
α-Amido Trifluoromethyl Xanthates: A New Class of RAFT/MADIX Agents

 ,
,
Abstract
1. Introduction
2. Results and Discussion
2.1. Xanthate Synthesis
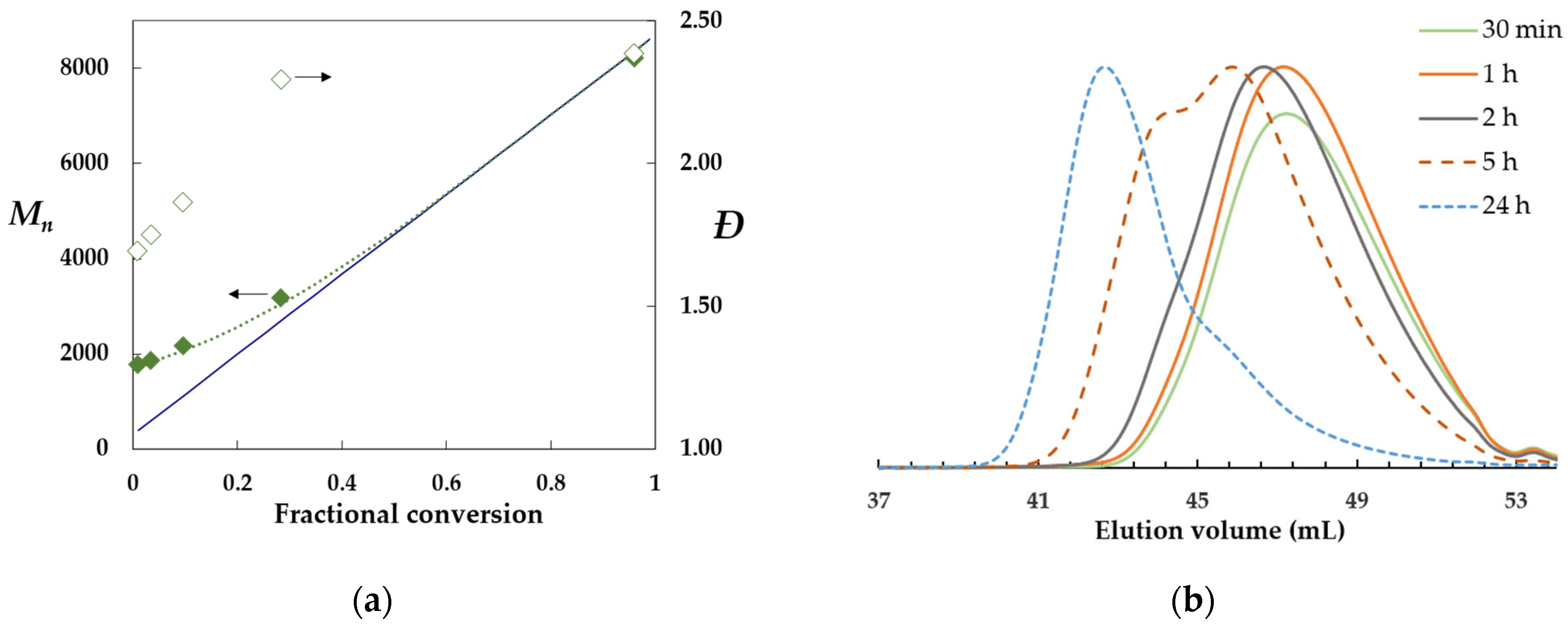
2.2. Structure–Reactivity Relationship of α-Amido Trifluoromethyl Xanthates
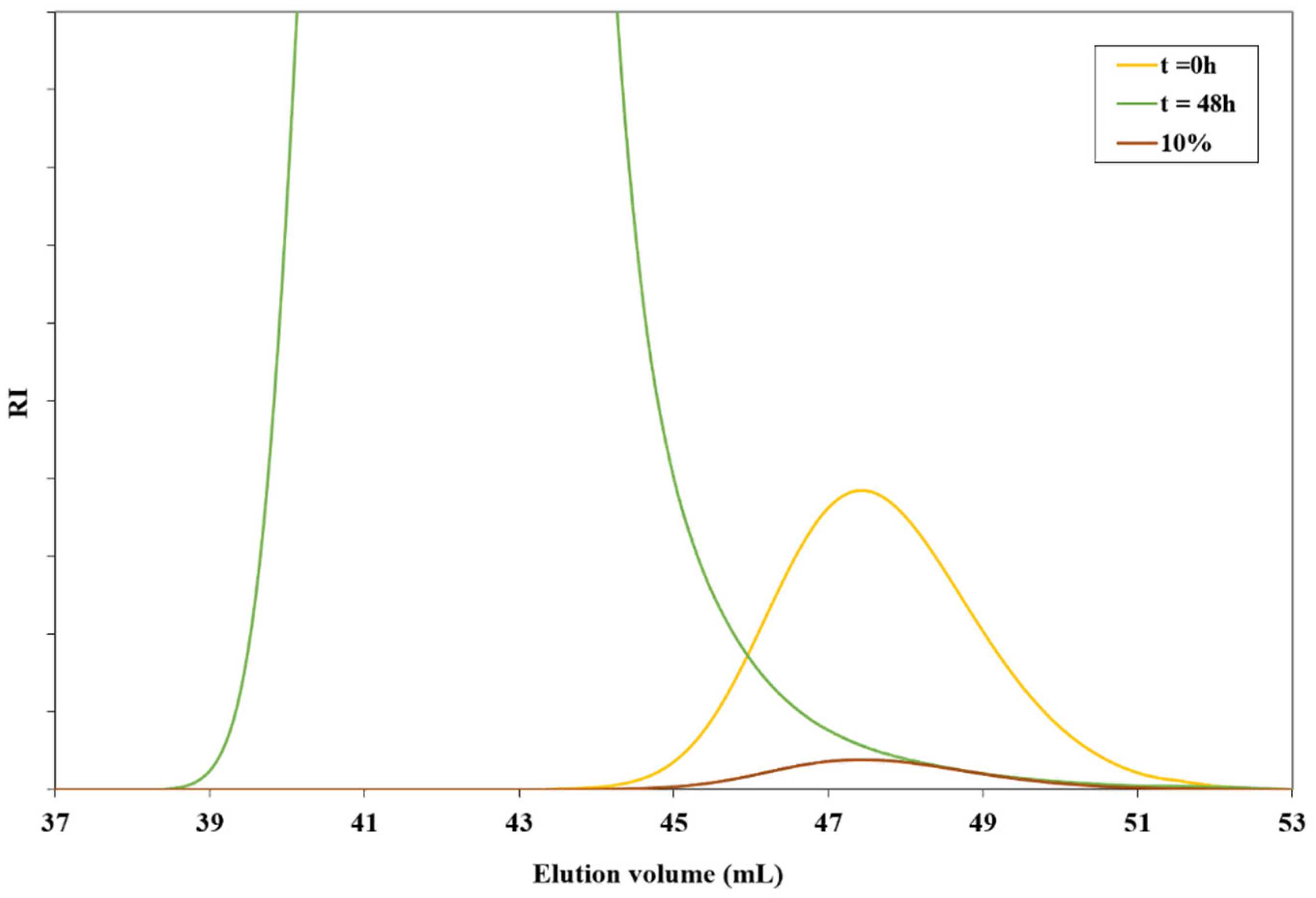
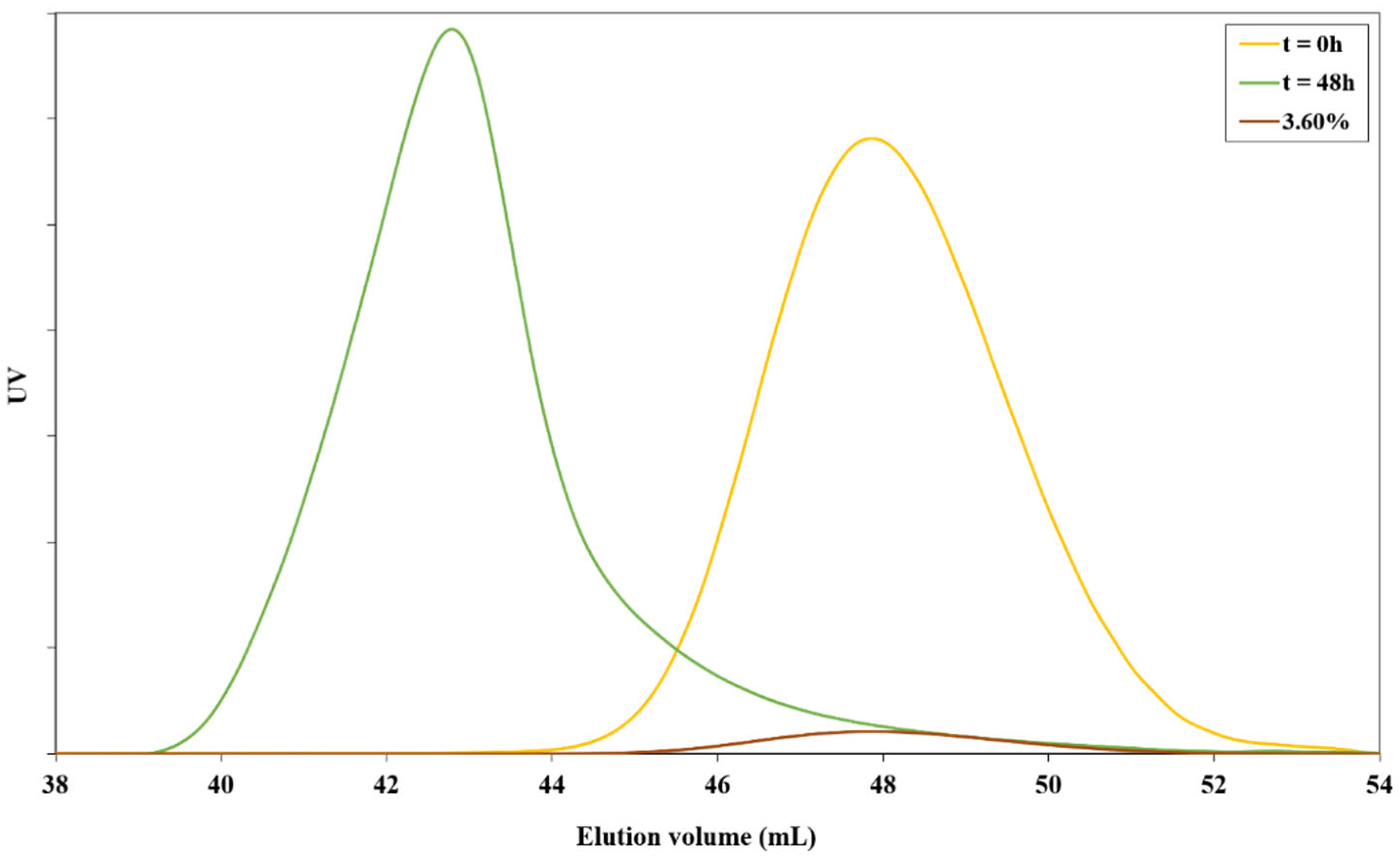
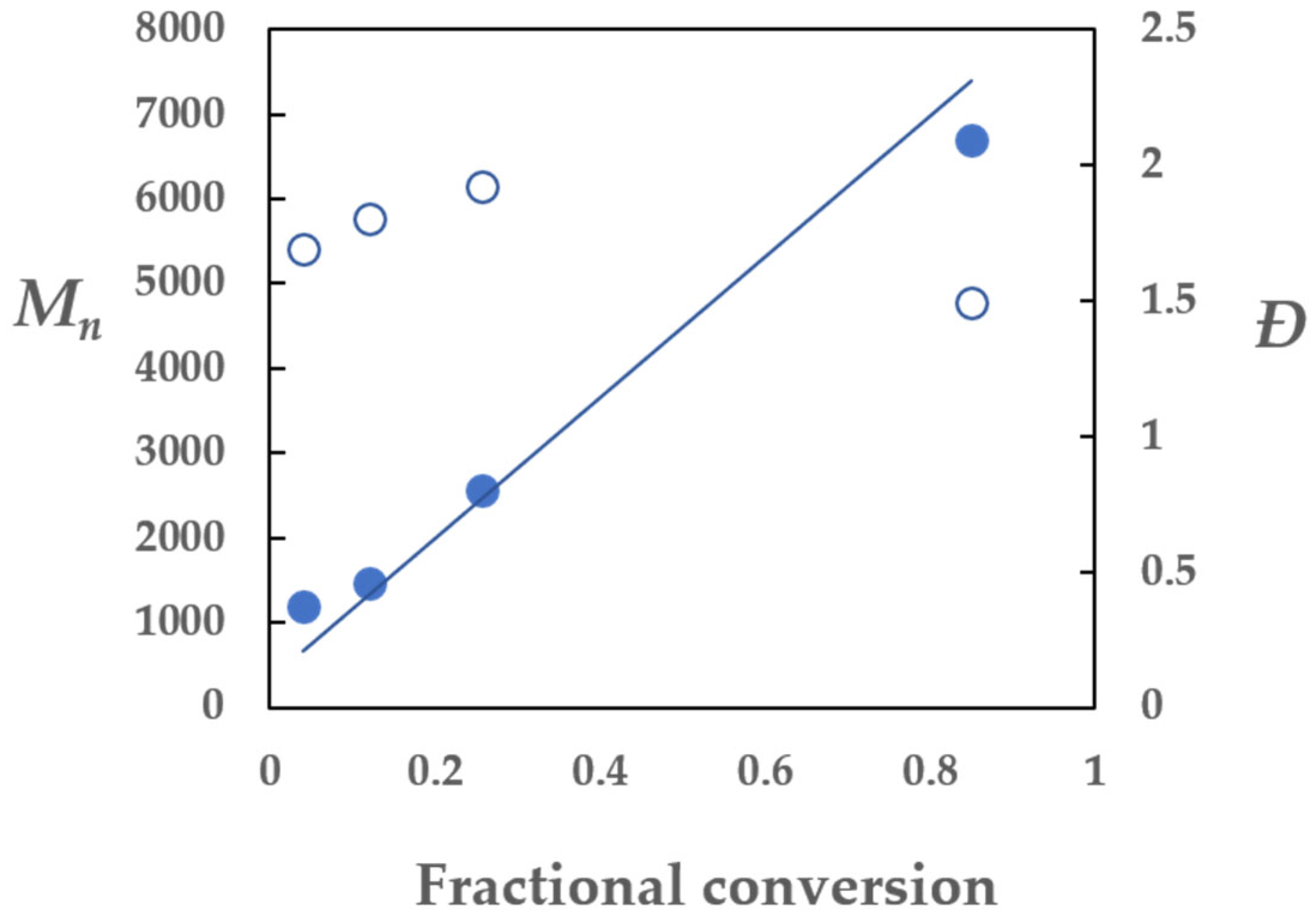
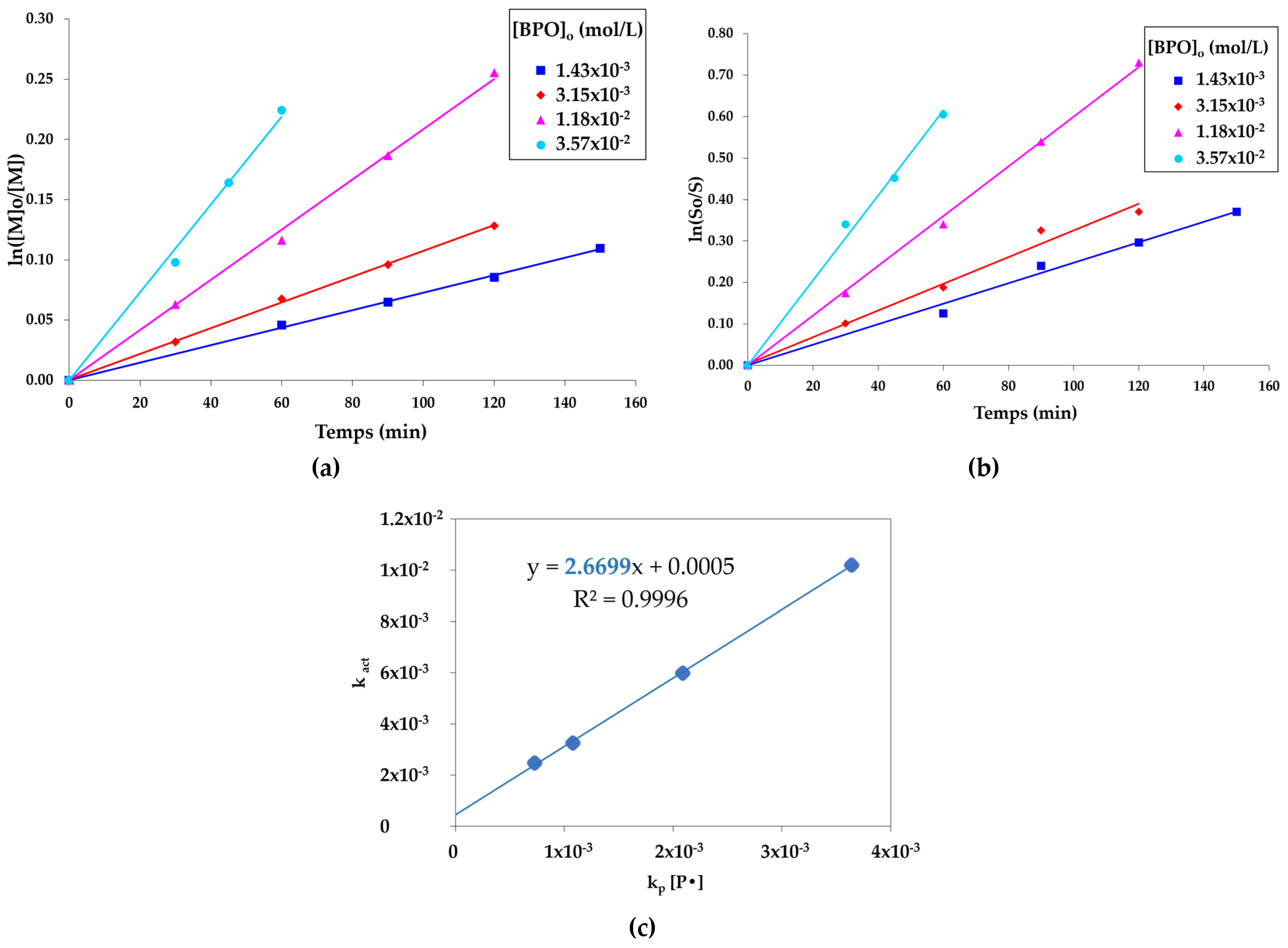
2.3. Determination of Cex for Z=OCH2CF3
3. Materials and Methods
3.1. Materials
3.2. Synthesis of the α-Amido Trifluoromethyl O-ethyl Xanthates
3.3. Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Calculation of the Fraction of Dead Chains Fdead in PSt-14
References
- Le, T.P.; Moad, G.; Rizzardo, E.; Thang, S.H. Polymerization with Living Characteristics 1998. E.I. Du Pont de Nemours and Company WO98/01478, 15 January 1998. [Google Scholar]
- Corpart, P.; Charmot, D.; Biadatti, T.; Zard, S.; Michelet, D. Procédé de Synthèse de Polymères à Blocs Par Polymérisation Radicalaire Contrôlée 1998. Rhodia Chimie WO98/58974, 30 December 1998. [Google Scholar]
