
Metabolism, Disposition, Excretion, and Potential Transporter Inhibition of 7–16, an Improving 5-HT2A Receptor Antagonist and Inverse Agonist for Parkinson’s Disease

Abstract
1. Introduction
2. Results and Discussion
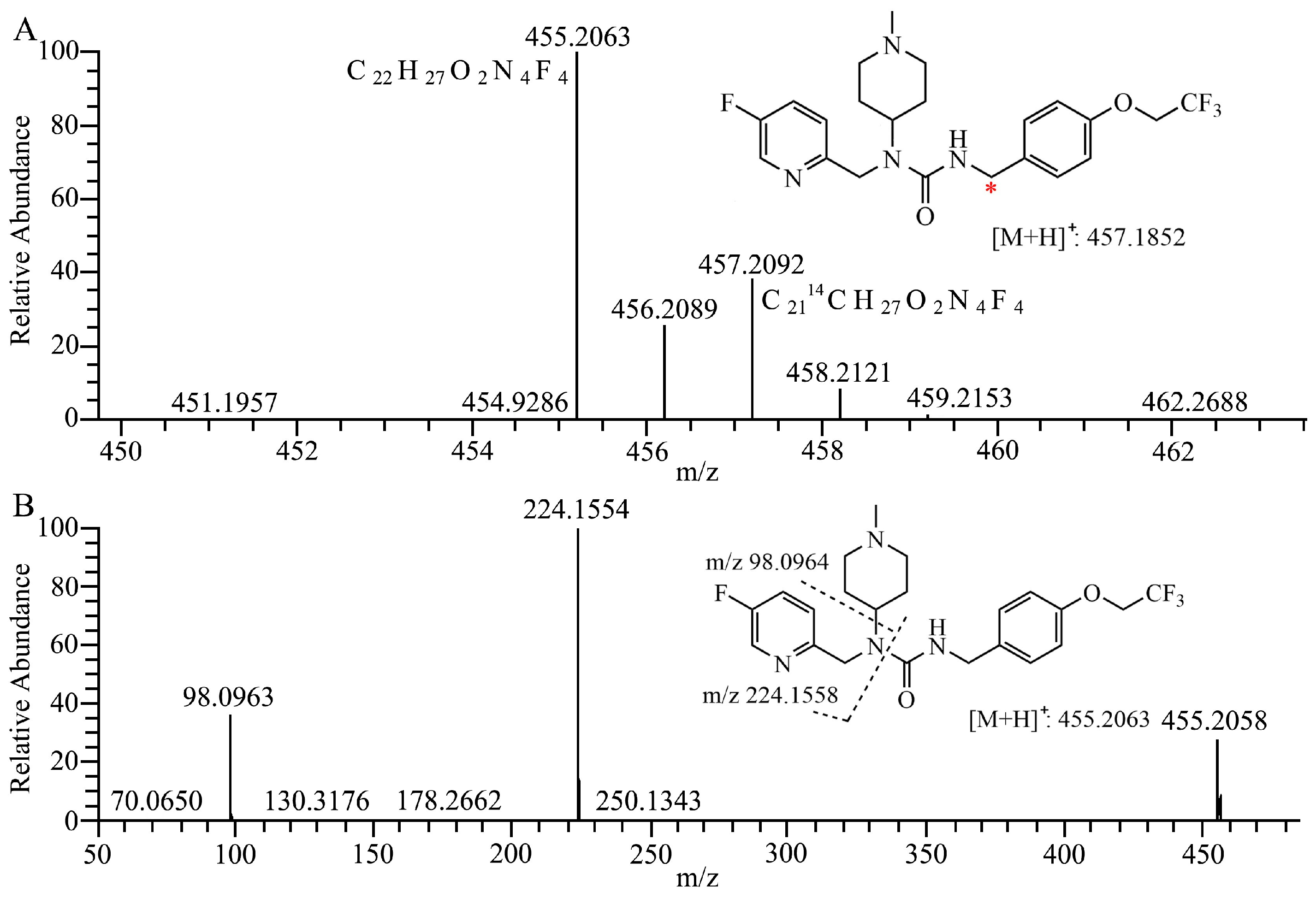
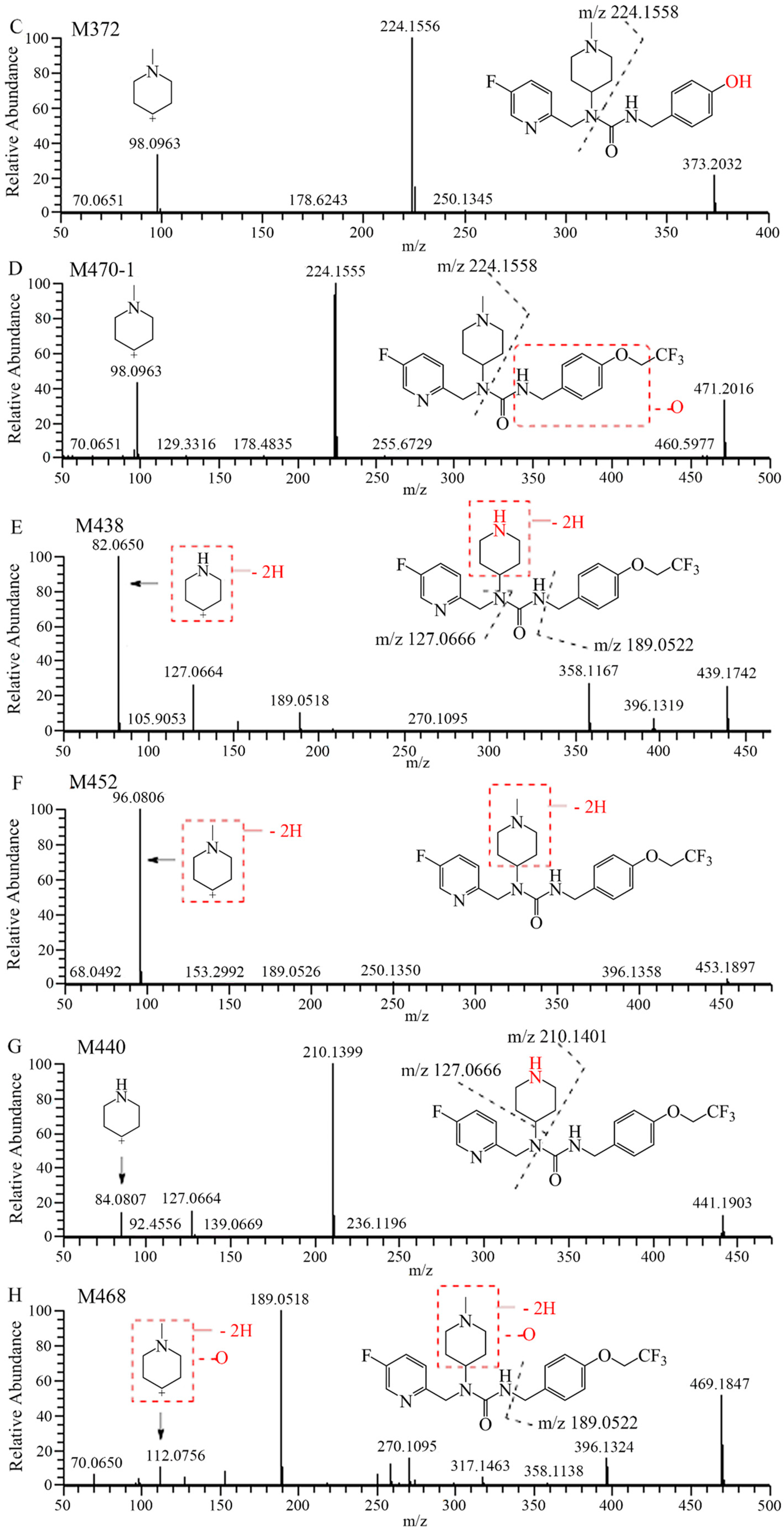
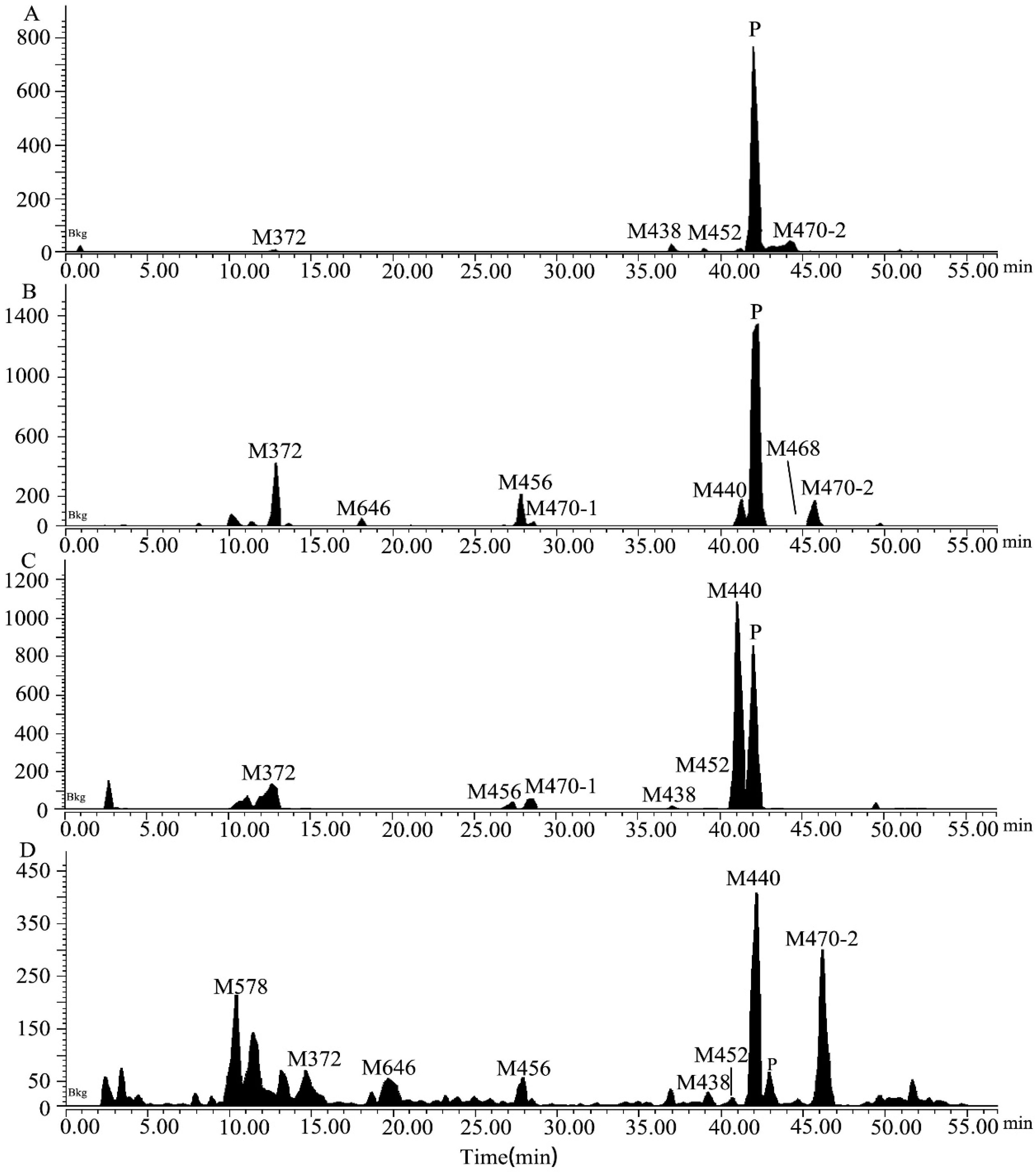
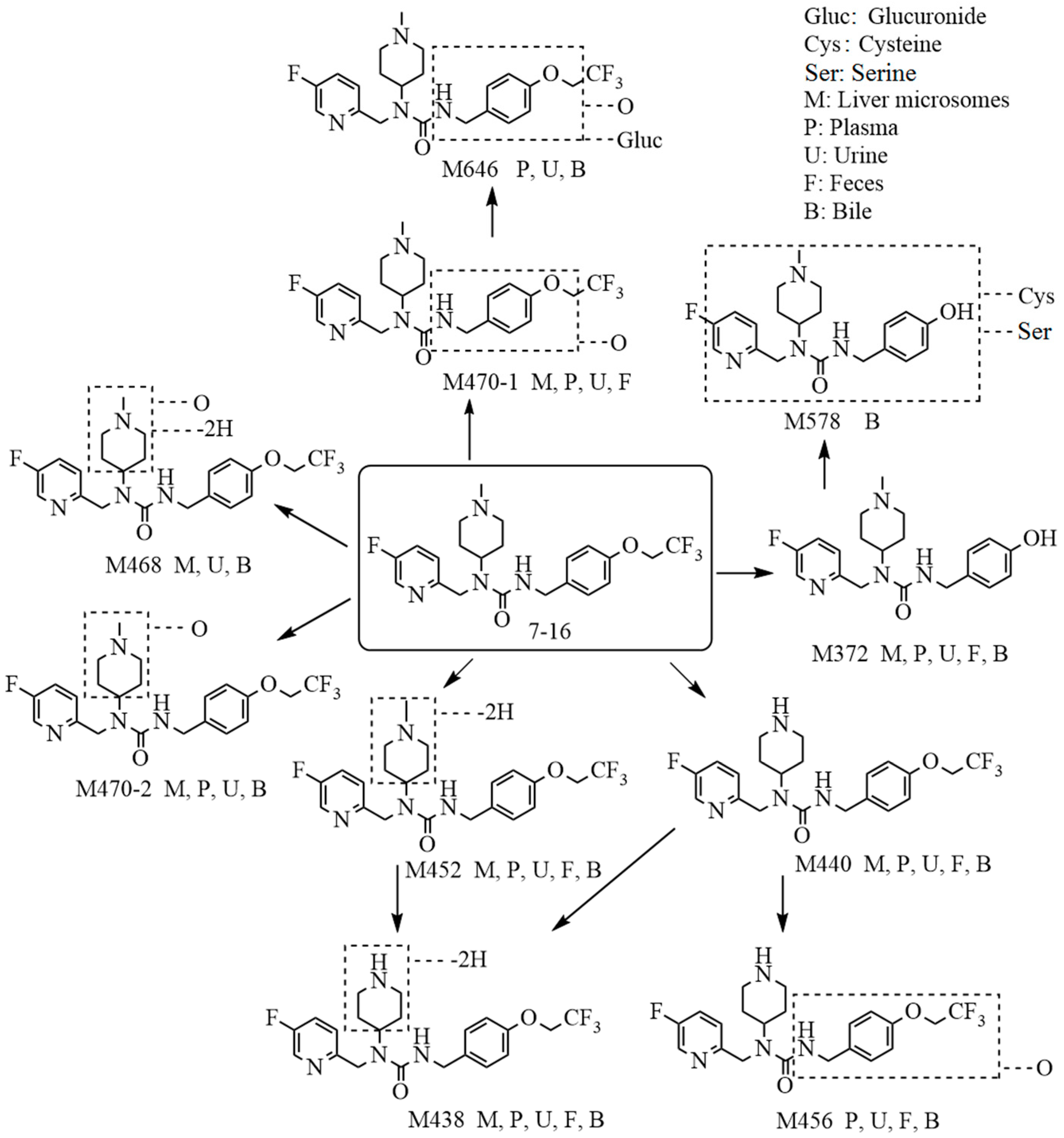
2.1. Metabolite Identification in Liver Microsomes
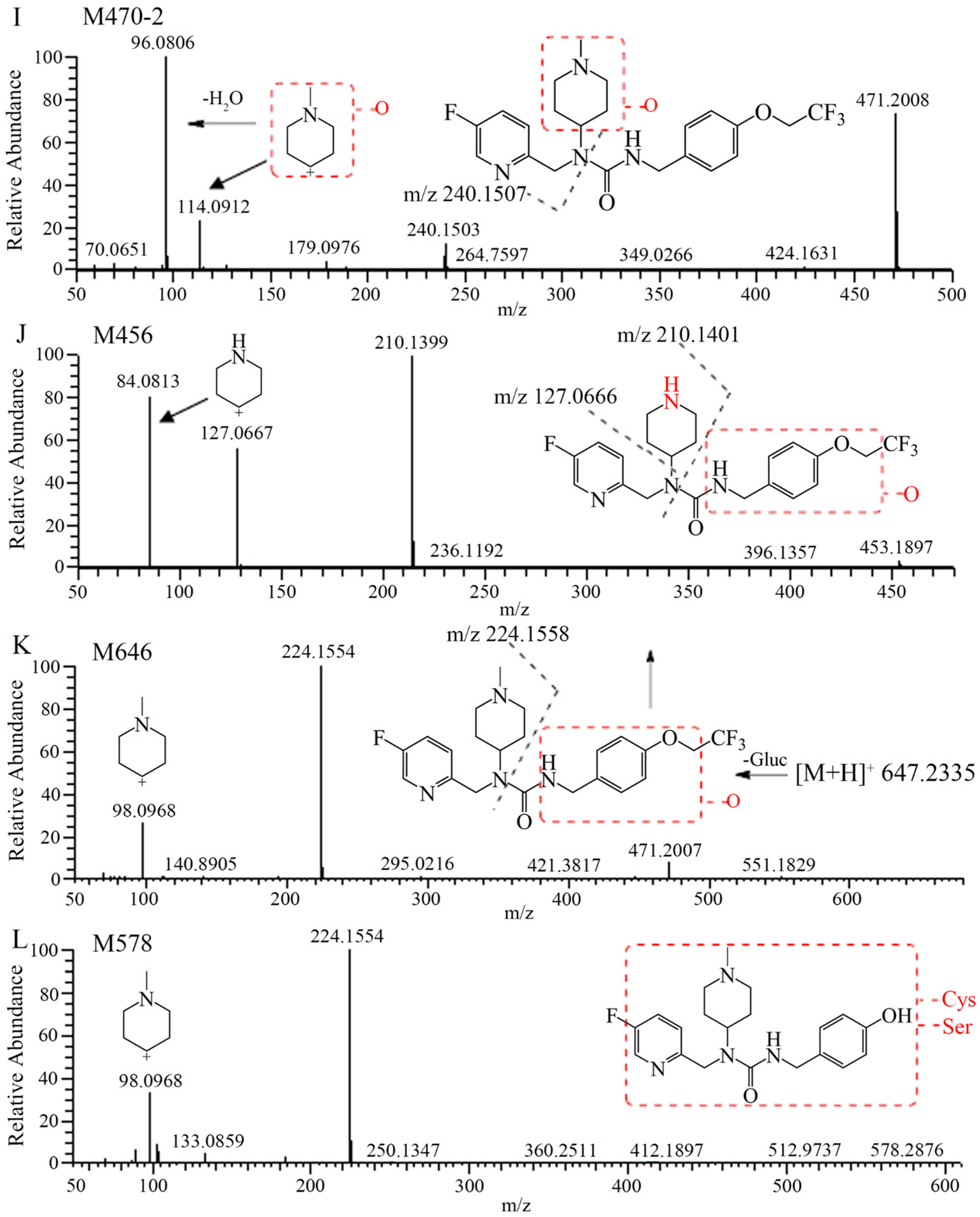
2.2. Metabolite Identification in Rat Plasma, Urine, Feces, and Bile
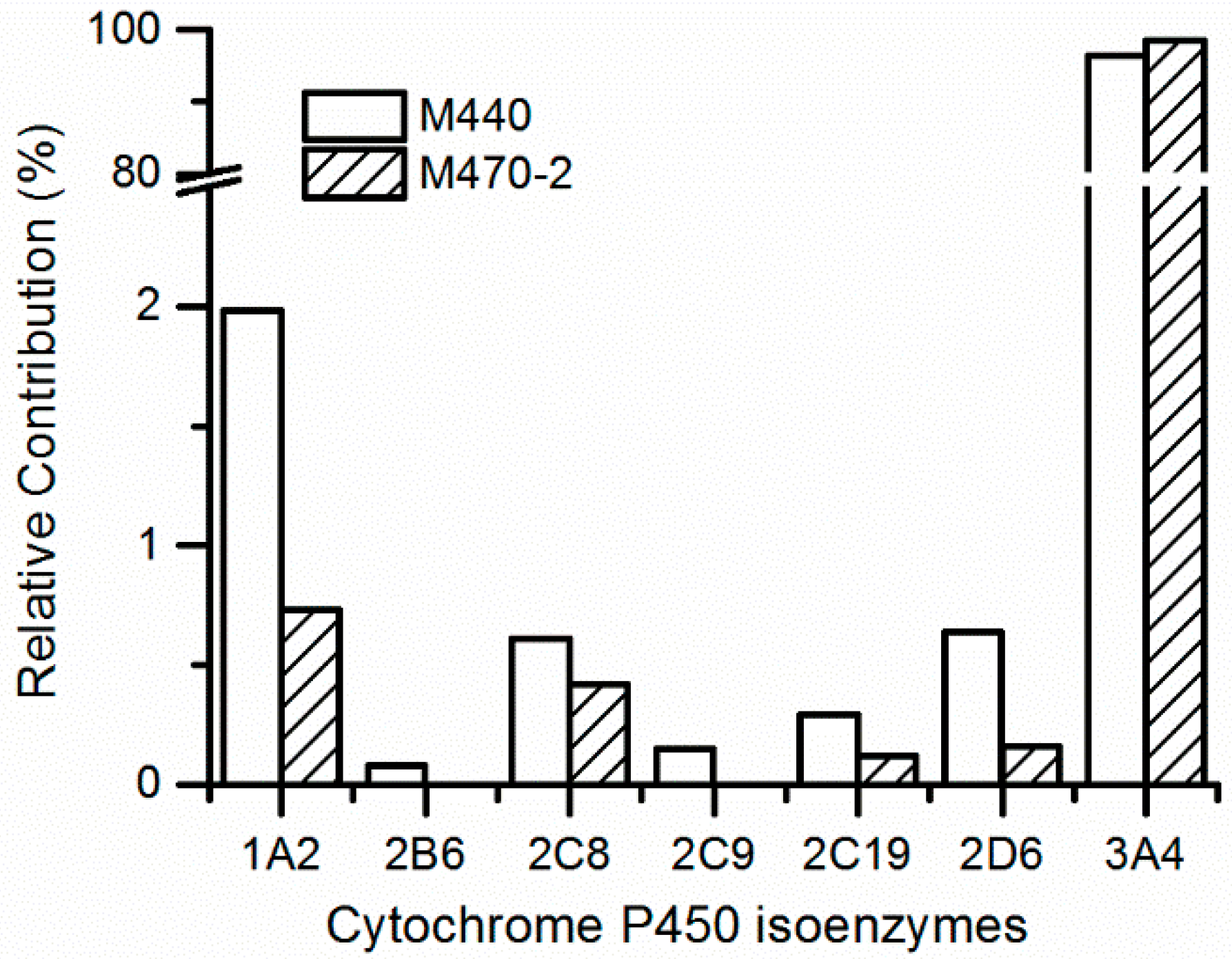
2.3. Metabolic Phenotype by P450 Enzymes
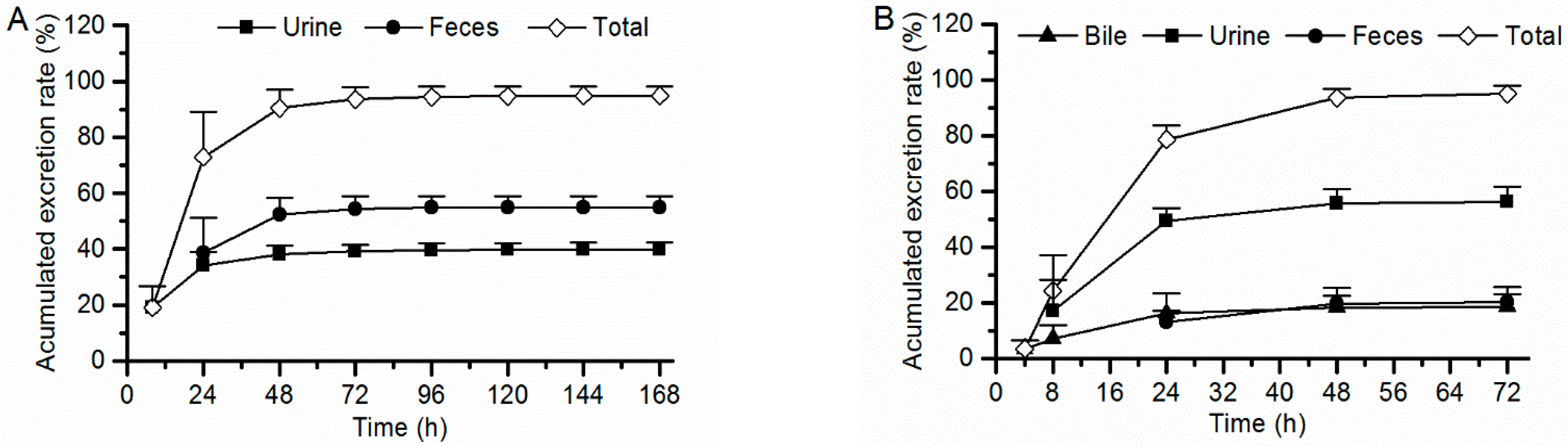
2.4. Excretion in Rats
2.5. Inhibitor Assay for the P-gp, OATP1B1, OATP1B3, OAT1, OAT3, and OCT2 Transporters
3. Materials and Methods
3.1. Materials
3.1.1. Chemicals and Reagents
3.1.2. Animals
3.2. Methods
3.2.1. Metabolite Identification in Liver Microsomes of Different Species
3.2.2. Metabolite Identification in Rat Plasma
3.2.3. Excretion in Rats and Metabolite Identification in Rat Urine, Feces, and Bile
3.2.4. Identification of P450 Isozymes Responsible for Metabolism
3.2.5. Transporter Inhibitor Assay
3.2.6. Data Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sveinbjornsdottir, S. The clinical symptoms of Parkinson’s disease. J. Neurochem. 2016, 139 (Suppl. S1), 318–324. [Google Scholar] [CrossRef]
