
Protic Processes in an Extended Pyrazinacene: The Case of Dihydrotetradecaazaheptacene

 ,
,  and
and 
Abstract
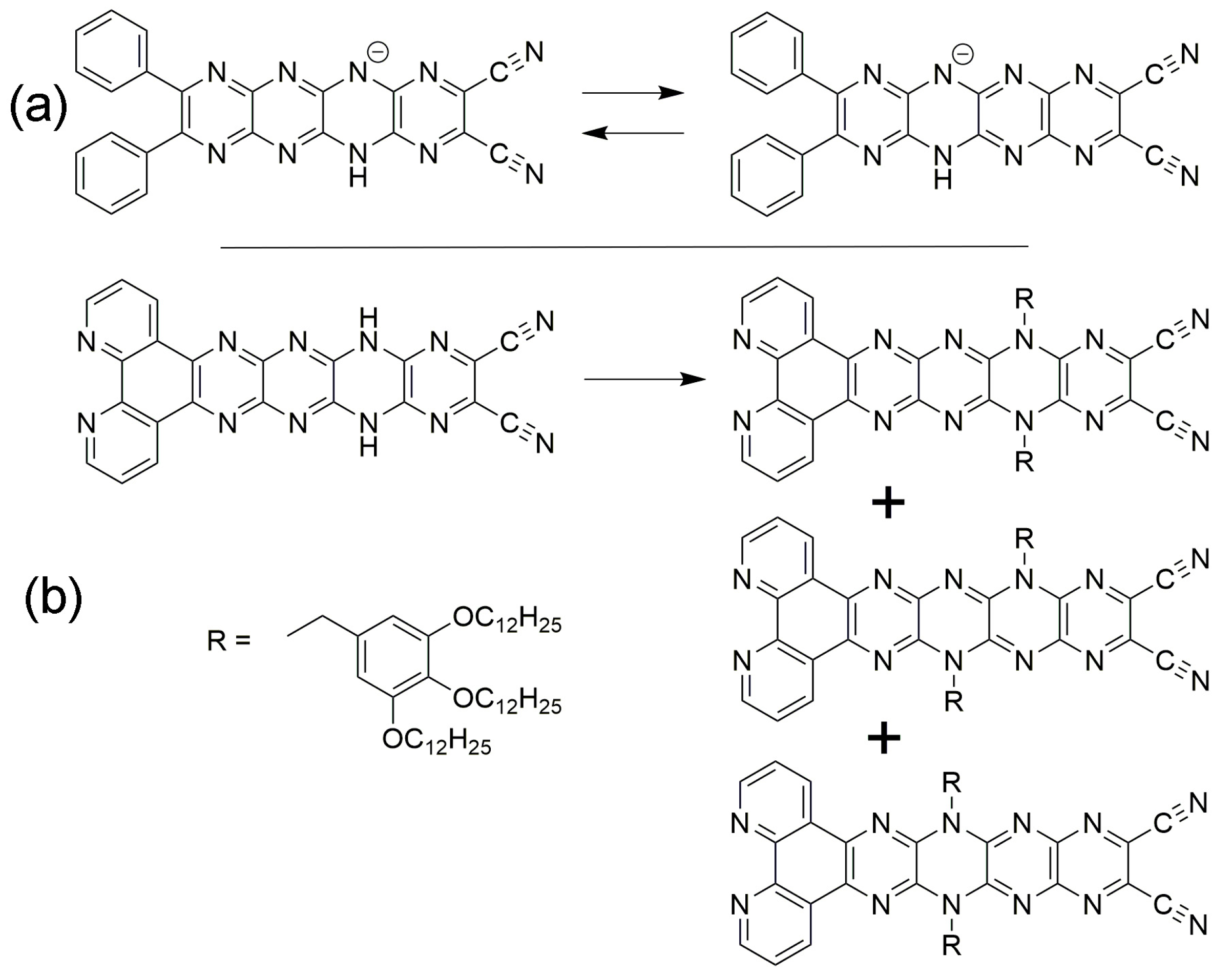
1. Introduction
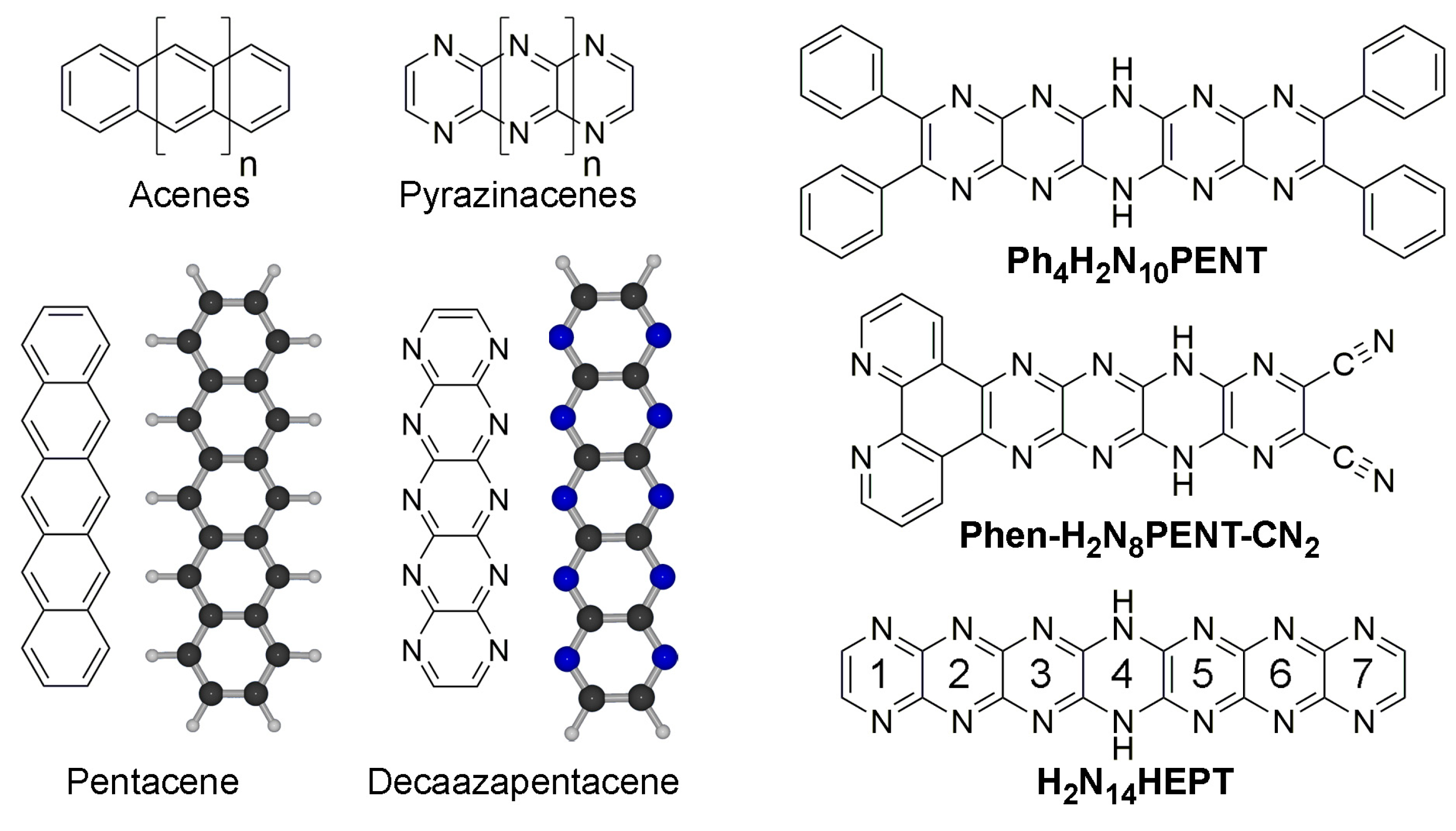
2. Results and Discussion
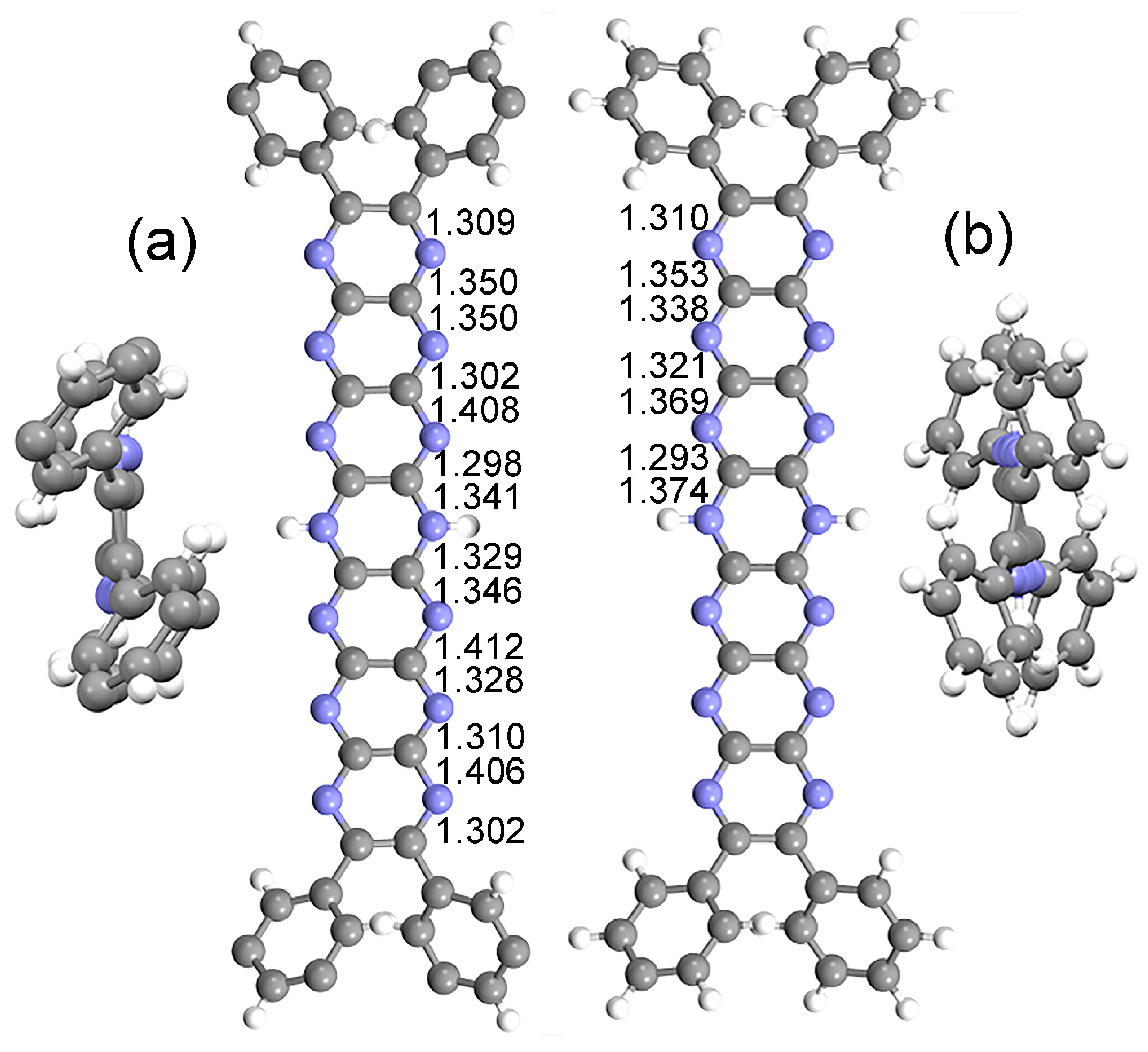
2.1. Experimental vs. Computed Structures
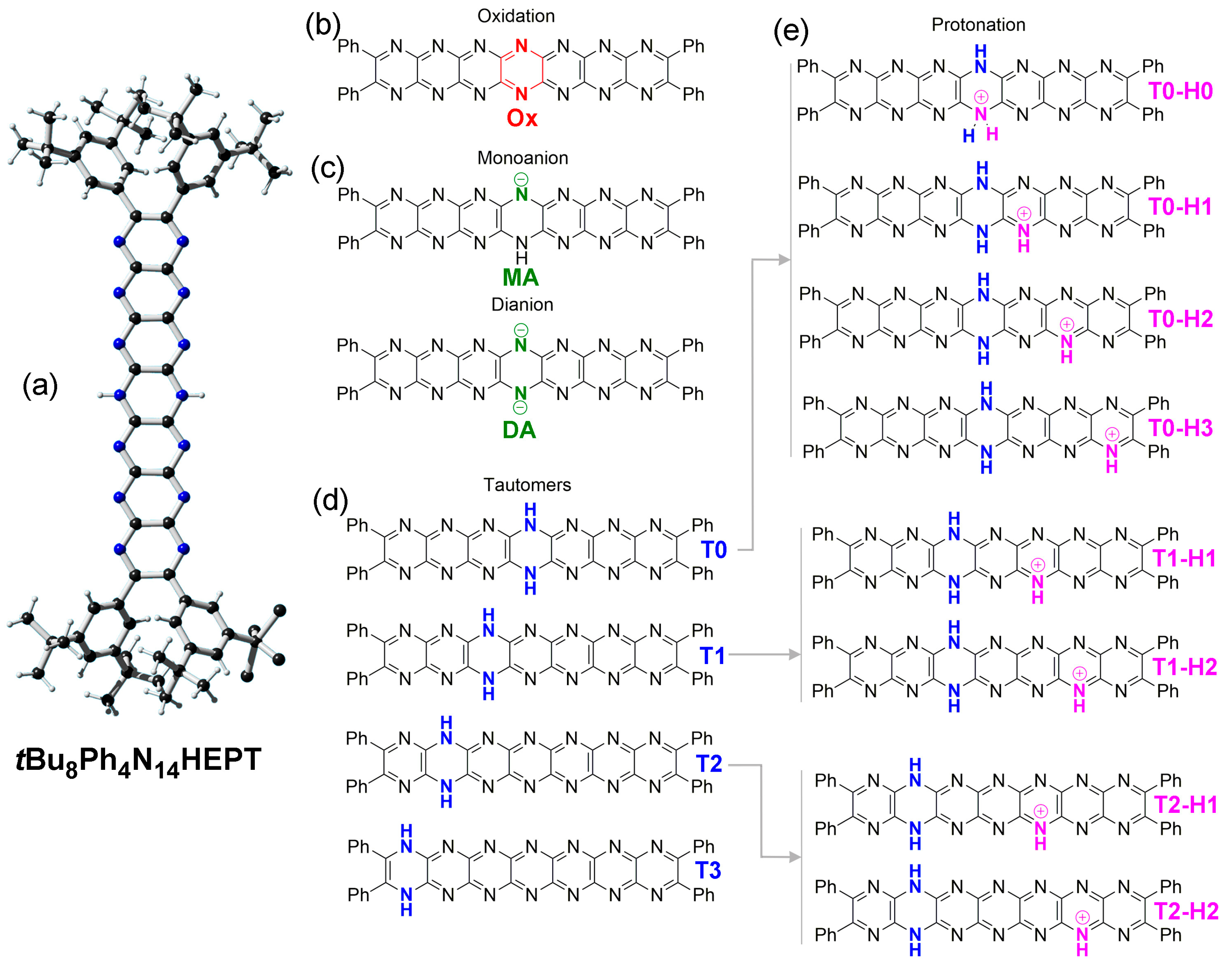
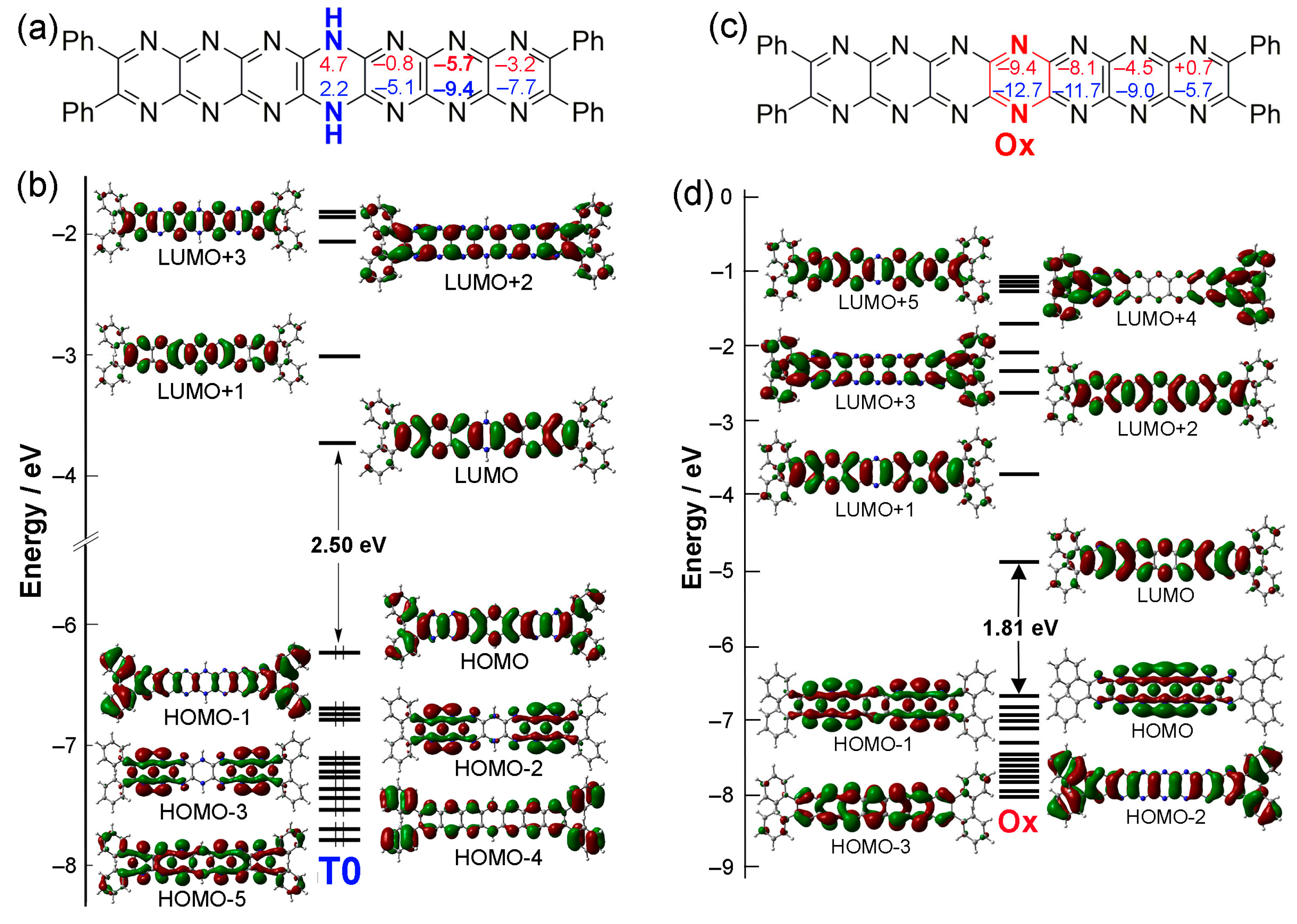
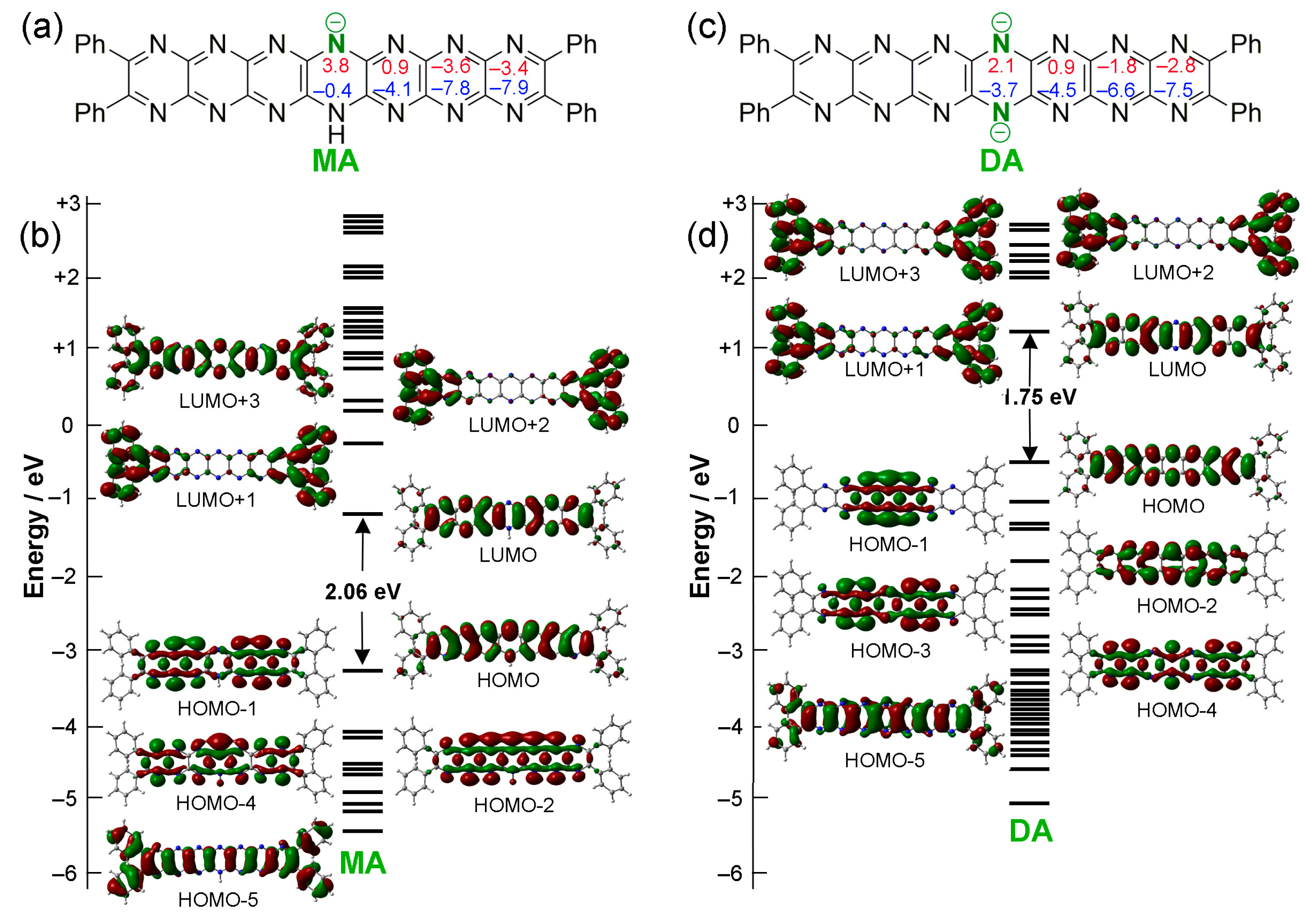
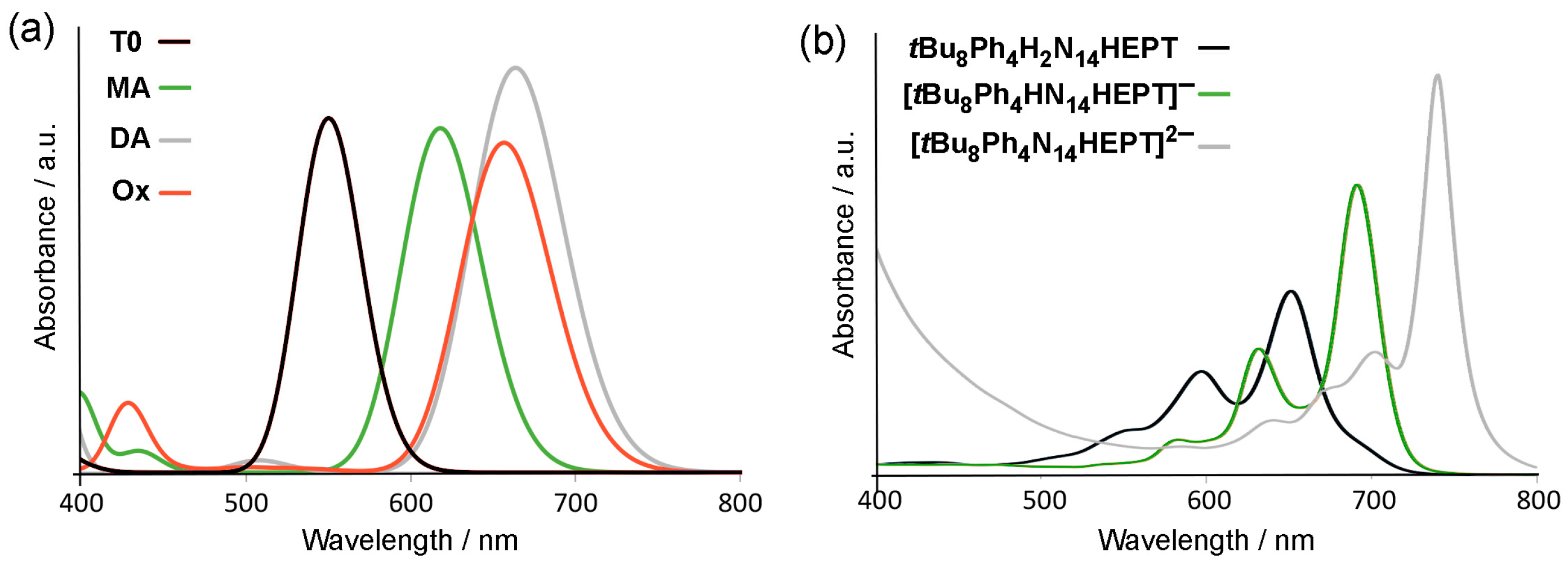
2.2. Oxidation and Deprotonation
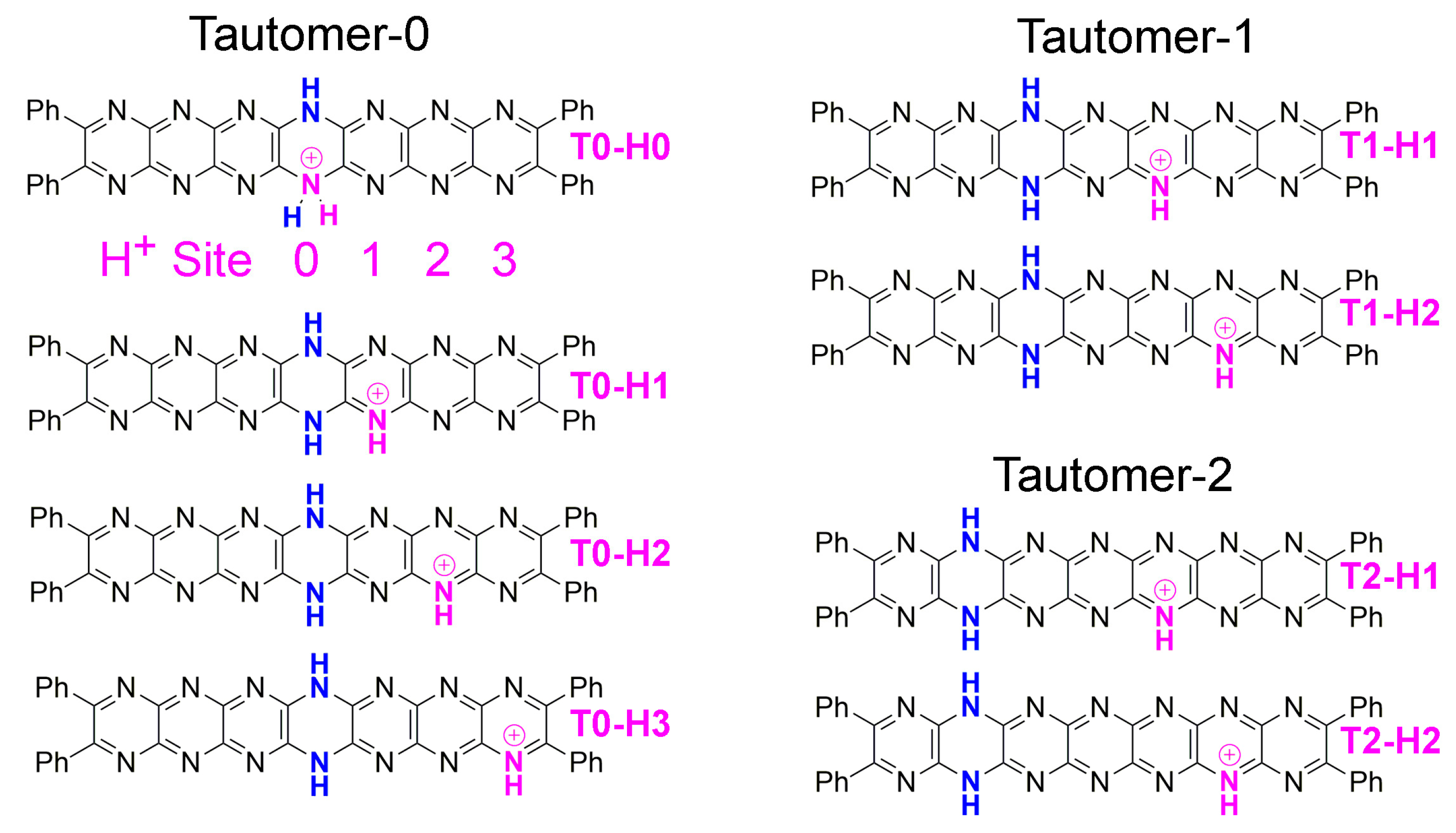
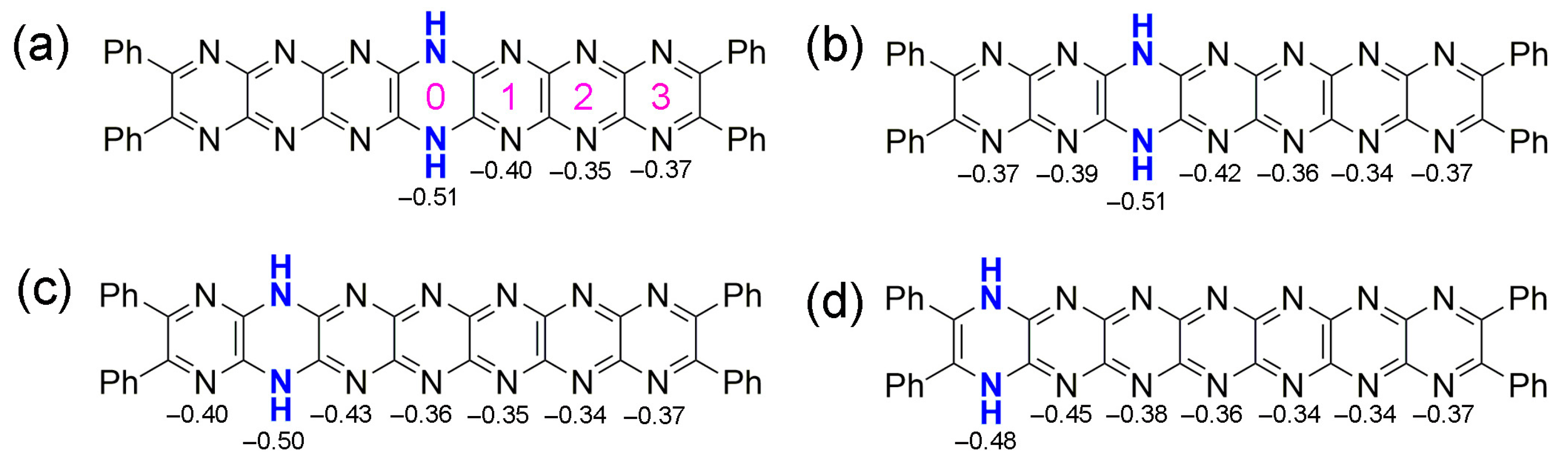
2.3. Tautomerization
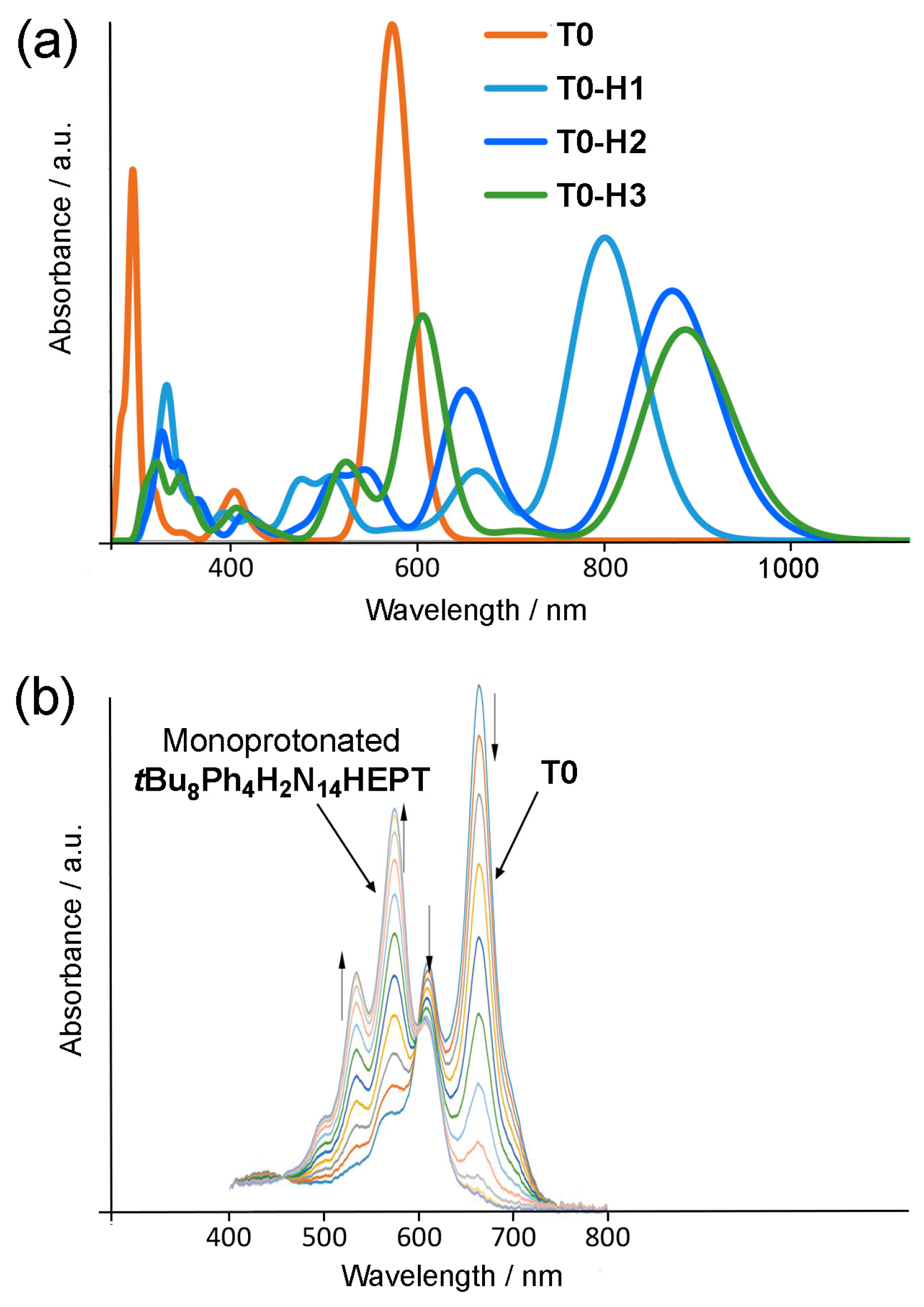
2.4. Protonation
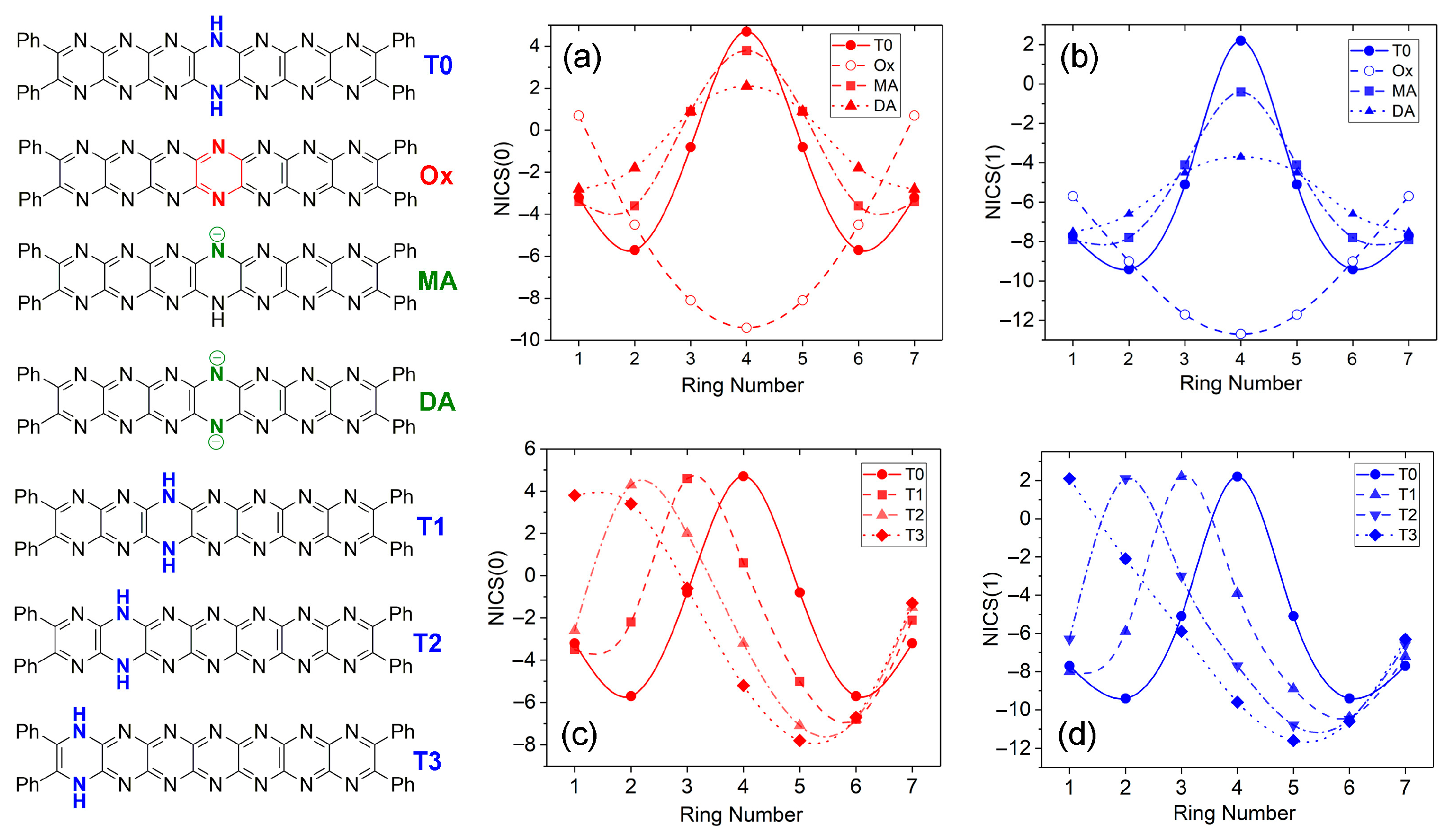
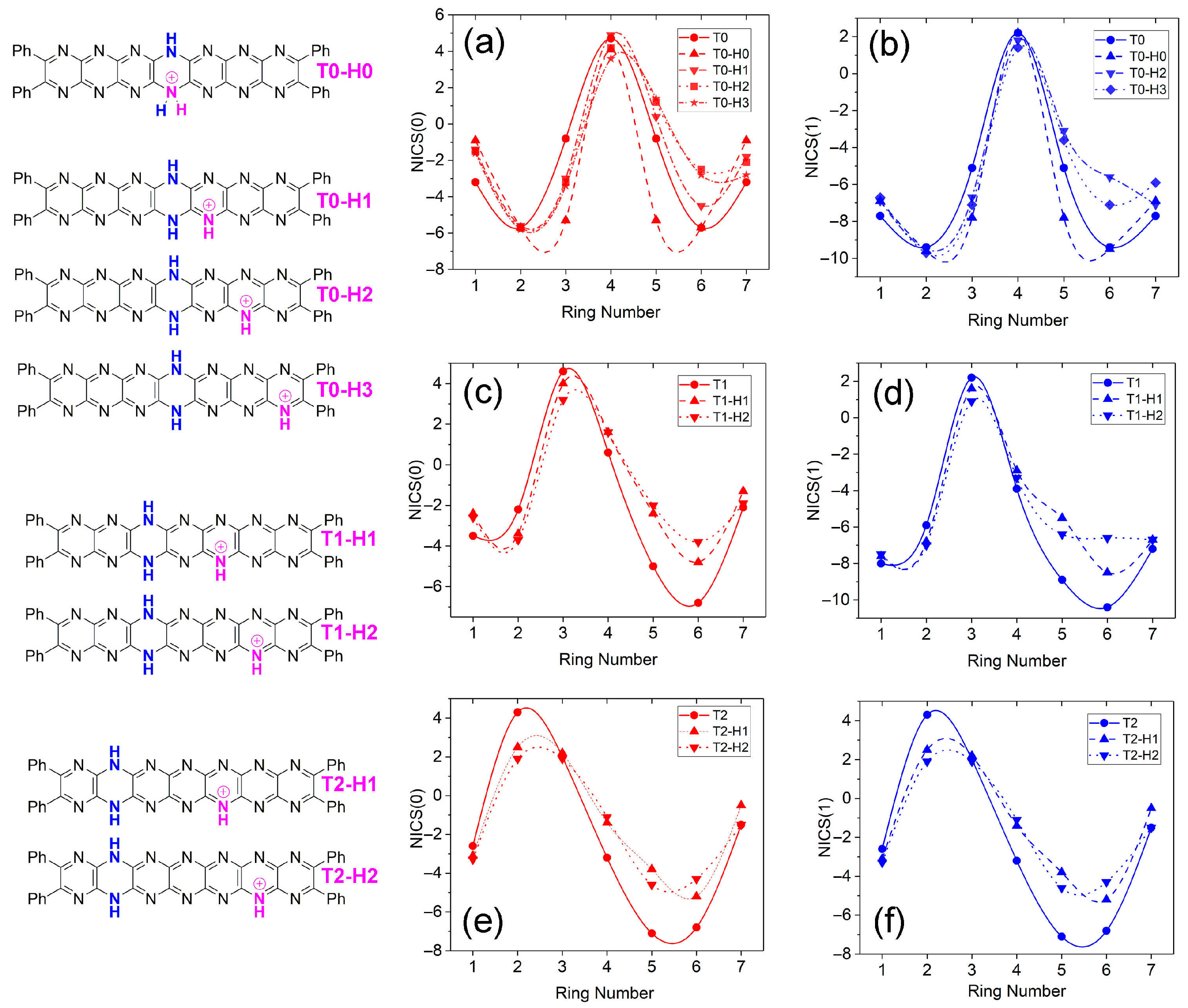
2.5. Trends in Aromaticity Based on NICS
3. Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Campbell, R.B.; Robertson, J.M.; Trotter, J. The Crystal and Molecular Structure of Pentacene. Acta Crystallogr. 1961, 14, 705–711. [Google Scholar] [CrossRef]
- Robertson, J.M.; Sinclair, V.C.; Trotter, J. The Crystal and Molecular Structure of Tetracene. Acta Crystallogr. 1961, 14, 697–704. [Google Scholar] [CrossRef]
- Watanabe, M.; Chang, Y.J.; Liu, S.-W.; Chao, T.-H.; Goto, K.; Islam, M.M.; Yuan, C.-H.; Tao, Y.-T.; Shinmyozu, T.; Chow, T.J. The Synthesis, Crystal Structure and Charge-Transport Properties of Hexacene. Nat. Chem. 2012, 4, 574–578. [Google Scholar] [CrossRef] [PubMed]
