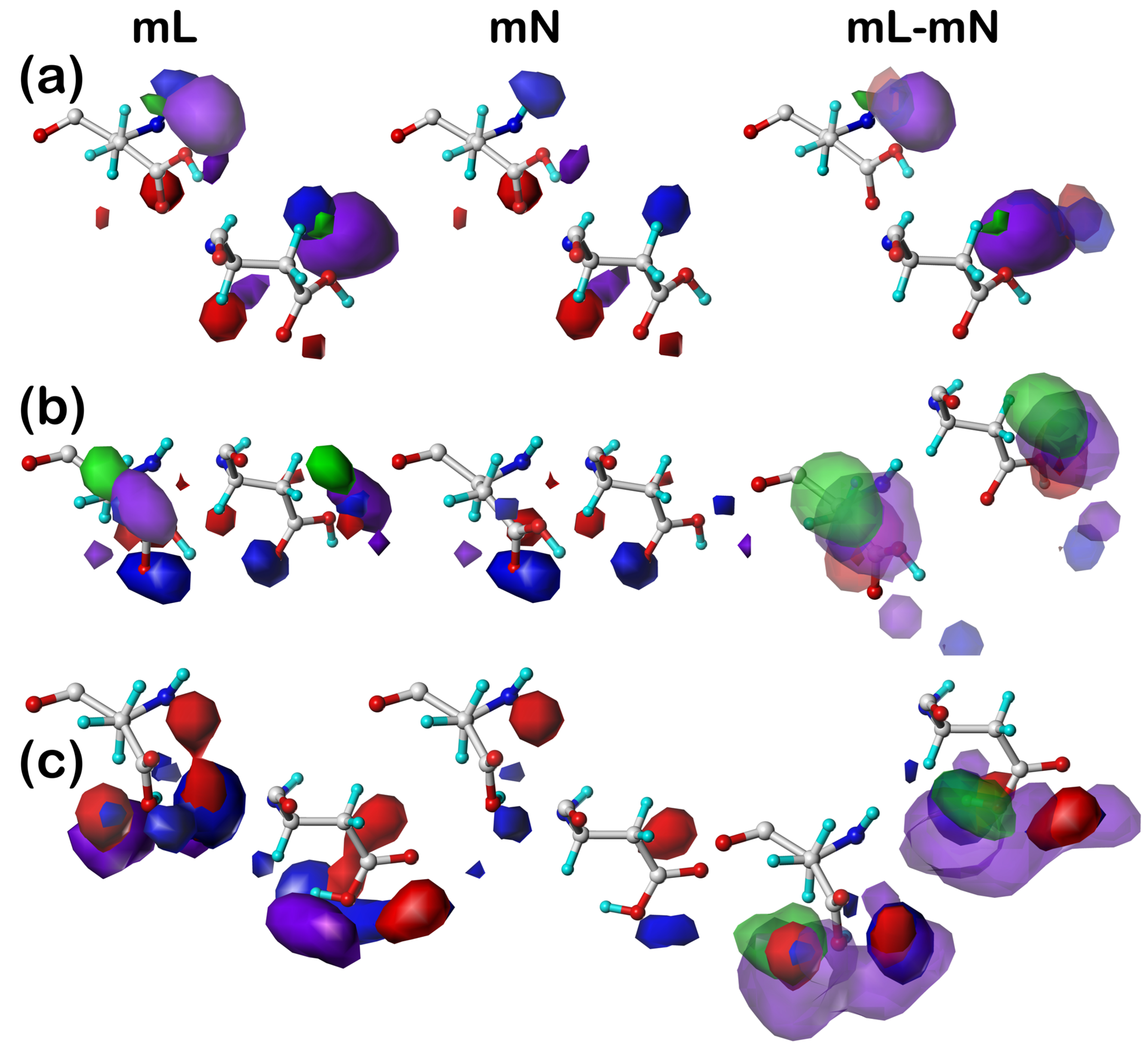
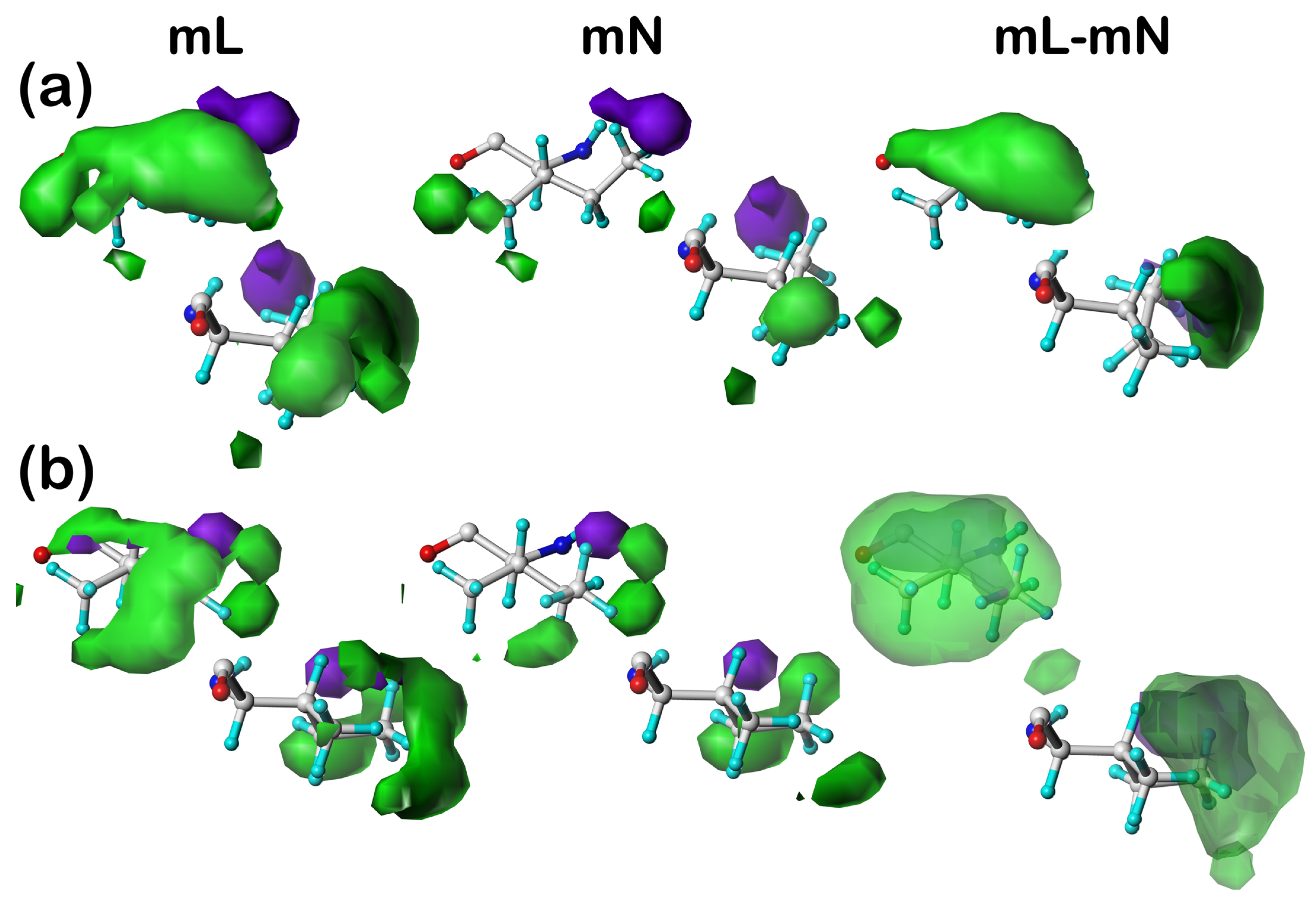
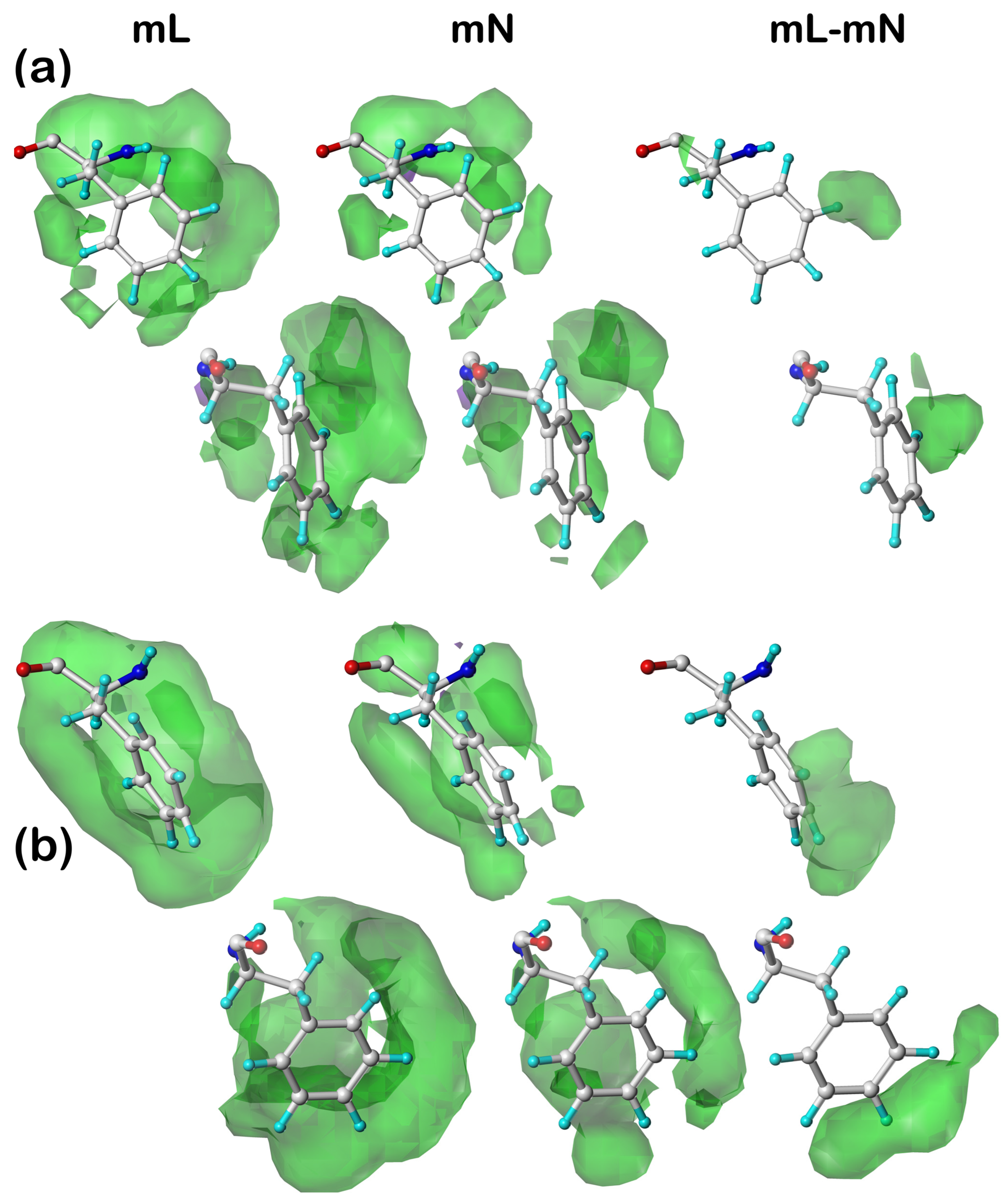
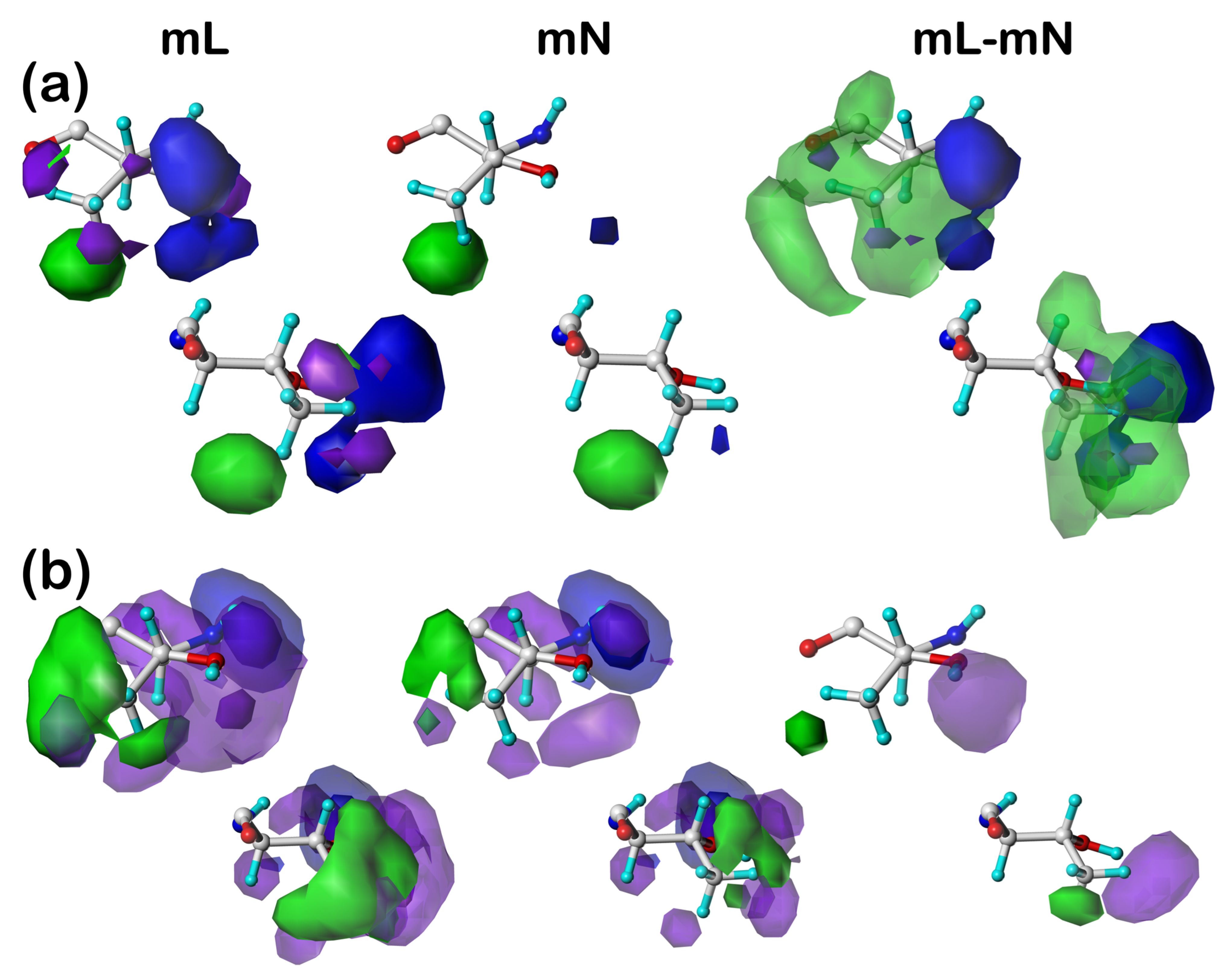
Table 4 focuses on the mL vs. mN results, where the former has interactions with both other protein residues and the lipids, and thus has both LASA and SASA data, while the latter has only interactions with the protein (i.e., ignoring the lipid) and has only SASA data. Not surprisingly, the residues in this environment generally have much more significant propensity for interaction with lipids (LASA) than they do with H
2O (SASA). However, this varies by residue type, chess square and cluster. For example, in ALA, the cluster
518 shows about 56% SASA, while
18 shows about 13%. Of the five residue types highlighted here, ASP has three clusters with higher SASA than LASA (
11,
72 and