- Fénelon, G.; Soulas, T.; Zenasni, F.; de Langavant, L.C. The changing face of Parkinson’s disease-associated psychosis: A cross-sectional study based on the new NINDS-NIMH criteria. Mov. Disord. 2010, 25, 763–766. [Google Scholar] [CrossRef]
- Combs, B.L.; Cox, A.G. Update on the treatment of Parkinson’s disease psychosis: Role of pimavanserin. Neuropsychiatr. Dis. Treat. 2017, 13, 737–744. [Google Scholar] [CrossRef]
- Hunter, N.S.; Anderson, K.C.; Cox, A. Pimavanserin. Drugs Today 2015, 51, 645–652. [Google Scholar] [CrossRef]
- Hacksell, U.; Burstein, E.S.; McFarland, K.; Mills, R.G.; Williams, H. On the discovery and development of pimavanserin: A novel drug candidate for Parkinson’s psychosis. Neurochem. Res. 2014, 39, 2008–2017. [Google Scholar] [CrossRef]
- Ma, M.; Yang, Y.; Du, G.; Dai, Y.; Zhu, X.; Wang, W.; Xu, H.; Zhang, J.; Zheng, L.; Zou, F.; et al. Improving the treatment of Parkinson’s disease: Structure-based development of novel 5-HT2A receptor antagonists/inverse agonists. Eur. J. Med. Chem. 2022, 234, 114246. [Google Scholar] [CrossRef]
- Ge, Y.; Yu, Y.; Zhang, Y.; Li, X.; Liu, Q. Characterization of the metabolite of AdipoRon in rat and human liver microsomes by ultra-high performance liquid chromatography combined with Q-Exactive Orbitrap tandem mass spectrometry. Biomed. Chromatogr. 2019, 33, e4645. [Google Scholar] [CrossRef]