- Goetz, K.P.; Li, Z.; Ward, J.W.; Bougher, C.; Rivnay, J.; Smith, J.; Conrad, B.R.; Parkin, S.R.; Anthopoulos, T.D.; Salleo, A.; et al. Effect of Acene Length on Electronic Properties in 5-, 6-, and 7-Ringed Heteroacenes. Adv. Mater. 2011, 23, 3698–3703. [Google Scholar] [CrossRef] [PubMed]
- Bettinger, H.F.; Tönshoff, C. The Longest Acenes. Chem. Rec. 2015, 15, 364–369. [Google Scholar] [CrossRef]
- Pascal, R.A. Twisted Acenes. Chem. Rev. 2006, 106, 4809–4819. [Google Scholar] [CrossRef] [PubMed]
- Lakshminarayana, A.N.; Ong, A.; Chi, C. Modification of Acenes for n-Channel OFET Materials. J. Mater. Chem. C 2018, 6, 3551–3563. [Google Scholar] [CrossRef]
- Müller, M.; Ahrens, L.; Brosius, V.; Freudenberg, J.; Bunz, U.H.F. Unusual Stabilization of Larger Acenes and Heteroacenes. J. Mater. Chem. C 2019, 7, 14011–14034. [Google Scholar] [CrossRef]
- Bendikov, M.; Wudl, F.; Perepichka, D.F. Tetrathiafulvalenes, Oligoacenenes, and Their Buckminsterfullerene Derivatives: The Brick and Mortar of Organic Electronics. Chem. Rev. 2004, 104, 4891–4946. [Google Scholar] [CrossRef]
- Anthony, J.E. The Larger Acenes: Versatile Organic Semiconductors. Angew. Chem. Int. Ed. 2008, 47, 452–483. [Google Scholar] [CrossRef]
- Zade, S.S.; Bendikov, M. Reactivity of Acenes: Mechanisms and Dependence on Acene Length. J. Phys. Org. Chem. 2012, 25, 452–461. [Google Scholar] [CrossRef]
