
A Triplet/Singlet Ground-State Switch via the Steric Inhibition of Conjugation in 4,6-Bis(trifluoromethyl)-1,3-phenylene Bisnitroxide

Abstract
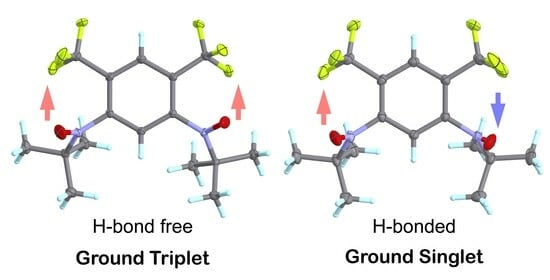
:
1. Introduction
2. Materials and Methods
3. Results
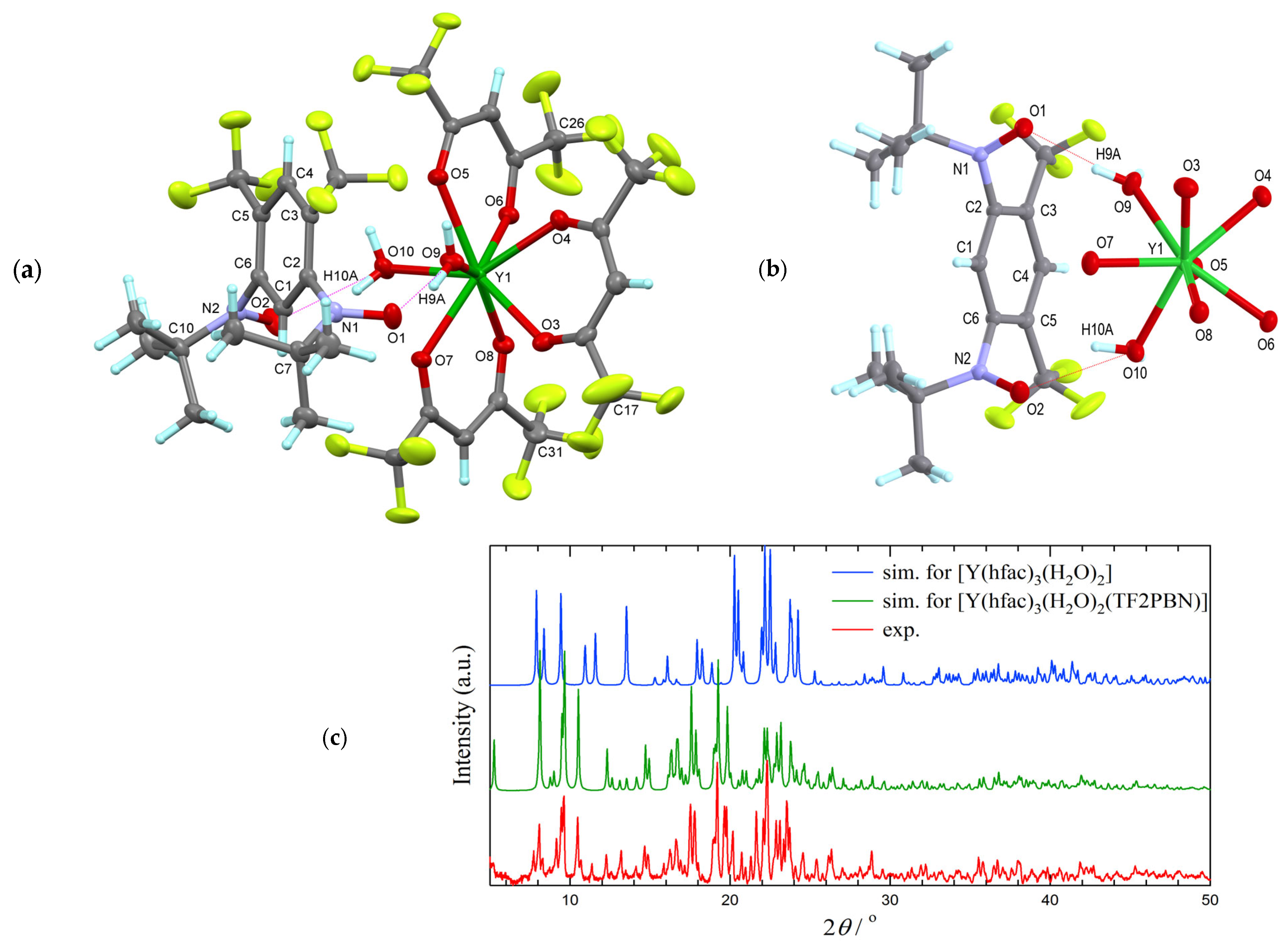
3.1. Synthesis and Structural Analysis
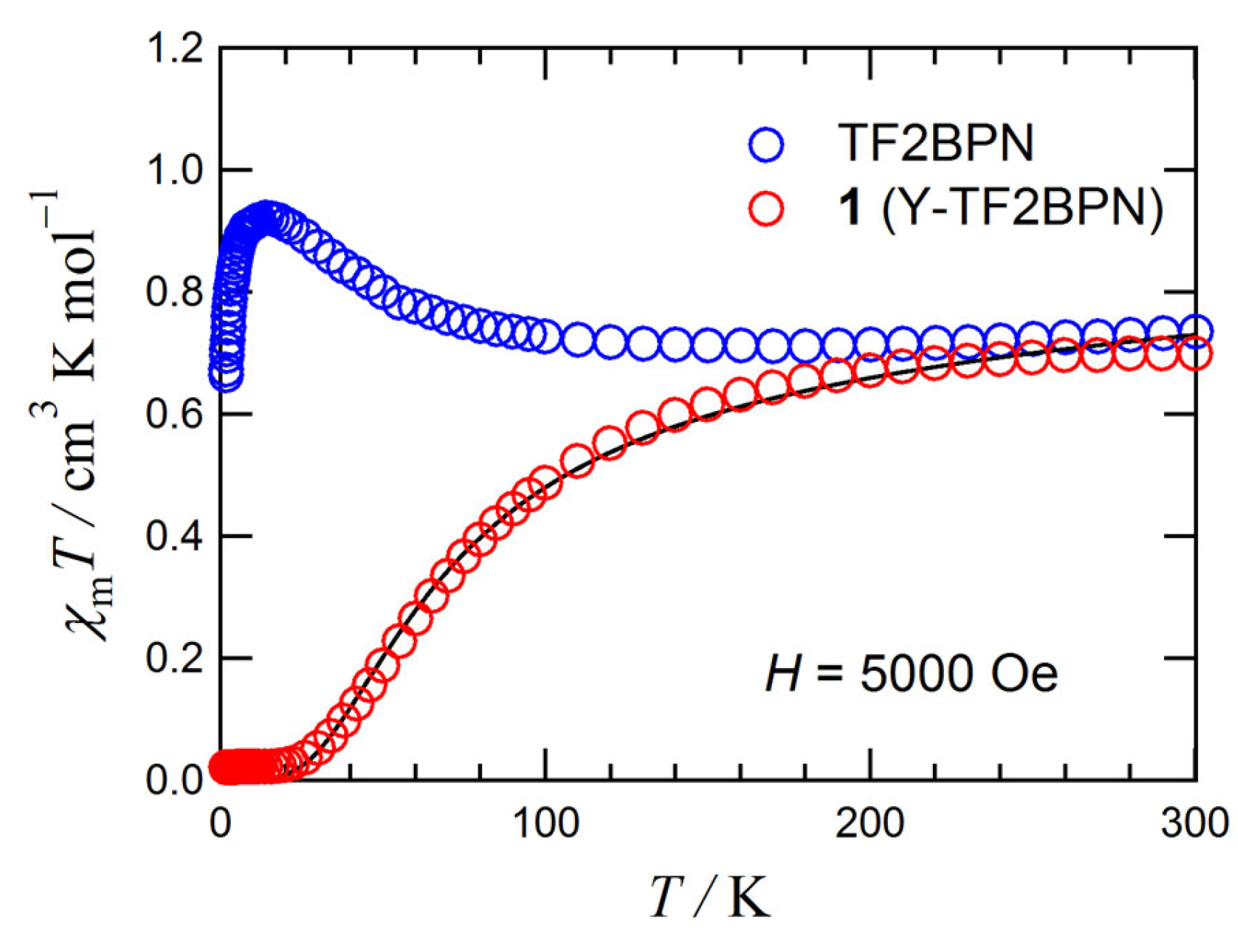
3.2. Magnetic Analysis
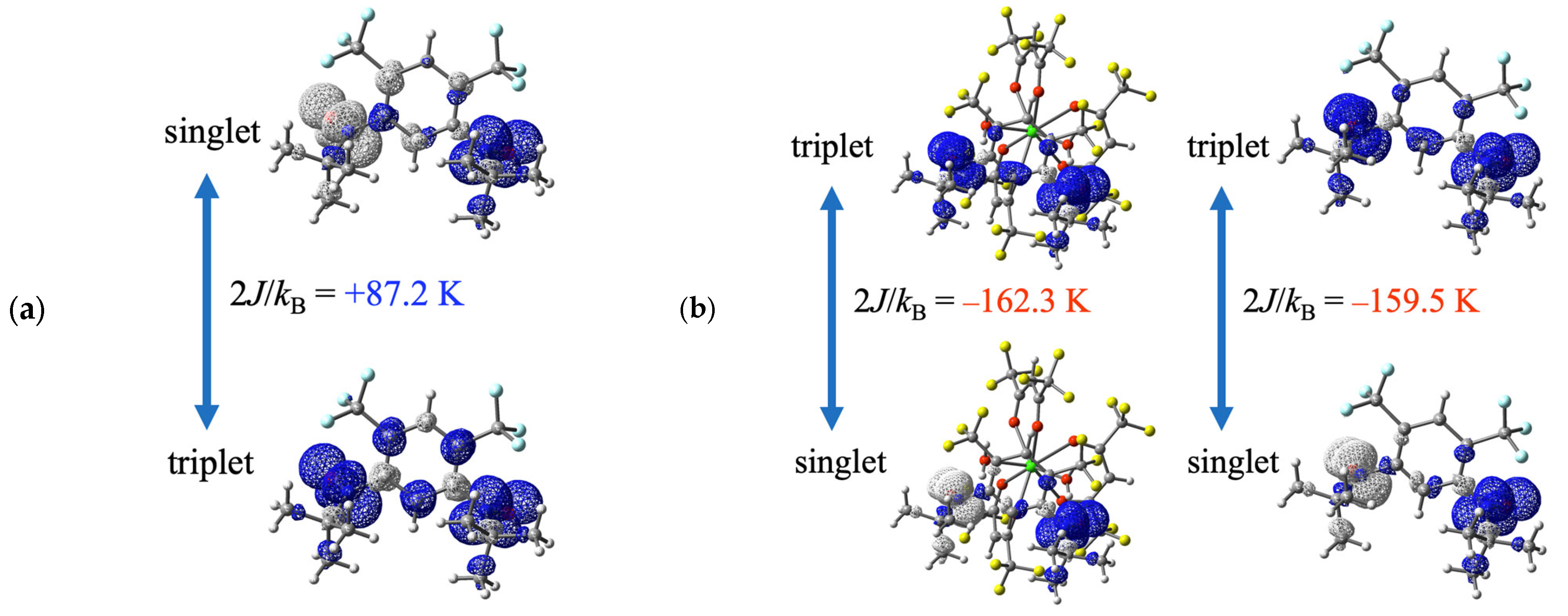
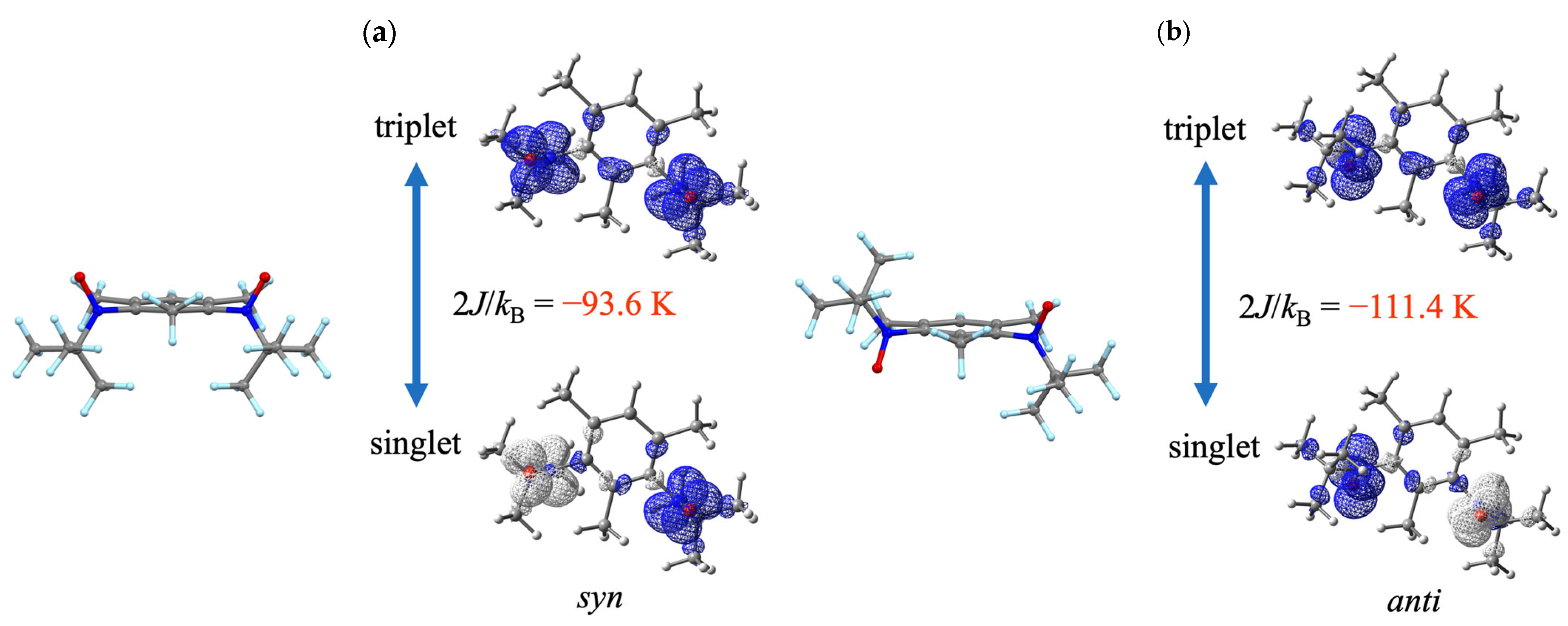
3.3. DFT Calculation Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Demir, S.; Jeon, I.R.; Long, J.R.; Harris, T.D. Radical ligand-containing single-molecule magnets. Coord. Chem. Rev. 2015, 289, 149–176. [Google Scholar] [CrossRef]
- Caneschi, A.; Gatteschi, D.; Sessoli, R.; Rey, P. Toward molecular magnets: The metal-radical approach. Acc. Chem. Res. 1989, 22, 392–398. [Google Scholar] [CrossRef]
- Tan, Y.; Hsu, S.-N.; Tahir, H.; Dou, L.; Savoie, B.M.; Boudouris, B.W. Electronic and Spintronic Open-Shell Macromolecules, Quo Vadis? J. Am. Chem. Soc. 2022, 144, 626–647. [Google Scholar] [CrossRef] [PubMed]
- Iwamura, H. What role has organic chemistry played in the development of molecule-based magnets? Polyhedron 2013, 66, 3–14. [Google Scholar] [CrossRef]
- Iwamura, H. High-spin organic molecules and spin alignment in organic molecular assemblies. Adv. Phys. Org. Chem. 1990, 26, 179–253. [Google Scholar]