- Coppola, P.; Andersson, A.; Cole, S. The importance of the human mass balance study in regulatory submissions. CPT Pharmacometrics Syst. Pharmacol. 2019, 8, 792–804. [Google Scholar] [CrossRef]
- Wu, W.; Chu, Y.; Wang, S.; Sun, X.; Zhang, J.; Wang, Y.; Chen, X. Investigation of metabolic profile of pimavanserin in rats by ultrahigh-performance liquid chromatography combined with Fourier transform ion cyclotron resonance mass spectrometry. Rapid Commun. Mass Spectrom. 2018, 32, 269–276. [Google Scholar] [CrossRef]
- Pánczél, J.; Schudok, M.; Schiell, M.; Riedel, J.; Kertesz, V. An Effective QWBA/UHPLC-MS/Tissue Punch Approach: Solving a Pharmacokinetic Issue via Quantitative Met-ID. Drug Metab. Lett. 2021, 14, 152–162. [Google Scholar] [CrossRef]
- Roffey, S.J.; Obach, R.S.; Gedge, J.I.; Smith, D.A. What is the objective of the mass balance study? A retrospective analysis of data in animal and human excretion studies employing radiolabeled drugs. Drug Metab. Rev. 2007, 39, 17–43. [Google Scholar] [CrossRef] [PubMed]
- Spracklin, D.K.; Chen, D.; Bergman, A.J.; Callegari, E.; Obach, R.S. Mini-review: Comprehensive drug disposition knowledge generated in the modern human radiolabeled ADME study. CPT Pharmacomet. Syst. Pharmacol. 2020, 9, 428–434. [Google Scholar] [CrossRef]