98), while ILE and PHE have none. While these results are fairly intuitive based on the general expectations of residue properties, the next section enumerates the actual average interaction character of each cluster.
2.4. Hydropathic Interaction Characters
The HINT formalism considers four interaction types: favorable polar, Pol(+), such as acid–base and hydrogen bonding; unfavorable polar, Pol(−), such as acid–acid and base–base; favorable hydrophobic, Hyd(+); and unfavorable hydrophobic, Hyd(−), i.e., hydrophobic-polar representing desolvation and related effects [
32,
43]. Each individual residue’s interaction map set was analyzed by summing its grid point values by interaction type; these are recorded as the interaction map character [
44].
Table 5 reports these on a residue type basis for all five datasets. For the purposes of clarity and to aid possible comparisons between the residues, the data in
Table 5 were normalized by the random coil SASA dataset out in
Table 2. These results are largely as expected, with hydrophobic residue sidechains being dominated by hydrophobic interactions, with the tendency for larger sidechains to have a larger fraction of their interactions to be favorable. The most productive residues for favorable polar interactions are the formally charged acids (ASP and GLU), amides (ASN and GLN), ARG (again, formally charged), and SER. GLY is an outlier because its data throughout this study are for the entire residue structure, not just the sidechain. As above for the SASA and LASA data,
Supplementary Tables S1–S5 contain the interaction character data at the chess square and cluster levels (but are not normalized).
Table 6 lists the (normalized) interaction character for the same set of residues in the soluble and mS datasets described in
Table 3.
Table 7 lists the (normalized) interaction character for the same set of residues in the mL and mN datasets described in
Table 4.
Comparisons between the SASA/LASA data and interaction character data (e.g.,
Table 3 vs.
Table 6 and
Table 4 vs.
Table 7) should be particularly informative. First, residue clusters with a high SASA + LASA make fewer measurable interactions (|Hyd(−)| + |Hyd(+)| + |Pol(−)| + |Pol(+)|), although interactions with water are incompletely measured: not all crystal structures in the soluble dataset have confirmed explicit waters. Second, even polar residues with a high LASA can have significant, often favorable, interactions with the DPPC lipid; see, for example, the ASP clusters
44 and
101 that have ~25–50% higher favorable polar interactions in the mL calculation than in the mN calculation. The THR clusters
6,
33 and
55 have similar trends. Such residues must be interacting with the lipid’s polar head groups. Third, as expected, hydrophobic residues, especially ILE in
Table 4 and
Table 7, make significant hydrophobic interactions with the lipid in clusters with a high LASA, but the relationship is multifactorial.