- Moad, G.; Rizzardo, E. A 20th Anniversary Perspective on the Life of RAFT (RAFT Coming of Age). Polym. Int. 2020, 69, 658–661. [Google Scholar] [CrossRef]
- Zard, S.Z. Discovery of the RAFT/MADIX Process: Mechanistic Insights and Polymer Chemistry Implications. Macromolecules 2020, 53, 8144–8159. [Google Scholar] [CrossRef]
- Keddie, D.J.; Moad, G.; Rizzardo, E.; Thang, S.H. RAFT Agent Design and Synthesis. Macromolecules 2012, 45, 5321–5342. [Google Scholar] [CrossRef]
- Nakabayashi, K.; Mori, H. Recent Progress in Controlled Radical Polymerization of N-Vinyl Monomers. Eur. Polym. J. 2013, 49, 2808–2838. [Google Scholar] [CrossRef]
- Yan, Y.; Zhang, W.; Qiu, Y.; Zhang, Z.; Zhu, J.; Cheng, Z.; Zhang, W.; Zhu, X. Universal Xanthate-Mediated Controlled Free Radical Polymerizations of the “Less Activated” Vinyl Monomers. J. Polym. Sci. Part Polym. Chem. 2010, 48, 5206–5214. [Google Scholar] [CrossRef]
- Wang, M.; Marty, J.-D.; Destarac, M. Xanthates in RAFT Polymerization. In RAFT Polymerization; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2021; pp. 493–548. ISBN 978-3-527-82135-8. [Google Scholar]