- Sato, O. Dynamic molecular crystals with switchable physical properties. Nat. Chem. 2016, 8, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Ratera, I.; Veciana, J. Playing with organic radicals as building blocks for functional molecular materials. Chem. Soc. Rev. 2012, 41, 303–349. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Li, Z. Recent progress in open-shell organic conjugated materials and their aggregated states. J. Mater. Chem. C 2022, 10, 2431–2449. [Google Scholar] [CrossRef]
- Xie, Y.; Zhang, K.; Yamauchi, Y.; Oyaizu, K.; Jia, Z. Nitroxide radical polymers for emerging plastic energy storage and organic electronics: Fundamentals, materials, and applications. Mater. Horiz. 2021, 8, 803–829. [Google Scholar] [CrossRef]
- Chen, Z.X.; Li, Y.; Huang, F. Persistent and stable organic radicals: Design, synthesis, and applications. Chem 2021, 7, 288–332. [Google Scholar] [CrossRef]
- Shu, C.; Yang, Z.; Rajca, A. From Stable Radicals to Thermally Robust High-Spin Diradicals and Triradicals. Chem. Rev. 2023, 123, 11954–12003. [Google Scholar] [CrossRef]
- Rajca, A. Magnetism of Nitroxides. In Nitroxides: Synthesis, Properties and Applications; The Royal Society of Chemistry: London, UK, 2021; Chapter 9. [Google Scholar]
- Datta, S.N.; Pal, A.K.; Panda, A. Design of Magnetic Organic Molecules and Organic Magnets: Experiment, Theory and Computation with Application and Recent Advances. Chem. Phys. Impact 2023, 7, 100379. [Google Scholar] [CrossRef]
- Baumgarten, M. High spin molecules directed towards molecular magnets. In EPR of Free Radicals in Solids II; Springer: Dordrecht, The Netherlands, 2012; pp. 205–244. [Google Scholar]
- Baumgarten, M. High spin organic molecules. In World Scientific Reference on Spin in Organics; Spin in Organics; World Scientific Publishing: Singapore, 2018; Volume 4, pp. 1–93. [Google Scholar]