- Clar, E. The Aromatic Sextet; Wiley: New York, NY, USA, 1972. [Google Scholar]
- Tönshoff, C.; Bettinger, H.F. Pushing the Limits of Acene Chemistry: The Recent Surge of Large Acenes. Chem. Eur. J. 2021, 27, 3193–3212. [Google Scholar] [CrossRef] [PubMed]
- Jancarik, A.; Levet, G.; Gourdon, A. A Practical General Method for the Preparation of Long Acenes. Chem. Eur. J. 2019, 25, 2366–2374. [Google Scholar] [CrossRef] [PubMed]
- Urgel, J.I.; Mishra, S.; Hayashi, H.; Wilhelm, J.; Pignedoli, C.A.; Di Giovannantonio, M.; Widmer, R.; Yamashita, M.; Hieda, N.; Ruffieux, P.; et al. On-Surface Light-Induced Generation of Higher Acenes and Elucidation of their Open-Shell Character. Nat. Commun. 2019, 10, 861. [Google Scholar] [CrossRef] [PubMed]
- Miklík, D.; Mousavi, S.F.; Burešova, Z.; Middleton, A.; Matsushita, Y.; Labuta, J.; Ahsan, A.; Buimaga-Iarinca, L.; Karr, P.A.; Bureš, F.; et al. Pyrazinacenes Exhibit On-Surface Oxidation-State-Dependent Conformational and Self-Assembly Behaviors. Commun. Chem. 2021, 4, 29. [Google Scholar] [CrossRef] [PubMed]
- Švec, P.; Webre, W.A.; Richards, G.J.; Labuta, J.; Wakayama, Y.; Miklík, D.; Karr, P.A.; Mori, T.; Ariga, K.; D’Souza, F.; et al. Phenanthroline-Fused Pyrazinacenes: One-Pot Synthesis, Tautomerization and a RuII(2,2′-Bpy)2 Derivative. Eur. J. Inorg. Chem. 2018, 2018, 2541–2548. [Google Scholar] [CrossRef]