- Zard, S.Z. On the Trail of Xanthates: Some New Chemistry from an Old Functional Group. Angew. Chem. Int. Ed. Engl. 1997, 36, 672–685. [Google Scholar] [CrossRef]
- Destarac, M.; Bzducha, W.; Taton, D.; Gauthier-Gillaizeau, I.; Zard, S.Z. Xanthates as Chain-Transfer Agents in Controlled Radical Polymerization (MADIX): Structural Effect of the O-Alkyl Group. Macromol. Rapid Commun. 2002, 23, 1049–1054. [Google Scholar] [CrossRef]
- Quémener, D.; Davis, T.P.; Barner-Kowollik, C.; Stenzel, M.H. RAFT and Click Chemistry: A Versatile Approach to Well-Defined Block Copolymers. Chem. Commun. 2006, 5051–5053. [Google Scholar] [CrossRef]
- Chong, Y.K.; Krstina, J.; Le, T.P.T.; Moad, G.; Postma, A.; Rizzardo, E.; Thang, S.H. Thiocarbonylthio Compounds [SC(Ph)S−R] in Free Radical Polymerization with Reversible Addition-Fragmentation Chain Transfer (RAFT Polymerization). Role of the Free-Radical Leaving Group (R). Macromolecules 2003, 36, 2256–2272. [Google Scholar] [CrossRef]