- Jala, A.; Ponneganti, S.; Vishnubhatla, D.S.; Bhuvanam, G.; Mekala, P.R.; Varghese, B.; Radhakrishnanand, P.; Adela, R.; Murty, U.S.; Borkar, R.M. Transporter-mediated drug-drug interactions: Advancement in models, analytical tools, and regulatory perspective. Drug Metab. Rev. 2021, 53, 285–320. [Google Scholar] [CrossRef]
- Rendic, S.P. Metabolism and interactions of Ivermectin with human cytochrome P450 enzymes and drug transporters, possible adverse and toxic effects. Arch. Toxicol. 2021, 95, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yu, D.; Zhu, X.; Du, G.; Wang, W.; Zou, F.; Wang, H.; Zhang, R.; Ye, L.; Tian, J. Synthesis and analysis of dihydrotetrabenazine derivatives as novel vesicular monoamine transporter 2 inhibitors. Eur. J. Med. Chem. 2021, 224, 113718. [Google Scholar] [CrossRef]
- Hosea, N.A.; Collard, W.T.; Cole, S.; Maurer, T.S.; Fang, R.X.; Jones, H.; Kakar, S.M.; Nakai, Y.; Smith, B.J.; Webster, R.; et al. Prediction of human pharmacokinetics from preclinical information: Comparative accuracy of quantitative prediction approaches. J. Clin. Pharmacol. 2009, 49, 513–533. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.A.; Hurst, S.I.; Bauman, J.; Jones, B.C.; Hyland, R.; Gibbs, J.P.; Obach, R.S.; Ball, S.E. Reaction phenotyping in drug discovery: Moving forward with confidence? Curr. Drug Metab. 2003, 4, 527–534. [Google Scholar] [CrossRef]