- Richards, G.J.; Cador, A.; Yamada, S.; Middleton, A.; Webre, W.A.; Labuta, J.; Karr, P.A.; Ariga, K.; D’Souza, F.; Kahlal, S.; et al. Amphiprotism-Coupled Near-Infrared Emission in Extended Pyrazinacenes Containing Seven Linearly Fused Pyrazine Units. J. Am. Chem. Soc. 2019, 141, 19570–19574. [Google Scholar] [CrossRef] [PubMed]
- Richards, G.J.; Hill, J.P.; Subbaiyan, N.K.; D’Souza, F.; Karr, P.A.; Elsegood, M.R.J.; Teat, S.J.; Mori, T.; Ariga, K. Pyrazinacenes: Aza Analogues of Acenes. J. Org. Chem. 2009, 74, 8914–8923. [Google Scholar] [CrossRef]
- Richards, G.J.; Hill, J.P. The Pyrazinacenes. Acc. Chem. Res. 2021, 54, 3228–3240. [Google Scholar] [CrossRef]
- Bunz, U.H.F.; Engelhart, J.U. The Palladium Way to N-Heteroacenes. Chem. Eur. J. 2016, 22, 4680–4689. [Google Scholar] [CrossRef]
- Kang, F.; Yang, J.; Zhang, Q. Recent Progress in Pyrazinacenes Containing Nonbenzenoid Rings: Synthesis, Properties and Applications. J. Mater. Chem. C 2022, 10, 2475–2493. [Google Scholar] [CrossRef]
- Richards, G.J.; Hill, J.P.; Mori, T.; Ariga, K. Putting the ‘N’ in Acene: Pyrazinacenes and their Structural Relatives. Org. Biomol. Chem. 2011, 9, 5005–5017. [Google Scholar] [CrossRef] [PubMed]
- Bunz, U.H.F.; Freudenberg, J. N-Heteroacenes and N-Heteroarenes as N-Nanocarbon Segments. Acc. Chem. Res. 2019, 52, 1575–1587. [Google Scholar] [CrossRef] [PubMed]