- Wood, M.R.; Duncalf, D.J.; Rannard, S.P.; Perrier, S. Selective One-Pot Synthesis of Trithiocarbonates, Xanthates, and Dithiocarbamates for Use in RAFT/MADIX Living Radical Polymerizations. Org. Lett. 2006, 8, 553–556. [Google Scholar] [CrossRef]
- Lai, J.T.; Shea, R. Controlled Radical Polymerization by Carboxyl- and Hydroxyl-Terminated Dithiocarbamates and Xanthates. J. Polym. Sci. Part Polym. Chem. 2006, 44, 4298–4316. [Google Scholar] [CrossRef]
- Bouhadir, G.; Legrand, N.; Quiclet-Sire, B.; Zard, S.Z. A New Practical Synthesis of Tertiary S-Alkyl Dithiocarbonates and Related Derivatives. Tetrahedron Lett. 1999, 40, 277–280. [Google Scholar] [CrossRef]
- Destarac, M.; Brochon, C.; Catala, J.-M.; Wilczewska, A.; Zard, S.Z. Macromolecular Design via the Interchange of Xanthates (MADIX): Polymerization of Styrene with O-Ethyl Xanthates as Controlling Agents. Macromol. Chem. Phys. 2002, 203, 2281–2289. [Google Scholar] [CrossRef]
- Adamy, M.; van Herk, A.M.; Destarac, M.; Monteiro, M.J. Influence of the Chemical Structure of MADIX Agents on the RAFT Polymerization of Styrene. Macromolecules 2003, 36, 2293–2301. [Google Scholar] [CrossRef]