- Gallagher, N.M.; Olankitwanit, A.; Rajca, A. High-spin organic molecules. J. Org. Chem. 2015, 80, 1291–1298. [Google Scholar] [CrossRef] [PubMed]
- Chapyshev, S.V.; Mendez-Vega, E.; Sander, W. Molecular Magnets: The Synthesis and Characterization of High-Spin Nitrenes. Chem. Eur. J. 2021, 27, 1258–1269. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Pink, M.; Wang, Y.; Rajca, S.; Rajca, A. High-spin S = 3/2 ground-state aminyl triradicals: Toward high-spin oligo-aza nanographenes. J. Am. Chem. Soc. 2022, 144, 19576–19591. [Google Scholar] [CrossRef] [PubMed]
- Pomikło, D.; Kaszyński, P. Blatter diradicals with a spin coupler at the N(1) position. Chem. Eur. J. 2023, 29, e202301069. [Google Scholar] [CrossRef] [PubMed]
- Tretyakov, E.V.; Ovcharenko, V.I.; Terent’ev, A.O.; Krylov, I.B.; Magdesieva, T.V.; Mazhukin, D.G.; Gritsan, N.P. Conjugated nitroxides. Russ. Chem. Rev. 2022, 91, RCR5025. [Google Scholar] [CrossRef]
- Zaytseva, E.V.; Mazhukin, D.G. Spirocyclic nitroxides as versatile tools in modern natural sciences: From synthesis to applications. Part I. Old and new synthetic approaches to spirocyclic nitroxyl radicals. Molecules 2021, 26, 677. [Google Scholar] [CrossRef] [PubMed]
- Likhtenshtein, G.I. (Ed.) Nitroxides: Brief History, Fundamentals, and Recent Developments; Springer: Cham, Switzerland, 2020. [Google Scholar]
- Bonucci, A.; Ouari, O.; Guigliarelli, B.; Belle, V.; Mileo, E. In-cell EPR: Progress towards structural studies inside cells. ChemBioChem 2020, 21, 451–460. [Google Scholar] [CrossRef]
