2.1. Geometric Parameters of Bis-HBX
All the geometric configurations of different states of bis-HBX (see
Figure 1 and
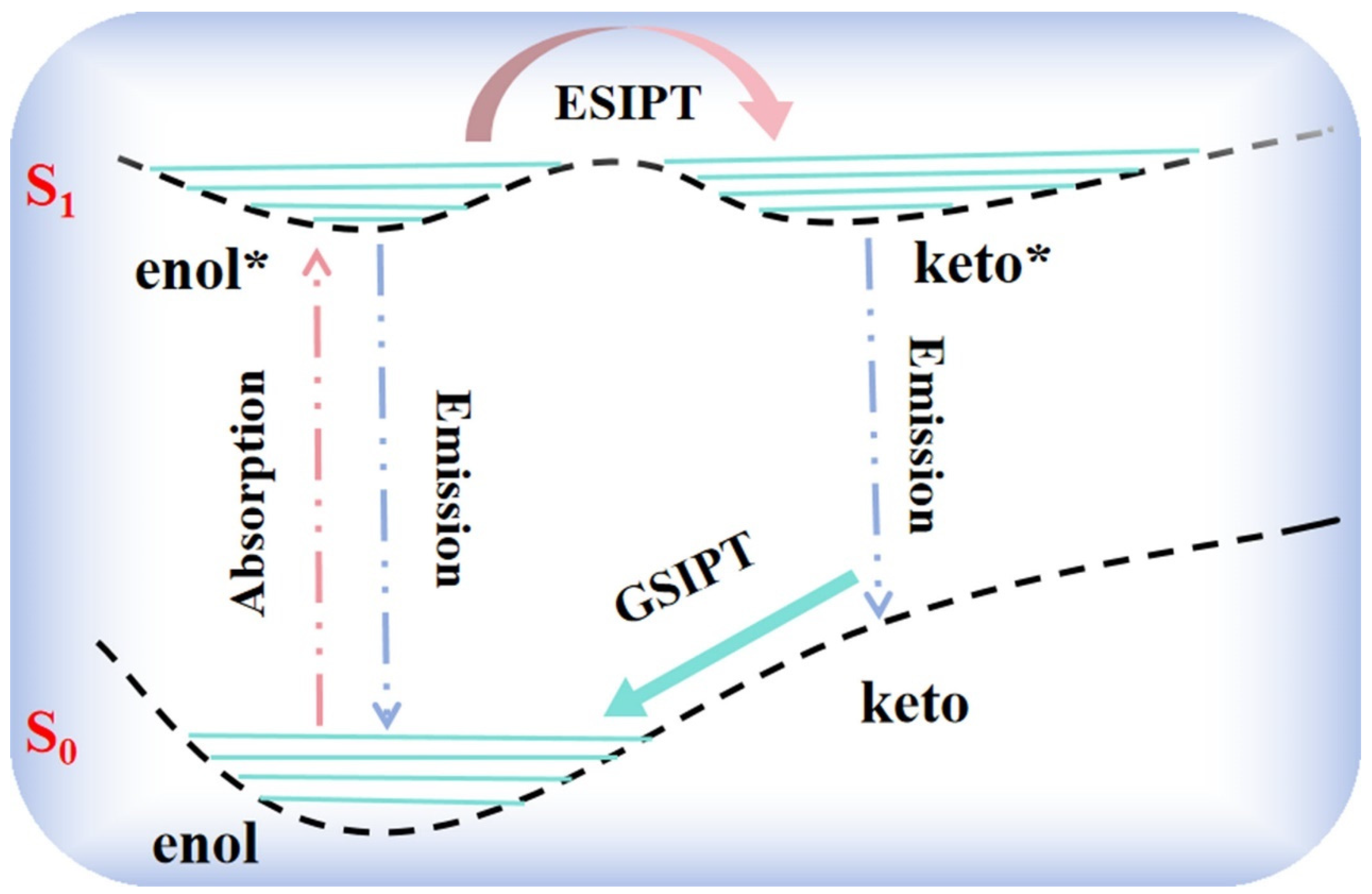
Figure S1) were optimized completely by the B3LYP-D3/6-31G+(d,p) method without any restrictions on bond lengths or bond angles. In the excited state, we labeled the enol* structure as the S
1 state and the keto* structure as the S
1′ state. Some of the significant geometric parameters are displayed in
Table 1, including bond lengths and bond angles.