- Anthony, J.E. Functionalized Acenes and Heteroacenes for Organic Electronics. Chem. Rev. 2006, 106, 5028–5048. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Miao, J.; Cao, J.; Zhu, J.; Liu, B.; Zhang, Q. Preparation and Photoelectrochemical Behavior of 1,4,6,8,11,13-Hexazapentacene (HAP). Chem. Commun. 2014, 50, 7656–7658. [Google Scholar] [CrossRef] [PubMed]
- Gu, P.-Y.; Wang, Z.; Zhang, Q. Azaacenes as Active Elements for Sensing and Bio Applications. J. Mater. Chem. B 2016, 4, 7060–7074. [Google Scholar] [CrossRef] [PubMed]
- Fleischhauer, J.; Zahn, S.; Beckert, R.; Grummt, U.-W.; Birckner, E.; Görls, H. A Way to Stable, Highly Emissive Fluorubine Dyes: Tuning the Electronic Properties of Azaderivatives of Pentacene by Introducing Substituted Pyrazines. Chem. Eur. J. 2012, 18, 4549–4557. [Google Scholar] [CrossRef] [PubMed]
- Tykwinski, R.R. Synthesis of Unsymmetrical Derivatives of Pentacene for Materials Applications. Acc. Chem. Res. 2019, 52, 2056–2069. [Google Scholar] [CrossRef]
- Winkler, M.; Houk, K.N. Nitrogen-Rich Oligoacenes: Candidates for n-Channel Organic Semiconductors. J. Am. Chem. Soc. 2007, 129, 1805–1815. [Google Scholar] [CrossRef]
- Bendikov, M.; Duong, H.M.; Starkey, K.; Houk, K.N.; Carter, E.A.; Wudl, F. Oligoacenes: Theoretical Prediction of Open-Shell Singlet Diradical Ground States. J. Am. Chem. Soc. 2004, 126, 7416–7417. [Google Scholar] [CrossRef]