- Kotake, Y.; Kuwata, K. Formation of Intramolecular Hydrogen Bond in Hydroxy-substituted Nitroxide Radicals as Evidenced by Electron Spin Resonance. Bull. Chem. Soc. Jpn. 1982, 55, 3686–3689. [Google Scholar] [CrossRef]
- Hicks, R.G. What’s new in stable radical chemistry? Org. Biomol. Chem. 2007, 5, 1321–1338. [Google Scholar] [CrossRef] [PubMed]
- Kubo, T. Synthesis, physical properties, and reactivity of stable, π-conjugated, carbon-centered radicals. Molecules 2019, 24, 665. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, G.; Ishida, T.; Nogami, T. Remarkably strong intermolecular antiferromagnetic couplings in the crystal of biphenyl-3,5-diyl bis(tert-butyl nitroxide). Chem. Phys. Lett. 2004, 392, 74–79. [Google Scholar] [CrossRef]
- Katoh, K.; Hosokoshi, Y.; Inoue, K.; Goto, T. Singlet ground states in an organic S = 1/2 spin ladder and a novel double spin chain of ferromagnetic dimers formed by an organic tetraradical. J. Phys. Soc. Jpn. 2000, 69, 1008–1011. [Google Scholar] [CrossRef]
- Iwamura, H.; Inoue, K. Spontaneous magnetization in a 2:3 complex formed by 3,4′,5-tris(N-oxy-tert-butylamino) biphenyl and manganese(II) bis(hexafluoroacetylacetonate). Adv. Mater. 1996, 8, 73–76. [Google Scholar] [CrossRef]
- Sekine, H.; Ishida, T. Unexpected Complexes of a [3+3] Cycloadduct from Biphenyl-3,5-diyl Bis(tert-butyl Nitroxide) with Gadolinium(III) 1,1,1,5,5,5-hexafluoropentane-2,4-dionate. Chem. Lett. 2018, 47, 74–77. [Google Scholar] [CrossRef]
- Ito, S.; Yoshitake, T.; Ishida, T. Ferromagnetic 2p-2p and 4f-2p Couplings in a Macrocycle from Two Biradicals and Two Gadolinium(III) Ions. Molecules 2022, 27, 4930. [Google Scholar] [CrossRef] [PubMed]