- Destarac, M.; Taton, D.; Zard, S.Z.; Saleh, T.; Six, Y. On the Importance of Xanthate Substituents in the MADIX Process. In Advances in Controlled/Living Radical Polymerization; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2003; Volume 854, pp. 536–550. ISBN 978-0-8412-3854-1. [Google Scholar]
- Gagosz, F.; Zard, S.Z. A Direct Approach to α-Trifluoromethylamines. Org. Lett. 2003, 5, 2655–2657. [Google Scholar] [CrossRef]
- Mayo, F.R. Chain Transfer in the Polymerization of Styrene: The Reaction of Solvents with Free Radicals1. J. Am. Chem. Soc. 1943, 65, 2324–2329. [Google Scholar] [CrossRef]
- Müller, A.H.E.; Zhuang, R.; Yan, D.; Litvinenko, G. Kinetic Analysis of “Living” Polymerization Processes Exhibiting Slow Equilibria. 1. Degenerative Transfer (Direct Activity Exchange between Active and “Dormant” Species). Application to Group Transfer Polymerization. Macromolecules 1995, 28, 4326–4333. [Google Scholar] [CrossRef]
- Brochon, C.; Catala, J.-M. Kinetic Studies of Free Radical Polymerization of Styrene with O-Ethyl Xanthates: Account for Multimodal Distribution. Polym. Prep. (Am. Chem. Soc. Div. Polym. Chem.) 2002, 43, 303–304. [Google Scholar]
- Goto, A.; Terauchi, T.; Fukuda, T.; Miyamoto, T. Gel Permeation Chromatographic Determination of Activation Rate Constants in Nitroxide-Controlled Free Radical Polymerization, 1. Direct Analysis by Peak Resolution. Macromol. Rapid Commun. 1997, 18, 673–681. [Google Scholar] [CrossRef]
- Tournier, L. Novel Applications of Xanthate Radical Chemistry: Synthesis of Organofluorine Molecules, Cyanodienes and Unsaturated δ-Lactones, and Various Heterocycles. Ph.D. Thesis, Ecole Polytechnique, Palaiseau, France, 12 December 2005. [Google Scholar]

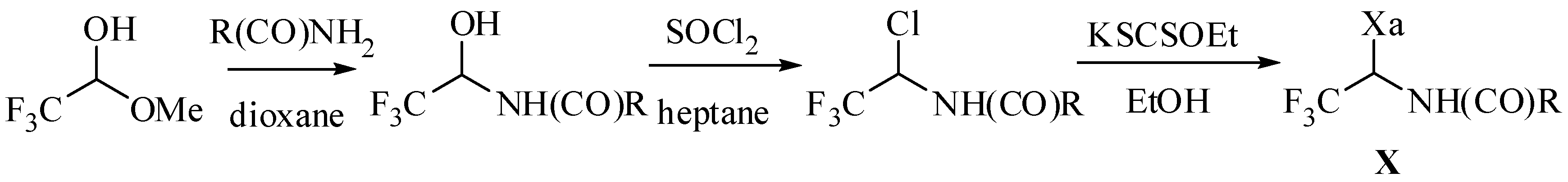


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| R | Alcohol (%) | Chloride (%) | Xanthate (%) |
|---|---|---|---|
| Me | 1 a (54) b | 5 (91) | 9 (87) |
| Bz | 2 (64) | 6 (71) | 10 (65) |
| tBu | 3 (40) | 7 (89) | 11 (79) |
| tBuO | 4 (36) | 8 (65) | 12 (52) |
| X | Ctr |
|---|---|