- Shi, Y.; Zhang, Y.; Cai, X. First-Principles Investigation of Electronic and Charge Transport Properties of Pyrazinacenes and Their Structural Relatives. Chem. Phys. Lett. 2021, 772, 138595. [Google Scholar] [CrossRef]
- Scipioni, R.; Hill, J.P.; Richards, G.J.; Boero, M.; Mori, T.; Ariga, K.; Ohno, T. Tautomers of Extended Reduced Pyrazinacenes: A Density-Functional-Theory Based Study. Phys. Chem. Chem. Phys. 2011, 13, 2145–2150. [Google Scholar] [CrossRef] [PubMed]
- Scipioni, R.; Boero, M.; Richards, G.J.; Hill, J.P.; Ohno, T.; Mori, T.; Ariga, K. Tautomerism in Reduced Pyrazinacenes. J. Chem. Theory Comput. 2010, 6, 517–525. [Google Scholar] [CrossRef]
- Miao, S.; Brombosz, S.M.; Schleyer, P.v.R.; Wu, J.I.; Barlow, S.; Marder, S.R.; Hardcastle, K.I.; Bunz, U.H.F. Are N,N-Dihydrodiazatetracene Derivatives Antiaromatic? J. Am. Chem. Soc. 2008, 130, 7339–7344. [Google Scholar] [CrossRef]
- Hele, T.J.H.; Fuemmeler, E.G.; Sanders, S.N.; Kumarasamy, E.; Sfeir, M.Y.; Campos, L.M.; Ananth, N. Anticipating Acene-Based Chromophore Spectra with Molecular Orbital Arguments. J. Phys. Chem. A 2019, 123, 2527–2536. [Google Scholar] [CrossRef] [PubMed]
- Schleyer, P.v.R.; Maerker, C.; Dransfeld, A.; Jiao, H.; van Eikema Hommes, N.J.R. Nucleus-Independent Chemical Shifts: A Simple and Efficient Aromaticity Probe. J. Am. Chem. Soc. 1996, 118, 6317–6318. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wannere, C.S.; Corminboeuf, C.; Puchta, R.; Schleyer, P.v.R. Nucleus-Independent Chemical Shifts (NICS) as an Aromaticity Criterion. Chem. Rev. 2005, 105, 3842–3888. [Google Scholar] [CrossRef]