- Calder, A.; Forrester, A.R. The stability of aryl-t-butylnitroxides. Chem. Commun. 1967, 682–684. [Google Scholar] [CrossRef]
- Chiarelli, R.; Gambarelli, S.; Rassat, A. Exchange interactions in nitroxide biradicals. Mol. Cryst. Liq. Cryst. 1997, 305, 455–478. [Google Scholar] [CrossRef]
- Ito, S.; Ishida, T. Practically Diamagnetic Macrocycle Consisting of Nickel-Biradical Heterospins with the Largest Out-of-Plane Torsion at Coordination Bonds. Chem. Lett. 2020, 49, 1062–1065. [Google Scholar] [CrossRef]
- Ito, S.; Takano, R.; Hatanaka, S.I.; Ishida, T. Rare-Earth (RE = Y, Gd, Tb, Dy, Ho, and Er) Chains Bridged with a Triplet Biradical and Magnetic Hysteresis Recorded for RE = Tb. Inorg. Chem. 2022, 61, 10619–10623. [Google Scholar] [CrossRef] [PubMed]
- Rajca, A.; Lu, K.; Rajca, S.; Ross, C.R., II. Singlet–triplet bistability in a 1,3-phenylene-based bis(aminoxyl) diradical. Chem. Commun. 1999, 13, 1249–1250. [Google Scholar] [CrossRef]
- Rajca, A.; Rajca, S.; Wongsriratanakul, J.; Ross, C.R., II. 4,6-Bis(trifluoromethyl)-N,N′-di-tert-butyl-1,3-phenylenebis(aminoxyl) and its bis(hexafluoroacetylacetonato)manganese(II) complex: Synthesis, X-ray crystallography, and magnetism. Polyhedron 2001, 20, 1669–1675. [Google Scholar] [CrossRef]
- Richardson, M.F.; Wagner, W.F.; Sands, D.E. Rare-earth trishexafluoroacetylacetonates and related compounds. J. Inorg. Nucl. Chem. 1968, 30, 1275–1289. [Google Scholar] [CrossRef]
- CrysAlisPRO; Oxford Diffraction/Agilent Technologies UK Ltd.: Yarnton, UK, 2021.