- Yuan, X.; Lu, H.; Zhao, A.; Ding, Y.; Min, Q.; Wang, R. Transcriptional regulation of CYP3A4 by nuclear receptors in human hepatocytes under hypoxia. Drug Metab. Rev. 2020, 52, 225–234. [Google Scholar] [CrossRef]
- Lin, J.H.; Yamazaki, M. Role of P-glycoprotein in pharmacokinetics: Clinical implications. Clin. Pharmacokinet. 2003, 42, 59–98. [Google Scholar] [CrossRef]
- Giacomini, K.M.; Huang, S.M. Transporters in drug development and clinical pharmacology. Clin. Pharmacol. Ther. 2013, 94, 3–9. [Google Scholar] [CrossRef]
- Nakanishi, T.; Tamai, I. Interaction of drug or food with drug transporters in intestine and liver. Curr. Drug Metab. 2015, 16, 753–764. [Google Scholar] [CrossRef]
- Lepist, E.I.; Ray, A.S. Beyond drug-drug interactions: Effects of transporter inhibition on endobiotics, nutrients and toxins. Expert Opin. Drug Metab. Toxicol. 2017, 13, 1075–1087. [Google Scholar] [CrossRef]
- Wang, W.; Du, G.; Lin, S.; Liu, J.; Yang, H.; Yu, D.; Ye, L.; Zou, F.; Wang, H.; Zhang, R.; et al. (+)-9-Trifluoroethoxy-α-Dihydrotetrabenazine as a highly potent vesicular monoamine transporter 2 inhibitor for tardive dyskinesia. Front. Pharmacol. 2021, 12, 770377. [Google Scholar] [CrossRef]
- Hop, C.E.; Wang, Z.; Chen, Q.; Kwei, G. Plasma-pooling methods to increase throughput for in vivo pharmacokinetic screening. J. Pharm. Sci. 1998, 87, 901–903. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | RT | Theoretical | Experimental | Error | Formula | Relative Abundance, UV Area% | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Min | Mass m/z | Mass m/z | ppm | [M + H]+ | Human | Mouse | Rat | Dog | Monkey | |