The intramolecular hydrogen bonding (IHB) parameters of bis-HBX from the S
0 to the S
1 state followed the similar varying tendency recorded in
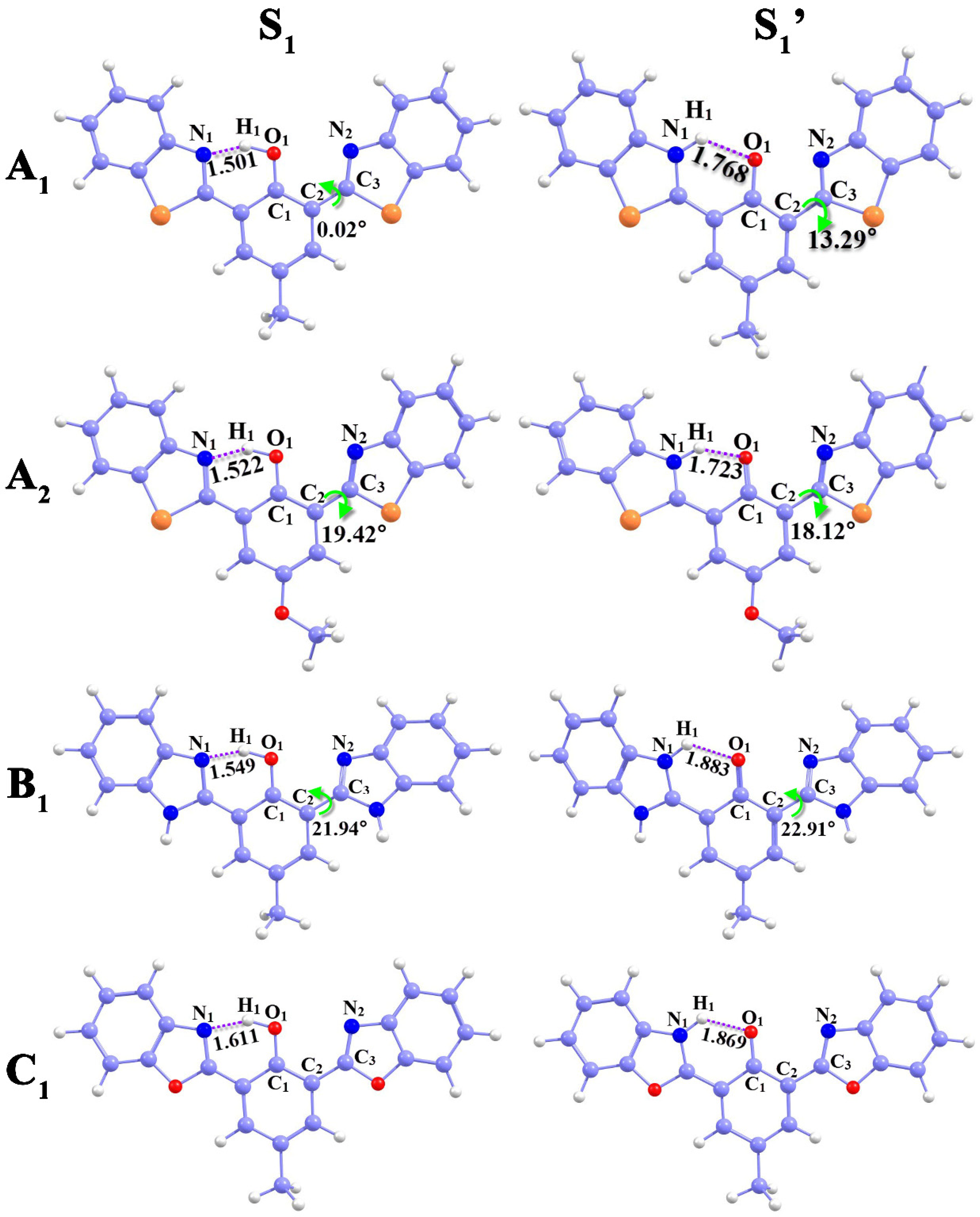
Table 1. We chose A
1 as a representative for ease of expounding and fully discussing the changing trend in IHB intensity upon photoexcitation. The bond length (O
1–H
1) of the A
1 molecule was elongated from 0.994 Å (S
0) to 1.058 Å (S
1), and N
1-H
1 was decreased from 1.720 Å (S
0) to 1.501 Å (S
1). Furthermore, the bond angle δ (O
1-H
1-N
1) was increased from 147.14° to 153.08°. It has been validated that the IHB (O
1-H
1…N
1) intensity is enhanced in the S
1 state, which is conducive to the ESIPT process [
28,
29,
30]. In addition, in the S
1′ state, we obtained tautomer configuration, and a new IHB (N
1-H
1…O
1) was formed with a bond length of 1.036 Å. The bis-HBX molecule underwent the ESIPT process, which was proved by the above-calculated results.