The key point is that the 3D interaction maps that we generated in our study, 14,158 for the soluble proteins and a total of 19,802 for the three subsets of the membrane proteins, are packed with information about the interaction preferences of the residues by type, backbone angle, and location. For those that are membrane-bound, we should have a handle on residue–lipid interactions as well as residue–residue interactions. Probing these maps by SASA/LASA or interaction character is inadequate to assess them.
2.6. Composite 3D Interaction Maps for Proteins
With the tens of thousands of backbone-, residue-, and environment-dependent distinct map clusters generated for this study, we can assemble an overall picture of non-covalent interactions within a protein, between proteins, between proteins and ligands, and of special interest here, between proteins in transmembrane regions and their lipid support. For this discussion, we arbitrarily chose one protein/lipid structure from the training set, pdbid: 4o6y, which is the X-ray diffraction structure (resolution of 1.70 Å) of cytochrome b561 from
Arabidopsis thaliana, a eukaryotic transmembrane ascorbate-dependent oxidoreductase [
45].
We traced each residue in the protein through our calculational procedure to identify its membrane protein subset, chess square, parse, and cluster membership. Next, we constructed three-dimensional grids large enough to encage the entire protein, 99 × 95 × 111 for mC residues, 153 × 127 × 113 for mL residues, and 135 × 125 × 145 for mS residues, all with 0.5 Å grid spacing. Then, the 3D clustered interaction maps for each residue in the protein were frame-shifted to that residue’s frame, and the interpolated values for each grid point in the residue-level maps (for favorable and unfavorable, hydrophobic and polar interactions) were accumulated in the protein level maps.
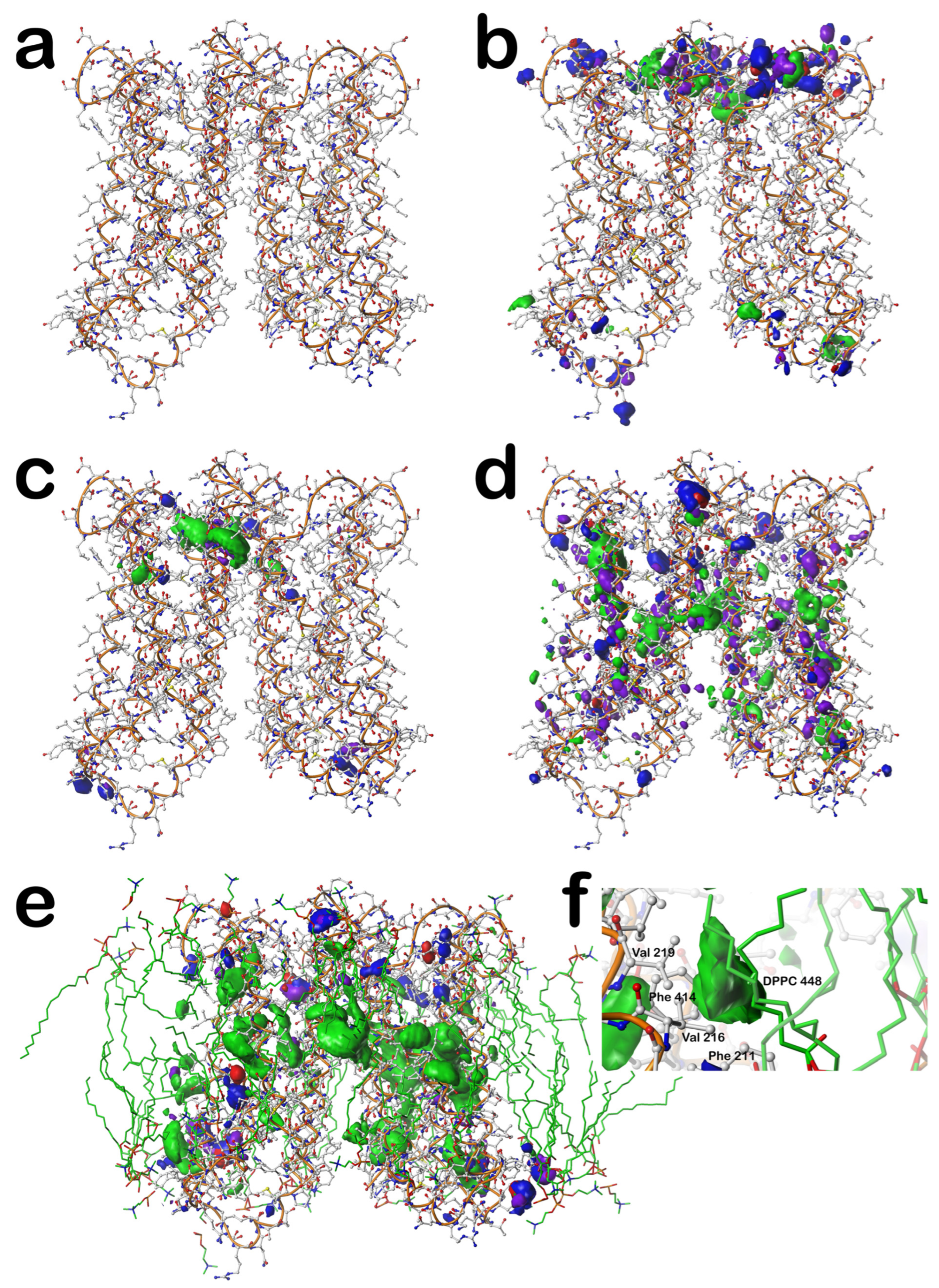
Figure 6 sets out these results.