| M372 | 9.91 | 373.2034 | 373.2025 | −2.4 | C20H26FN4O2 | 0.60 | 0.61 | 1.10 | 1.73 | 2.52 |
| M470-1 | 17.34 | 471.2014 | 471.2003 | −2.3 | C22H27F4N4O3 | + | + | + | + | + |
| M438 | 19.85 | 439.1752 | 439.1743 | −2.0 | C21H23O2N4F4 | 0.20 | 1.06 | 0.99 | 1.53 | 0.76 |
| M452 | 20.32 | 453.1908 | 453.1898 | −2.2 | C22H25F4N4O2 | 0.32 | 1.13 | 1.92 | 1.14 | 0.43 |
| M440 | 21.02 | 441.1908 | 441.1903 | −1.1 | C21H25O2N4F4 | 0.78 | 0.39 | 1.29 | 0.47 | 1.73 |
| 7–16 | 21.15 | 455.2065 | 455.2054 | −2.4 | C22H27F4N4O2 | 95.2 | 91.3 | 86.1 | 77.2 | 90.6 |
| M468 | 21.27 | 469.1857 | 469.1847 | −2.1 | C22H25F4N4O3 | 0.40 | 0.48 | + | 0.59 | 1.11 |
| M470-2 | 22.02 | 471.2014 | 471.2007 | −1.5 | C22H27F4N4O3 | 2.53 | 5.05 | 8.63 | 17.3 | 2.81 |
| No. | RT | Theoretical | Experimental | Error | Formula | TRA% a (F/M), or Dose% b | |||
|---|---|---|---|---|---|---|---|---|---|
| Min | Mass m/z | Mass m/z | ppm | [M + H]+ | Plasma | Urine | Feces | Bile | |
| M578 | 11.27 | 581.2428 | 581.2430 | 0.3 | C2514CH36FN6O6S | −/− | −/− | −/− | 1.60/2.32 |
| M372 | 13.42 | 375.2066 | 375.2064 | −0.5 | C1914CH26O2N4F | 1.11/2.50 | 4.34/5.75 | 4.97/5.42 | 0.41/0.83 |
| M646 | 18.70 | 649.2367 | 649.2362 | −0.8 | C2714CH35O9N4F4 | +/0.05 | 0.02/0.44 | −/− | 1.08/1.06 |