It is noteworthy that, in contrast to C1, the dihedral angle δ (C1–C2–C3–N2) of A1, A2, and B1 existed a torsion between the central benzene ring and the right five-membered ring. The δ (C1–C2–C3–N2) of A1, called the dihedral angle, enlarged from 47.02°(S0) to 13.29° (S1′), with a torsion of 60.31°. Similarly, both the A2 and B1 molecules underwent torsions of 28.16° and 24.69°, respectively. All the alterations indicated that A1, A2, and B1 underwent the ESIPT process with distortions in molecular configuration.
2.2. Infrared (IR) Vibrational Spectra
Due to the IR vibrational spectra providing direct evidence for the formation and fracture of hydrogen bonds (HBs) during the ESIPT process [
31], we simulated the IR vibrational spectra of four probes in different electronic states.
From the simulated IR spectra (
Figure 2), we found that the stretching vibration frequencies of O
1-H
1 in A
1, A
2, B
1, and C
1 were separately redshifted from 3244 cm
−1, 3279 cm
−1, 3193 cm
−1, and 3322 cm
−1 in the S
0 state to 2148 cm
−1, 2266 cm
−1, 2359 cm
−1, and 2654 cm
−1 in the S
1 state, accompanied with redshifts of 1096 cm
−1, 1013 cm
−1, 798 cm
−1, and 668 cm
−1, respectively. The above data indicated that the redshift value followed the order: A
1 > A
2 > B
1 > C
1, as we all know the weakening and strengthening of the IHB can be explained by the blue- and redshifts in the IR vibrational frequencies of the O-H bond [
32,
33,
34], respectively, so we can infer that the ESIPT process was more prone to occur in A
1. Meanwhile, in the S
1′ state, a new vibrational peak was observed, corresponding to the stretching vibration frequency of N
1–H
1, which was located around 3225 cm
−1 (A
1), 3125 cm
−1 (A
2), 3425 cm
−1 (B
1), and 3374 cm
−1 (C
1). The above discussion confirmed that the four probe molecules underwent the ESIPT process after photoexcitation.
2.3. RDG Scatter Plot and Topological Analyses
Considering that using the scatter plot of RDG versus Sign(I
2)r can effectively reveal IHB interactions in real space [
35], and Lu et al. recently proposed the IRI isosurface based on RDG versus Sign(I
2)r, the expression of RDG function was as shown in Equation (1) [
36,
37]:
The obtained electron density matrix equation was Equation (2):
As clarified in
Figures S2 and S3, the blue section of the color level corresponds to the HB effect, and the intensity of HB gradually increased with the deepening of the color. In addition, the spatial interactions and van der Waals forces were represented by the red and green areas, respectively. Through analyzing the RDG versus Sign(I
2)r scatter plot, the relationship of IHB intensity in A
1, A
2, B
1, and C
1 was clear-cut and unambiguous. As shown in
Figure 3, with the substitution modes of C
1, B
1, A
2, and A
1, the Sign(I
2)r values became more and more negative, unveiling that the strength of IHB in different surroundings obeys the order of A
1 > A
2 > B
1 > C
1. In the S
0 state, the prong peaks of A
1, A
2, B
1, and C
1 were located at −0.050, −0.049, −0.050, and −0.044, respectively. After photoexcitation, the prong peaks of A
1, A
2, B
1, and C
1 shifted to the more negative region (−0.087, −0.081, −0.075, and −0.063, respectively). The interaction type was judged to be an IHB interaction, corresponding to O
1–H
1…N
1. The IHB intensity of the four compounds was enhanced after photoexcitation, as indicated by the above-mentioned data, which is conducive to the ESIPT process. Moreover, comparison of the left-shifted peak positions in the four compounds revealed that the strength of IHB followed the order of A
1 > A
2 > B
1 > C
1 in the S
1 state. This was consistent with our previous analysis conclusion, revealing that different substitutions at multiple sites did affect the ESIPT process.