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.; Puschmann, H. Olex2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Bain, G.A.; Berry, J.F. Diamagnetic corrections and Pascal’s constants. J. Chem. Educ. 2008, 85, 532–536. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2019. [Google Scholar]
- Neese, F. Prediction of molecular properties and molecular spectroscopy with density functional theory: From fundamental theory to exchange-coupling. Coord. Chem. Rev. 2009, 253, 526–563. [Google Scholar] [CrossRef]
- Noodleman, L.; Norman, J.G., Jr. The Xα valence bond theory of weak electronic coupling. Application to the low-lying states of Mo2Cl84−. J. Chem. Phys. 1979, 70, 4903–4906. [Google Scholar] [CrossRef]
- Noodleman, L. Valence bond description of antiferromagnetic coupling in transition metal dimers. J. Chem. Phys. 1981, 74, 5737–5743. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Toyoda, Y.; Fueno, T. A generalized MO (GMO) approach to unstable molecules with quasi-degenerate electronic states: Ab initio GMO calculations of intramolecular effective exchange integrals and designing of organic magnetic polymers. Synth. Met. 1987, 19, 81–86. [Google Scholar] [CrossRef]
- Soda, T.; Kitagawa, Y.; Onishi, T.; Takano, Y.; Shigeta, Y.; Nagao, H.; Yoshioka, Y.; Yamaguchi, K. Ab initio computations of effective exchange integrals for H–H, H–He–H and Mn2O2 complex: Comparison of broken-symmetry approaches. Chem. Phys. Lett. 2000, 319, 223–230. [Google Scholar] [CrossRef]
- Nakamoto, K.; Margoshes, M.; Rundle, R.E. Stretching frequencies as a function of distances in hydrogen bonds. J. Am. Chem. Soc. 1955, 77, 6480–6486. [Google Scholar] [CrossRef]
- Ishida, T.; Ito, S.; Homma, Y.; Kyoden, Y. Molecular S = 2 High-Spin, S = 0 Low-Spin and S = 0 ⇄ 2 Spin-Transition/-Crossover Nickel(II)-Bis(nitroxide) Coordination Compounds. Inorganics 2021, 9, 10. [Google Scholar] [CrossRef]
- Lluncll, M.; Casanova, D.; Circra, J.; Bofill, J.M.; Alcmany, P.; Alvarez, S.; Pinsky, M.; Avnir, D. SHAPE, Version 2.1; University of Barcelona: Barcelona, Spain; Hebrew University of Jerusalem: Jerusalem, Israel, 2005. [Google Scholar]
- Lee, J.H.; Jung, Y.S.; Sohn, Y.S.; Kang, S.-J. Synthesis and Characterization of Holmium Complexes Containing β-Diketonate Ligands. Bull. Korean Chem. Soc. 1998, 19, 231–235. [Google Scholar]
- Tan, R.H.C.; Motevalli, M.; Abrahams, I.; Wyatt, P.B.; Gillin, W.P. Quenching of IR Luminiescence of Erbium, Neodymium and Ytterbium β-diketonate Complexes by Ligand C-H and C-D bonds. J. Phys. Chem. B 2006, 110, 24476–24479. [Google Scholar] [CrossRef] [PubMed]
- Shannon, R. Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides. Acta Crystallogr. Sect. A Cryst. Phys. Diffr. Theor. Gen. Crystallogr. 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Bleaney, B.; Bowers, K.D. Anomalous paramagnetism of copper acetate. Proc. Royal Soc. Ser. A 1952, 214, 451–465. [Google Scholar] [CrossRef]
- Nakamura, T.; Ishida, T. Strong exchange couplings in lanthanide complexes with an aliphatic nitroxide radical 1,1,3,3-tetramethylisoindolin-2-oxyl. Polyhedron 2015, 87, 302–306. [Google Scholar] [CrossRef]
- Murakami, R.; Nakamura, T.; Ishida, T. Doubly TEMPO-coordinated gadolinium(III), lanthanum(III), and yttrium(III) complexes. Strong superexchange coupling across rare earth ions. Dalton Trans. 2014, 43, 5893–5898. [Google Scholar] [CrossRef]
- Baumgarten, M.; Karabunarliev, S. Reverting the effect of magnetic couplers in bridged di-and polyradicals. Chem. Phys. 1999, 244, 35–47. [Google Scholar] [CrossRef]
- Zeng, Z.; Sung, Y.M.; Bao, N.; Tan, D.; Lee, R.; Zafra, J.L.; Lee, B.S.; Ishida, M.; Ding, J.; López Navarrete, J.T.; et al. Stable Tetrabenzo-Chichibabin’s Hydrocarbons: Tunable Ground State and Unusual Transition between Their Closed-Shell and Open-Shell Resonance Forms. J. Am. Chem. Soc. 2012, 134, 14513–14525. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.I.; van Eikema Hommes, N.J.; Lenoir, D.; Bachrach, S.M. The quest for a triplet ground-state alkene: Highly twisted C=C double bonds. J. Phys. Org. Chem. 2019, 32, e3965. [Google Scholar] [CrossRef]