First, in
Figure 6a, the structure for the cytochrome b561 protein is shown; it has two chains, each possessing six transmembrane helices. There are a relatively small number of extramembrane residues, largely on the extracellular side (top,
Figure 6a). Also, the gap between the two chains is noticeably smaller on the extracellular side than on the intracellular side. In
Figure 6b, the composite interaction maps for the mS residues outside the membrane surfaces (extracellular on top and intracellular on the bottom) are shown (see caption for display details). As suggested by the relative number of extramembrane residues (top and bottom), the extracellular region has a much richer set of interactions than the intracellular. Each displayed contour corresponds to one or more interactions between the residue sidechains that impact the structure: the blue isovalue surfaces represent favorable polar interactions like hydrogen bonds, just as described above for the individual residue-level maps; red = unfavorable polar; green = favorable hydrophobic; and purple = unfavorable hydrophobic.
In the transmembrane (helix) region, we first display the interactions between the core-facing mC residues and their neighbors in
Figure 6c. It is important to point out that all of these map figures (
Figure 1,
Figure 2,
Figure 3,
Figure 4,
Figure 5 and
Figure 6) were manually contoured, i.e., the isovalues were chosen to maximize reader viewability and information accessibility. The digital information within the clustered map grids themselves is the key result. With that in mind,
Figure 6c could be contoured such that every atom pair is associated with an interaction or that no interactions are shown at all. Thus, this figure shows that there are numerous favorable and relatively significant interactions at or near the junction between the two six-helical bundles. Of the 17 residues identified as mC in this protein, there are 2 ARGmC, 4 HISmC, 4 LYSmC, and 1 TRPmC, all very polar and large. More research is called for, beyond the scope of this work, but these results are suggestive that the mapping strategy may provide mechanistic insights into the structure/function in membrane proteins. For example, our pH-dependent map data may be particularly informative.
Figure 6d focuses on the “internal” residue–residue interactions between the lipid-facing residues, i.e., the mL dataset, excluding interactions with lipids, which is designated mN. It highlights the hydrogen bonds and favorable/unfavorable hydrophobic interactions and very few unfavorable polar interactions, which characterize this protein’s structure. Clearly, there are a large number of hydrogen bonds, and as should be expected since over half of the residues in mL are hydrophobic, a substantial volume of hydrophobic interactions is also seen. The prominent unfavorable hydrophobic, although technically an energetic cost, always tracks with the favorable hydrophobic due to interactions between hydrophobic sidechains and nearby polar residues or ubiquitous backbone atoms within the protein.
Shown in
Figure 6e is the map for residue–lipid interactions, i.e., mL–mN. This figure also includes the molecular model of the associated DPPC lipids (with green-rendered carbons) extracted from the simulation reported in the MemProtMD database [
40]. Unsurprisingly, the contours shown are dominated by those that are favorable hydrophobic, especially in the middle where the lipid tails are found. Clearly, however, the lipid headgroups are also recognized by the protein’s transmembrane helixes, and their sequence is quite obviously designed by Nature to anchor within the cellular membrane with both polar and hydrophobic interactions. There are literally thousands of specific protein–lipid interactions comprising this map. To illustrate the information content (and complexity) of this composite map, we isolated just one favorable hydrophobic contour in
Figure 6f. This shows that the lipid tails of DPPC 448 (nomenclature from MemProtMD) interact with the hydrophobic sidechains of Phe 211, Val 216, Val 219, and Phe 414.
While this protein’s analysis is of an existing structure, there clearly is very significant scope for using this information in structure prediction applications. Strategies for this are described in the next section.