Topological analysis, first proposed by Bader [
38], is also one of the most common methods for measuring the strength of HBs [
39]. The obvious bond paths and bond critical points (BCPs) [
40] in the IHB region are captured in
Figure S4, and we have listed the relevant parameters in
Table 2. As is known to all, ρ(r) and ν(r) can gauge the intensity of IHBs with advantages, while the larger ρ(r) is and the more negative ν(r) is, the stronger IHB intensity will be. It could clearly be seen that the IHBs of the four molecules were all enhanced in the S
1 state. Moreover, a horizontal comparison of the absolute values of
EHB in bis-HBX showed that the
EHB values followed the order of
EHB(BCP2) >
EHB(BCP4) >
EHB(BCP6) >
EHB(BCP8), indicating that the IHB intensity of A
1 was the strongest of the four molecules in the S
1 state. It is well known that the stronger the IHB intensity is, the more favorable the ESIPT process is. Therefore, through the analysis of the above methods, we believe that the stronger the IHBs interaction, the easier the ESIPT process.
2.4. Fluorescence Attribution and Mechanism Analysis
We performed electron–hole analysis on bis-HBX molecules in the S
1 state, as shown in
Figure 4. Furthermore, they were intuitively analyzed by utilizing the inter-fragment charge transfer (IFCT) method. During the emission process, from the right side to the central benzene ring direction, 0.776 electrons, 0.691 electrons, 0.675 electrons, and 0.188 electrons were transmitted, respectively. Moreover, the characteristics of hole and electron distribution indexes are included in
Table 3. It was well known that for analogs, the lower the Sr index and the more positive the t index, the more complete the separation of electrons and holes. In the light of the t and Sr indexes, a significant separation of holes and electrons was implied through the S
0→S
1 transition in A
1 and B
1, which proved that A
1 and B
1 had significant ICT (intramolecular charge transfer) characteristics. Furthermore, combined with the configuration of bis-HBX, the dihedral angles δ (C
1–C
2–C
3–N
2) of A
1 and B
1 had abnormal twists of 47 degrees and 24.69 degrees, respectively, during photoexcitation. Perspicuously, it can be confirmed that the typical twisted intramolecular charge transfer (TICT) [
41,
42] process existed in A
1 and B
1. From the above analysis results, it can be seen that A
1 and B
1 first underwent the TICT process in the S
1 state, and then underwent the ESIPT process. Thus, we know that the TICT process in A
1 and B
1 facilitated the ESIPT process.
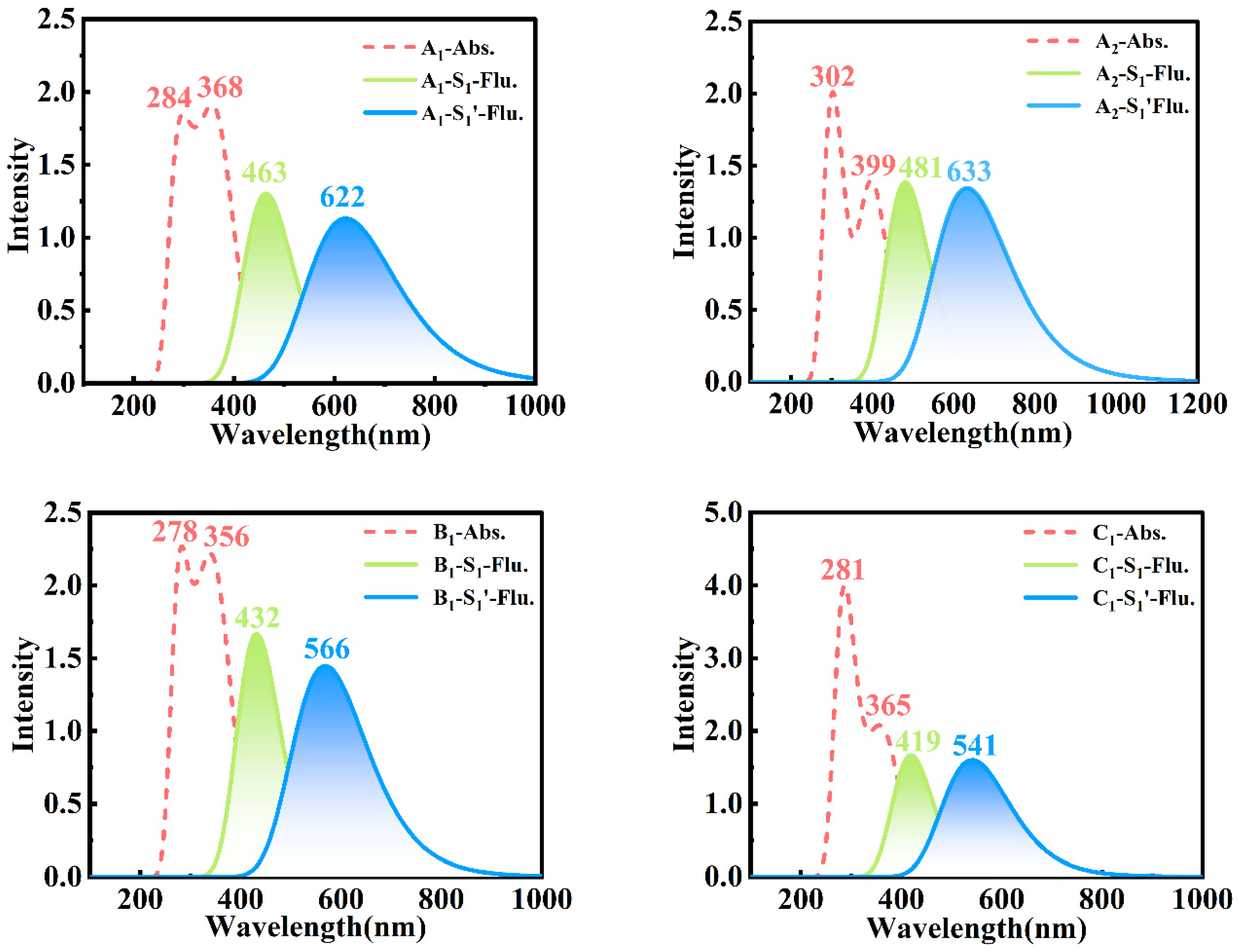
In order to gain a deeper understanding of the effects of different substitutions at multiple sites on photophysical properties in bis-HBX, we simulated the absorption and fluorescence spectra of bis-HBX (see
Figure 5), which were obtained based on the optimized S
0, S
1, and S
1′ configurations. As shown in