- Shimizu, A.; Morikoshi, T.; Sugisaki, K.; Shiomi, D.; Sato, K.; Takui, T.; Shintani, R. Synthesis and Isolation of a Kekulé Hydrocarbon with a Triplet Ground State. Angew. Chem. Int. Ed. 2022, 61, e202205729. [Google Scholar] [CrossRef] [PubMed]
- Nishimaki, H.; Mashiyama, S.; Yasui, M.; Nogami, T.; Ishida, T. Bistable Polymorphs Showing Diamagnetic and Paramagnetic States of an Organic Crystalline Biradical Biphenyl-3,5-diyl Bis(tert-butylnitroxide). Chem. Mater. 2006, 18, 3602–3604. [Google Scholar] [CrossRef]
- Nishimaki, H.; Ishida, T. Organic two-step spin-transition-like behavior in a linear S = 1 array: 3’-methylbiphenyl-3,5-diyl bis(tert-butylnitroxide) and related compounds. J. Am. Chem. Soc. 2010, 132, 9598–9599. [Google Scholar] [CrossRef] [PubMed]
- Konno, T.; Kudo, H.; Ishida, T. Intermediate-paramagnetic phases with a half and a quarter spin entities in fluorinated biphenyl-3,5-diyl bis(tert-butyl nitroxides). J. Mater. Chem. C 2015, 3, 7813–7818. [Google Scholar] [CrossRef]
- Yoshitake, T.; Kudo, H.; Ishida, T. Thermally Activated Paramagnets from Diamagnetic Polymers of Biphenyl-3,5-diyl Bis(tert-butyl Nitroxides) with Methyl and Fluoro Groups at the 2’- and 5’-Positions. Crystals 2016, 6, 30. [Google Scholar] [CrossRef]
- Kanno, F.; Inoue, K.; Koga, N.; Iwamura, H. 4,6-Dimethoxy-1,3-phenylenebis(N-tert-butyl nitroxide) with a singlet ground state. Formal violation of a rule that m-phenylene serves as a robust ferromagnetic coupling unit. J. Am. Chem. Soc. 1993, 115, 847–850. [Google Scholar] [CrossRef]
- Fujita, J.; Tanaka, M.; Suemune, H.; Koga, N.; Matsuda, K.; Iwamura, H. Antiferromagnetic exchange interaction among the three spins placed in an isosceles triangular configuration in 2,4-dimethoxy-1,3,5-benzenetriyltris(N-tert-butyl nitroxide). J. Am. Chem. Soc. 1996, 118, 9347–9351. [Google Scholar] [CrossRef]
- Dvolaitzky, M.; Chiarelli, R.; Rassat, A. Stable N,N′-Di-tert-butyl-meta-phenylene-bisnitroxides—Unexpected Ground-State Singlets. Angew. Chem. Int. Ed. Engl. 1992, 31, 180–181. [Google Scholar] [CrossRef]
- Yoshizawa, K.; Kuga, T.; Sato, T.; Hatanaka, M.; Tanaka, K.; Yamabe, T. Through-bond and through-space interactions of organic radicals coupled by m-phenylene. Bull. Chem. Soc. Jpn. 1996, 69, 3443–3450. [Google Scholar] [CrossRef]
- Fang, S.; Lee, M.S.; Hrovat, D.A.; Borden, W.T. Ab initio calculations show why m-phenylene is not always a ferromagnetic coupler. J. Am. Chem. Soc. 1995, 117, 6727–6731. [Google Scholar] [CrossRef]
- Barone, V.; Cacelli, I.; Ferretti, A.; Monti, S.; Prampolini, G. An integrated protocol for the accurate calculation of magnetic interactions in organic magnets. J. Chem. Theory Comput. 2011, 7, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Barone, V.; Boilleau, C.; Cacelli, I.; Ferretti, A.; Prampolini, G. Conformational effects on the magnetic properties of an organic diradical: A computational study. J. Chem. Theory Comput. 2013, 9, 1958–1963. [Google Scholar] [CrossRef] [PubMed]
- Venkataraman, L.; Klare, J.E.; Nuckolls, C.; Hyberstsen, M.S.; Steigerwald, M.L. Dependence of single-molecule junction conductance on molecular conformation. Nature 2006, 442, 904–907. [Google Scholar] [CrossRef] [PubMed]
- Vonlanthen, D.; Mishchenko, A.; Elbing, M.; Neuburger, M.; Wandlowski, T.; Mayor, M. Chemically controlled conductivity: Torsion-angle dependence in a single-molecule biphenyldithiol junction. Angew. Chem. Int. Ed. 2009, 48, 8886–8890. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa, S.; Hasegawa, J.Y.; Matsuda, K. Theoretical investigation of the dependence of exchange interaction on dihedral angle between two aromatic rings in a wire unit. Chem. Lett. 2014, 43, 530–532. [Google Scholar] [CrossRef]