- Portella, G.; Poater, J.; Bofill, J.M.; Alemany, P.; Solà, M. Local Aromaticity of [n]Acenes, [n]Phenacenes, and [n]Helicenes (n = 1–9). J. Org. Chem. 2005, 70, 2509–2521. [Google Scholar] [CrossRef]
- Wu, J.I.; Wannere, C.S.; Mo, Y.; Schleyer, P.v.R.; Bunz, U.H.F. 4n π Electrons but Stable: N,N-Dihydrodiazapentacenes. J. Org. Chem. 2009, 74, 4343–4349. [Google Scholar] [CrossRef]
- Benkyi, I.; Tapavicza, E.; Fliegl, H.; Sundholm, D. Calculation of Vibrationally Resolved Absorption Spectra of Acenes and Pyrene. Phys. Chem. Chem. Phys. 2019, 21, 21094–21103. [Google Scholar] [CrossRef]
- Blight, B.A.; Hunter, C.A.; Leigh, D.A.; McNab, H.; Thomson, P.I.T. An AAAA–DDDD Quadruple Hydrogen-Bond Array. Nat. Chem. 2011, 3, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. B 1964, 136, 864–871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phy. Rev. A 1965, 140, 1133–1138. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate Spin-Dependent Electron Liquid Correlation Energies for Local Spin Density Calculations: A Critical Analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-Fitting Basis Sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- EMSL. Basis Set Library. Available online: https://www.basissetexchange.org/ (accessed on 15 May 2024).