Table 4, the relevant photophysical parameters were recorded, and the absorption peak, oscillator strength, and corresponding orbital transition contributions were also included.
The bis-HBX had double absorption peaks that could be distinctly observed from the data listed in
Table S1, in which the calculated maximum absorption peak was very consistent with the experimental value, proving the feasibility of our method [
26]. In addition, as shown in
Figure 5, the short-wavelength emission peaks of A
2, A
1, B
1, and C
1 were 481 nm, 463 nm, 432 nm, and 419 nm, showing a blueshift from A
2 to C
1, respectively. The proton transfer tautomers in A
2, A
1, B
1, and C
1 corresponding to the longer wavelength fluorescence peaks were 633 nm, 622 nm, 566 nm, and 541 nm, respectively. Obviously, with the introduction of different substitution forms, the significant redshift was produced in the longer wavelength emission peaks of A
2, A
1, B
1, and C
1, and the degree of redshift followed the order of A
1 > A
2 > B
1 > C
1. However, only one short-wavelength fluorescence was observed in A
1 in accord with the emission peak in the experiment; thus, we attributed it to the S
1 (TICT) state.
Moreover, in order to study the fluorescence properties in bis-HBX more comprehensively, we used Equations (3) and (4) [
36,
37]:
We calculated the fluorescence lifetime and rate in the bis-HBX and listed them in
Table 5. In the formula,
represents the oscillator strength and
delegates the wavenumber. It can be observed that A
1 exhibited the greatest fluorescence lifetime, which may be due to its lower oscillation intensity. Similarly, a larger fluorescence lifetime resulted in a lower fluorescence rate, with A
1 exhibiting the lowest fluorescence rate. All the above analyses indicated that A
1 was the fluorescence probe molecule with the strongest luminous efficiently in bis-HBX, and we were also able to confirm that different substitutions at multiple sites did affect the spectral characteristics of bis-HBX.
Finally, we conducted a detailed analysis of the excited-state decay process in bis-HBX. We have provided a diagrammatic sketch of the excitation and emission processes in bis-HBX (
Figure 6) according to
Table 4. Perspicuously, from the oscillator strength in
Table 3, the excited states of A
1, A
2, and B
1 were separated from each other. It was interesting that the excited states of S
1 (0.4634) and S
5 (0.4930) in C
1 were coupled together and formed an intersection point where the internal conversion (IC) took place. The vertical transition S
1→S
0 that occurred at the Franck–Condon region of A
1, A
2, B
1, and C
1 was accompanied by shorter wavelength fluorescence measurements of 463 nm, 481 nm, 432 nm, and 419 nm, respectively. Subsequently, in the S
1 state, from the higher vibration level to the lowest vibration level, a vibration relaxation process occurred in which A
1, A
2, B
1, and C
1 recovered from the tautomer structure to the S
0 state, while at 622 nm, 633 nm, 566 nm, and 541 nm, respectively, they emitted long-wavelength fluorescence.