2.7. Reconstruction of Protein Structure Using 3D Interaction Maps
The approach to reconstruct the maps into a structure prediction is simply to match the maps’ patterns such that as many structural interaction features within sidechains as possible are reinforced. Thus, as each individual residue interaction map encodes a specific set of interactions representing a plausible environment, the “ideal” interacting residue or residues would have map features with the same character at the same points in space when all residue maps are aligned in the “protein frame”. As was seen in many of the individual residue interaction maps (
Figure 1,
Figure 2,
Figure 3,
Figure 4 and
Figure 5), as well as in the 4o6y composite map (
Figure 6), this does not mean that all interactions in a stable protein structure are favorable.
Most maps include several to a dozen distinct interaction features that are, in principle, at the midpoint between the two (or more) interacting functional groups or atoms. This suggests a surfeit of information for self-consistently reassembling the protein’s sidechain conformations. The advantage here over conventional technology, such as backbone-dependent rotamer libraries [
46,
47,
48,
49], and codes, such as SCWRL [
50,
51,
52], is that this optimization is not just geometric, or including just hydrogen bonds, but inventories the entire hydropathic interaction network in its optimization.
However, reconstruction remains a combinatorial problem, albeit a smaller one: (1) instead of a near infinite number of backbone conformations, our chessboard schema defines only sixty-four, of which, except for GLY, less than half are significantly populated; (2) instead of a near infinite number of sidechain conformations for most residues, we systematized each to an accessible number—between one (no χ1) and nine (χ1 and χ2), for each of which there are between four and eighteen maps; and (3) as the interaction map clusters are quite unevenly distributed amongst the residue types, backbone, and sidechain conformations, dictated by the populations of residues in the experimental structural data, consequently predicting structure for new cases will follow the same rules, i.e., the more common residues and conformations will have more extensive and validated training data than rare or potentially Ramachandran-disallowed backbone conformations.
To proceed, 3D grids enclosing the protein or its region of interest can be constructed, much as described above for the creation of
Figure 6. In this case, the grids will contain “scores” assessing the quality of interaction feature matches at the positions in space associated with each grid point. Encoding match scores as well as the molecular interaction features in 3D maps has operational computing advantages. They are easily parallelizable, perhaps even more than “embarrassingly” so, and are similarly completely amenable to GPU computing. Many popular scoring functions for docking use grid-based algorithms [
53,
54,
55].
2.7.1. Building a Structural Model from Interaction Maps
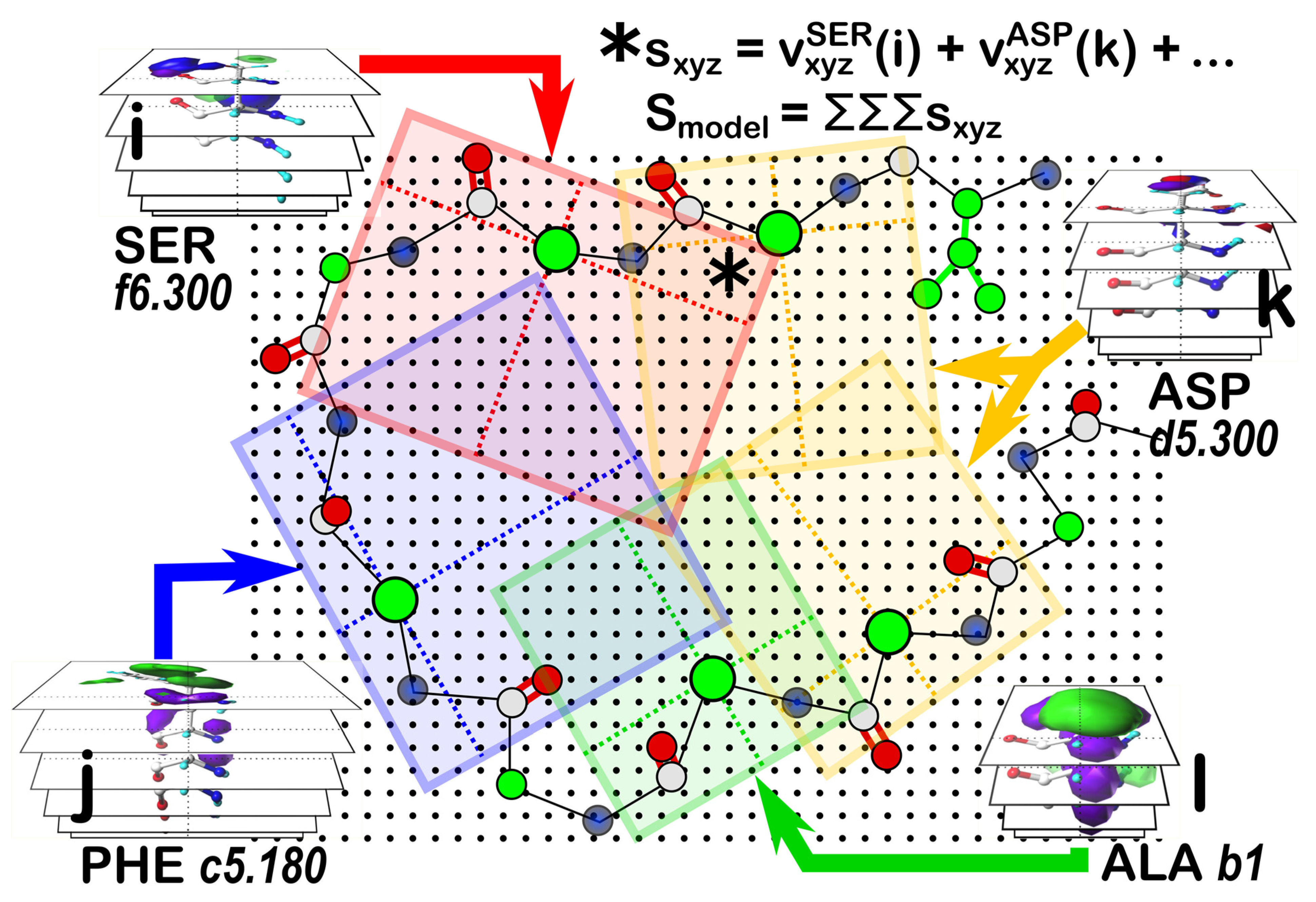
Applying our strategy for building a sidechain-optimized structural model by matching interaction features is illustrated by the cartoon of
Figure 7. The score grid, similar to the map composite grid, holds at each grid point (xyz) the sum of all interaction maps from residues (i, j, k l, …) that have been frame-shifted to align with the protein’s backbone. The total score (S