| 9 | 2.1 a (3.1) b |
| 10 | 2.5 (3.2) |
| 11 | 2.2 (2.9) |
| 13 | 6.1 (6.1) |
| 14 | (15) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Destarac, M.; Ruchmann-Sternchuss, J.; Van Gramberen, E.; Vila, X.; Zard, S.Z. α-Amido Trifluoromethyl Xanthates: A New Class of RAFT/MADIX Agents. Molecules 2024, 29, 2174. https://doi.org/10.3390/molecules29102174
Destarac M, Ruchmann-Sternchuss J, Van Gramberen E, Vila X, Zard SZ. α-Amido Trifluoromethyl Xanthates: A New Class of RAFT/MADIX Agents. Molecules. 2024; 29(10):2174. https://doi.org/10.3390/molecules29102174
Chicago/Turabian StyleDestarac, Mathias, Juliette Ruchmann-Sternchuss, Eric Van Gramberen, Xavier Vila, and Samir Z. Zard. 2024. "α-Amido Trifluoromethyl Xanthates: A New Class of RAFT/MADIX Agents" Molecules 29, no. 10: 2174. https://doi.org/10.3390/molecules29102174
APA StyleDestarac, M., Ruchmann-Sternchuss, J., Van Gramberen, E., Vila, X., & Zard, S. Z. (2024). α-Amido Trifluoromethyl Xanthates: A New Class of RAFT/MADIX Agents. Molecules, 29(10), 2174. https://doi.org/10.3390/molecules29102174