2.5. Frontier Molecular Orbitals and NBO Population
It is particularly acknowledged that frontier molecular orbitals (FMOs) are an effective means to reflect charge distribution and recombination [
43]. Since the first single transition of the target compound is mainly related to the highest occupied orbital (HOMO) and the lowest unoccupied orbital (LUMO),
Figure 7 only renders these two orbitals. During the photoexcitation process, the electronic clouds on the O
1 and N
1 atoms were drastically reduced and increased, respectively. This result indicated that there was a strong binding ability between the N
1 atom and the H
1 proton, which provided the driving force for the ESIPT process. For the purpose of studying the changes in electron density distribution on O
1 and N
1 quantitatively, the NBO charge distributions of bis-HBX are recorded in
Table 6. The negative charges on the O
1 shrank from −0.693 a. u., −0.707 a. u., −0.693 a. u., and −0.696 a. u. in the ground state to −0.677 a. u., −0.685 a. u., −0.684 a. u., and −0.676 a. u. in the excited state, while that of the N
1 atom was enlarged during the photo-absorption process, which was confirmed by the analytical results of the FMOs.
In addition, we used the Hirshfeld method to calculate the electron density components in Fragment 1 and Fragment 2 in bis-HBX (see
Figure 7). In Fragment 1, the electron density components of A
1, A
2, and B
1 increased from 80.897%, 88.180%, 53.063%, and 57.988% in HOMO to 93.816%, 93.110%, 92.774%, and 86.433% in LUMO, respectively. Regarding Fragment 2, the electron density components were 19.103%, 11.820%, 46.937%, and 42.012% in HOMO, while they separately decreased to 6.184%, 6.890%, 7.226%, and 13.562% in LUMO. Meanwhile, as mentioned in the geometric parameters, the obtained dihedral angles δ (C
1–C
2–C
3–N
2) of A
1, A
2, B
1, and C
1 underwent torsions of 47°, 28.16°, 24.69°, and 0°, respectively, which demonstrated that A
1 has obvious TICT characteristics.
2.6. Potential Energy Curves (PECs)
With the aim of uncovering the mechanism of the ESIPT process with different substitutions at multiple sites more intuitively, the potential energy curves (PECs) in bis-HBX were scanned via prolonging the O
1–H
1 bond length from 0.9 Å to 1.9 Å in increments of 0.1 Å based on the optimized structures in the gas phase [
44].
As shown in
Figure 8, the PECs of the four probe molecules showed an ascending trend in the S
0 state. This demonstrates that it was difficult for the proton transfer (PT) process to occur. After photoexcitation, the energy barriers in the four probe molecules were diminished to 0.24 kcal/mol (A
1), 0.55 kcal/mol (A
2), 0.67 kcal/mol (B
1), and 1.15 kcal/mol (C
1), respectively. It is obvious that it was more possible for the four probe molecules to undergo the PT process in the S
1 state. It is a remarkable fact that the order of energy barriers in bis-HBX followed the order of A
1 < A
2 < B
1 < C
1, which verified that A
1 can occur the ESIPT process more easily. Although the B3LYP/6-31+G (d,p) can provide qualitative descriptions of the hypersurface, it may underestimate some of the energy differences and energy barriers. We recalculated the PECs using Cam-B3LYP/TZVP [
45,
46] theory (see
Figure 8; the blue font is the corrected energy barrier), taking the corrected results as the reference. It was very clear and concise that the energy barriers calculated using Cam-B3LYP/TZVP theory were consistent with the order of A
1 < A
2 < B
1 < C
1. In addition, to further confirm the ESIPT process mechanism, we also performed transition-state (TS) calculations and obtained the intrinsic reaction coordinate (IRC) to validate the reasonableness and accuracy. Meanwhile, we have listed the ESIPT barriers via the IRC paths for bis-HBX in
Table 7, which is consistent with the simulation results of the PECs. We further confirmed that A
1 can cause the ESIPT process to occur more easily. To date, we have explained the effects of different substitutions at multiple sites in the ESIPT process of bis-HBX molecules and reasonably elucidated the whole kinetic process of the studied molecules. This provides important reference values for the application of fluorescent probe molecules.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}