- Barone, V.; Cacelli, I.; Cimino, P.A.; Ferretti, S. Monti and G. Prampolini, Magnetic Interactions in Phenyl-Bridged Nitroxide Diradicals: Conformational Effects by Multireference and Broken Symmetry DFT Approaches. J. Phys. Chem. A 2009, 113, 15150–15155. [Google Scholar] [CrossRef]
- Shultz, D.A.; Fico, R.M.; Lee, H.; Kampf, J.W.; Kirschbaum, K.; Pinkerton, A.A.; Boyle, P.D. Mechanisms of exchange modulation in trimethylenemethane-type biradicals: The roles of conformation and spin density. J. Am. Chem. Soc. 2003, 125, 15426–15432. [Google Scholar] [CrossRef]
- Shultz, D.A.; Fico, R.M.; Bodnar, S.H.; Kumar, R.K.; Vostrikova, K.E.; Kampf, J.W.; Boyle, P.D. Trends in exchange coupling for trimethylenemethane-type bis(semiquinone) biradicals and correlation of magnetic exchange with mixed valency for cross-conjugated systems. J. Am. Chem. Soc. 2003, 125, 11761–11771. [Google Scholar] [CrossRef]
- Karplus, M. Vicinal proton coupling in nuclear magnetic resonance. J. Am. Chem. Soc. 1963, 85, 2870–2871. [Google Scholar] [CrossRef]
- Heller, C.; McConnell, H.M. Radiation damage in organic crystals. II. Electron spin resonance of (CO2H)CH2CH(CO2H) in β-succinic acid. J. Chem. Phys. 1960, 32, 1535–1539. [Google Scholar] [CrossRef]
- Kahn, O. Molecular Magnetism; VCH: New York, NY, USA, 1993. [Google Scholar]
- Zheng, X.; Bu, Y. Hydrogen-Bonding-Assisted Substituent Engineering for Modulating Magnetic Spin Couplings and Switching in m-Phenylene Nitroxide Diradicals. J. Phys. Chem. A 2023, 127, 7443–7451. [Google Scholar] [CrossRef] [PubMed]
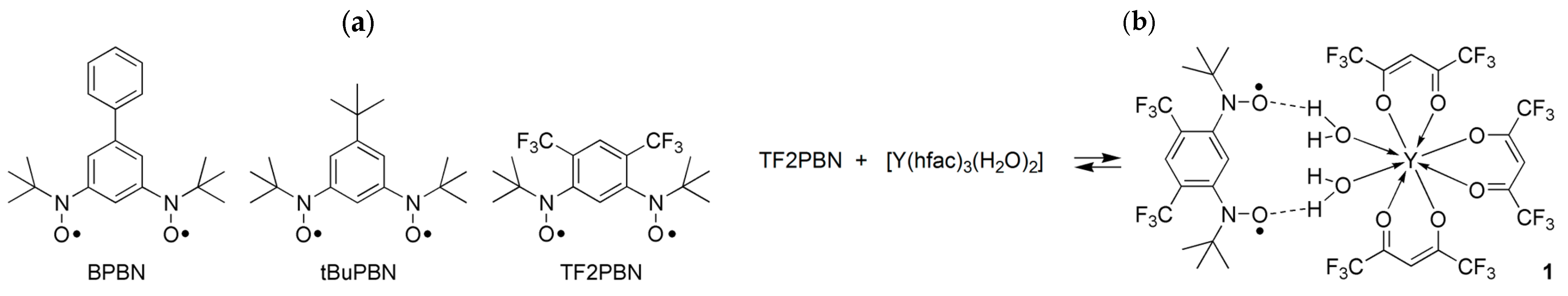





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | TF2PBN 1 | 1 | [Mn(hfac)2(TF2PBN)2] |
|---|---|---|---|
| N1–O1 | 1.2863(19) | 1.2842(16) | 1.294(3) |
| N2–O2 | 1.2826(13) | 1.2850(16) | 1.2779(17) |
| N1–C2 | 1.423(8) | 1.4389(16) | 1.421(3) |
| N2–C6 | 1.4361(14) | 1.4393(16) | 1.433(2) |
| N1–C7 | 1.505(3) | 1.5043(17) | 1.512(5) |
| N2–C10 | 1.5051(16) | 1.5004(17) | 1.499(4) |
| O1–N1–C2 | 115.80(15) | 116.44(11) | 115.9(2) |
| O2–N2–C6 | 116.58(9) | 115.83(11) | 115.74(15) |
| O1–N1–C7 | 117.8(3) | 118.29(11) | 117.59(18) |
| O2–N2–C10 | 119.81(8) | 117.73(11) | 119.63(12) |
| C2–N1–C7 | 126.36(18) | 124.33(11) | 126.47(12) |
| C6–N2–C10 | 123.58(9) | 124.12(11) | 123.56(13) |
| N1–C2–C1 | 119.55(9) | 117.96(12) | 118.23(19) |
| N1–C2–C3 | 121.0(3) | 121.94(12) | 121.83(13) |
| N2–C6–C1 | 118.85(9) | 118.30(12) | 118.84(19) |
| N2–C6–C5 | 121.4(3) | 121.53(11) | 121.15(15) |
| O1–N1–C2–C1 | 126.69(11) 2 | 101.2262(14) | 130.08(16) 3 |
| O1–N1–C2–C3 | −48.78(14) 2 | −74.9312(14) | −46.05(19) 3 |
| O2–N2–C6–C1 | −107.53(14) 2 | −91.264(2) | −94.9(2) 3 |
| O2–N2–C6–C5 | 69.65(15) 2 | 84.7612(19) | 81.4(2) 3 |
| O1⋯O2 | 6.339(16) 2 | 5.8868(1) | 6.231(11) 3 |
| reference | ref. [36] | this work | Ref. [37] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haga, N.; Ishida, T. A Triplet/Singlet Ground-State Switch via the Steric Inhibition of Conjugation in 4,6-Bis(trifluoromethyl)-1,3-phenylene Bisnitroxide. Molecules 2024, 29, 70. https://doi.org/10.3390/molecules29010070
Haga N, Ishida T. A Triplet/Singlet Ground-State Switch via the Steric Inhibition of Conjugation in 4,6-Bis(trifluoromethyl)-1,3-phenylene Bisnitroxide. Molecules. 2024; 29(1):70. https://doi.org/10.3390/molecules29010070
Chicago/Turabian StyleHaga, Nagito, and Takayuki Ishida. 2024. "A Triplet/Singlet Ground-State Switch via the Steric Inhibition of Conjugation in 4,6-Bis(trifluoromethyl)-1,3-phenylene Bisnitroxide" Molecules 29, no. 1: 70. https://doi.org/10.3390/molecules29010070
APA StyleHaga, N., & Ishida, T. (2024). A Triplet/Singlet Ground-State Switch via the Steric Inhibition of Conjugation in 4,6-Bis(trifluoromethyl)-1,3-phenylene Bisnitroxide. Molecules, 29(1), 70. https://doi.org/10.3390/molecules29010070