| M456 | 27.92 | 459.1889 | 459.1887 | −0.4 | C2014CH25F4N4O3 | +/0.20 | 2.12/4.94 | 0.72/0.42 | +/0.05 |
| M470-1 | 29.15 | 473.2046 | 473.2042 | −0.8 | C2114CH27O3N4F4 | +/1.20 | 0.42/2.41 | 1.35/1.41 | −/− |
| M438 | 37.86 | 441.1784 | 441.1784 | −0.5 | C2014CH23O2N4F4 | 2.51/1.50 | 0.02/0.02 | 0.32/0.58 | 0.10/0.25 |
| M452 | 39.79 | 455.1940 | 455.1938 | −0.4 | C2114CH25O2N4F4 | 1.17/1.50 | +/+ | 0.23/0.15 | 0.05/0.06 |
| M440 | 41.79 | 443.1940 | 443.1935 | −1.1 | C2014CH25O2N4F4 | 1.40/3.70 | 2.66/5.77 | 21.1/24.2 | 2.71/3.83 |
| [14C]7–16 | 42.82 | 457.2097 | 457.2092 | −1.1 | C2114CH27O2N4F4 | 81.9/57.3 | 23.6/9.79 | 16.8/5.29 | 0.76/0.53 |
| M468 | 44.98 | 471.1889 | 471.1884 | −1.1 | C2014CH25O3N4F4 | ND | 0.02/0.58 | −/− | 0.24/0.20 |
| M470-2 | 46.42 | 473.2046 | 473.2046 | −0.0 | C2114CH27O3N4F4 | 8.63/27.2 | 2.50/1.21 | −/− | 2.39/2.86 |
| Transporters | Substrates | Inhibition IC50 (μM) | |
|---|---|---|---|
| 7–16 | Control (Positive Inhibitor) | ||
| P-gp | Digoxin | 30.6 | 1.57 (Verapamil) |
| OATP1B1 | Estrone 3-sulfate | >100 | 0.0556 (Cyclosporin A) |
| OATP1B3 | β-Estradiol 17-(β-d-glucuronide) | 89.5 | 0.734 (Ritonavir) |
| OAT1 | p-Aminohippurate | >100 | 3.80 (Probenecid) |
| OAT3 | Estrone 3-sulfate | >100 | 1.43 (Probenecid) |