- Feller, D. The Role of Databases in Support of Computational Chemistry Calculations. J. Comp. Chem. 1996, 17, 1571–1586. [Google Scholar] [CrossRef]
- Schuchardt, K.L.; Didier, B.T.; Elsethagen, T.; Sun, L.; Gurumoorthi, V.; Chase, J.; Li, J.; Windus, T.L. Basis Set Exchange: A Community Database for Computational Sciences. J. Chem. Inf. Model. 2007, 47, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Landis, C.R.; Weinhold, F. NBO 7.0; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, USA, 2013; Available online: http://nbo6.chem.wisc.edu (accessed on 15 May 2024).
- Runge, E.; Gross, E.K.U. Density-Functional Theory for Time-Dependent Systems. Phys. Rev. Lett. 1984, 52, 997–1000. [Google Scholar] [CrossRef]
- Adamo, C.; Jacquemin, D. The Calculations of Excited-State Properties with Time-Dependent Density Functional Theory. Chem. Soc. Rev. 2013, 42, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Gorelsky, S.I. Swizard, Revision 4.5. Available online: https://www.sg-chem.net/aomix/ (accessed on 15 May 2024).
- Gorelsky, S.I. AOMix: Program for Molecular Orbital Analysis, Version 6.5. 2019. Available online: http://www.sg-chem.net/swizard/ (accessed on 15 May 2024).
- Gorelsky, S.I.; Lever, A.B.P. Electronic Structure and Spectra of Ruthenium Diimine Complexes by Density Functional Theory and INDO/S. Comparison of the Two Methods. J. Organomet. Chem. 2001, 635, 187–196. [Google Scholar] [CrossRef]
- Eisenhut, F.; Kühne, T.; Garcia, F.; Fernández, S.; Guitián, E.; Pérez, D.; Trinquier, G.; Cuniberti, G.; Joachim, C.; Peña, D.; et al. Dodecacene Generated on Surface: Reopening of the Energy Gap. ACS Nano 2020, 14, 1011–1017. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Average Bond Length (Å) | X-ray Structure (tBu8Ph4H2N14HEPT) | Optimized Structure (T0 (Ph4H2N14HEPT)) |
|---|---|---|
| C-N | 1.348 | 1.337 |
| Acene C-C | 1.439 | 1.448 |
| Ph C-C | 1.393 | 1.392 |
| N-H | 0.880 | 1.013 |
| Compound | HOMO-LUMO Gap (eV) | λabs (nm), Eabs (eV) | fabs | Transition | λem (nm), Eem (eV) | fem |
|---|---|---|---|---|---|---|
| T0 | 2.50 | 550 (2.25) | 2.39 | HOMO → LUMO(98%) | 585 (2.12) | 2.42 |
| Ox (singlet) | 1.81 | 657 (1.89) | 2.23 | HOMO-2 → LUMO(98%) |
| Compound | HOMO-LUMO Gap (eV) | λabs (nm), Eabs (eV) | fabs | Transition | λem (nm), Eem (eV) | fem |
|---|---|---|---|---|---|---|
| T0 | 2.50 | 550 (2.25) | 2.39 | HOMO → LUMO(98%) | 585 (2.12) | 2.42 |
| MA | 2.06 | 618 (2.01) | 2.33 | HOMO → LUMO(99%) | 654 (1.90) | 1.95 |
| DA | 1.75 | 664 (1.87) | 2.73 | HOMO → LUMO(100%) | 695 (1.78) | 1.83 |
| Compound | HOMO-LUMO Gap (eV) | ΔE (kcal/mol) a | ΔE0 (kcal/mol) b | ΔG (kcal/mol) |
|---|---|---|---|---|
| T0 | 2.499 | 0.000 | 0.000 | 0.000 |
| T1 | 2.388 | +2.592 | +2.503 | +2.454 |
| T2 | 2.204 | +11.566 | +10.236 | +13.409 |
| T3 | 2.029 | +31.629 | +31.058 | +30.512 |
| Compound | λabs (nm), Eabs (eV) | fabs | Transition | λem (nm), Eem (eV) | fem |
|---|---|---|---|---|---|
| T0 | 550 (2.25) | 2.39 | HOMO → LUMO (98%) | 585 (2.12) | 2.42 |
| T1 | 569 (2.18) | 2.19 | HOMO → LUMO (98%) | 647 (1.92) | 1.84 |
| T2 | 606 (2.05) | 2.07 | HOMO → LUMO (98%) | 690 (1.80) | 1.75 |
| T3 | 628 (1.97) | 2.28 | HOMO → LUMO (98%) |
| Compound | HOMO-LUMO Gap (eV) | ΔE (kcal/mol) a | ΔE0 (kcal/mol) b | ΔG (kcal/mol) |
|---|---|---|---|---|
| T0-H0 | 2.355 | +51.212 | +50.571 | +48.799 |
| T0-H1 | 1.763 | +17.952 | +17.461 | +17.195 |
| T0-H2 | 1.565 | +2.932 | +2.908 | +2.847 |
| T0-H3 | 1.529 | +4.485 | +4.188 | +4.518 |
| T1-H1 | 1.531 | +1.952 | +1.906 | +1.806 |
| T1-H2 | 1.403 | 0.000 | 0.000 | 0.000 |
| T2-H1 | 1.432 | +4.242 | +4.045 | +3.988 |
| T2-H2 | 1.349 | +6.027 | +5.753 | +5.643 |
| Compound | HOMO-LUMO Gap (eV) | λabs (nm), Eabs (eV) | fabs | Transition | λem (nm), Eem (eV) | fem |
|---|---|---|---|---|---|---|
| Tautomer-0 (Ph4N14Ph4H2) | 2.50 | 550 (2.25) | 2.39 | HOMO → LUMO (+98%) | 585 (2.12) | 2.42 |
| T0-H0 | 2.36 | Too unstable to be considered (+50 kcal.mol−1) | ||||
| T0-H1 | 1.76 | 777 (1.60) | 1.40 | HOMO → LUMO (100%) | ||
| T0-H2 | 1.57 | 849 (1.46) | 1.16 | HOMO → LUMO (99%) | ||
| T0-H3 | 1.53 | 863 (1.44) | 0.98 | HOMO → LUMO (99%) | ||
| T1-H1 | 1.53 | 853 (1.45) | 1.46 | HOMO → LUMO (99%) | 1002 (1.24) | 1.35 |
| T1-H2 | 1.40 | 907 (1.37) | 1.27 | HOMO → LUMO (99%) | 1071 (1.16) | 1.26 |
| T2-H1 | 1.43 | 891 (1.39) | 1.43 | HOMO → LUMO (99%) | ||
| T2-H2 | 1.35 | 932 (1.33) | 1.28 | HOMO → LUMO (99%) | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cador, A.; Kahlal, S.; Richards, G.J.; Halet, J.-F.; Hill, J.P. Protic Processes in an Extended Pyrazinacene: The Case of Dihydrotetradecaazaheptacene. Molecules 2024, 29, 2407. https://doi.org/10.3390/molecules29102407
Cador A, Kahlal S, Richards GJ, Halet J-F, Hill JP. Protic Processes in an Extended Pyrazinacene: The Case of Dihydrotetradecaazaheptacene. Molecules. 2024; 29(10):2407. https://doi.org/10.3390/molecules29102407
Chicago/Turabian StyleCador, Aël, Samia Kahlal, Gary J. Richards, Jean-François Halet, and Jonathan P. Hill. 2024. "Protic Processes in an Extended Pyrazinacene: The Case of Dihydrotetradecaazaheptacene" Molecules 29, no. 10: 2407. https://doi.org/10.3390/molecules29102407
APA StyleCador, A., Kahlal, S., Richards, G. J., Halet, J.-F., & Hill, J. P. (2024). Protic Processes in an Extended Pyrazinacene: The Case of Dihydrotetradecaazaheptacene. Molecules, 29(10), 2407. https://doi.org/10.3390/molecules29102407