model) would be the sum of all values (s
xyz) in the score grid. The difference between this score grid and the composite display maps is that S
model is a fitness function that must be optimized to define the best set of sidechain backbone conformations. Since SER has six maps per parse, PHE has twelve, each ASP has twelve, and ALA has four, the cartoon example suggests that there are 41,472 unique models to be scored, which is a problem amenable to various machine learning or evolutionary algorithm solutions, especially for the more biologically relevant cases of the tens-to-hundreds of residues to be modeled. We are working on a genetic algorithm solution to this problem [
56].
2.7.2. Underlying a Residue’s Molecular Structure
Each 3D interaction cluster map is associated with two molecular structures or coordinate sets: one being that of the specific residue responsible for the cluster exemplar and the other an average of all members of the cluster, weighted for the Euclidian distance from the cluster’s centroid [
34]. For each cluster’s average structure, we have access to RMSDs for each of its atoms as well as average sidechain dihedral angles with standard deviations. Thus, depending on the scope of the problem and the quality of experimental data, the number of residue maps to be explored can be controlled. For example, if the sidechains are well resolved, the structural filters taking into account the now lesser uncertainties of atomic positions and dihedral angles can reduce the pool size of residue maps to be auditioned. In contrast, a poorly resolved structure or one missing a few or all sidechains might require auditioning larger sets of residue maps, e.g., the trial map set for serine may include all three χ
1 parses.
2.7.3. Fitting Lipids to a Membrane Protein Structure
A relatively small number of experimental membrane protein structures deposited in the protein data bank or other databases include any or more than a small handful of structural lipids in the transmembrane region, let alone their native set [
19,
20,
57,
58,
59,
60,
61]. This experimental deficit has led to a number of proposed methodologies to simulate the lipid structures [
62,
63,
64] or to predict or simulate protein structure within implicit [
65,
66,
67,
68] or explicit [
69,
70,
71,
72] membrane environments.
The observation by Qin et al. [
9] that there were conserved lipid binding sites on transmembrane proteins led us to believe that, just as the three-dimensional hydropathic interaction maps that we have been developing, and illustrated here, represent conserved interaction motifs possessing conserved character, loci, strength of interactions, etc., they may also include lipid interaction information if constructed from an appropriate training set. Furthermore, they would represent a means to develop an alternative, structure-based, approach for building reliable protein–lipid assemblies at a modest cost compared to many molecular dynamics-based approaches.
Simply, referring back to
Figure 6 and
Section 2.7.1, if the lipid-facing amino acid residues in the transmembrane region are modeled and optimized with the mN dataset (similar, visually, to
Figure 6d), their mL (or mL-mN, as in
Figure 6e) analogues spotlight where hydrophobic, acid, base, etc. features on lipid molecules need to be placed to produce the indicated interactions indicated by the maps.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}