| OCT2 | Metformin | 72.5 | 11.3 (Verapamil) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, Z.; Wang, W.; Yang, H.; Zhao, F.; Sha, C.; Mi, W.; Yin, S.; Wang, H.; Tian, J.; Ye, L. Metabolism, Disposition, Excretion, and Potential Transporter Inhibition of 7–16, an Improving 5-HT2A Receptor Antagonist and Inverse Agonist for Parkinson’s Disease. Molecules 2024, 29, 2184. https://doi.org/10.3390/molecules29102184
Hu Z, Wang W, Yang H, Zhao F, Sha C, Mi W, Yin S, Wang H, Tian J, Ye L. Metabolism, Disposition, Excretion, and Potential Transporter Inhibition of 7–16, an Improving 5-HT2A Receptor Antagonist and Inverse Agonist for Parkinson’s Disease. Molecules. 2024; 29(10):2184. https://doi.org/10.3390/molecules29102184
Chicago/Turabian StyleHu, Zhengping, Wenyan Wang, Huijie Yang, Fengjuan Zhao, Chunjie Sha, Wei Mi, Shuying Yin, Hongbo Wang, Jingwei Tian, and Liang Ye. 2024. "Metabolism, Disposition, Excretion, and Potential Transporter Inhibition of 7–16, an Improving 5-HT2A Receptor Antagonist and Inverse Agonist for Parkinson’s Disease" Molecules 29, no. 10: 2184. https://doi.org/10.3390/molecules29102184
APA StyleHu, Z., Wang, W., Yang, H., Zhao, F., Sha, C., Mi, W., Yin, S., Wang, H., Tian, J., & Ye, L. (2024). Metabolism, Disposition, Excretion, and Potential Transporter Inhibition of 7–16, an Improving 5-HT2A Receptor Antagonist and Inverse Agonist for Parkinson’s Disease. Molecules, 29(10), 2184. https://doi.org/10.3390/molecules29102184