
Opioid/Dopamine Receptor Binding Studies, NMR and Molecular Dynamics Simulation of LENART01 Chimera, an Opioid-Bombesin-like Peptide


 ,
,  , , ,
, , ,  ,
,  ,
, 

Abstract
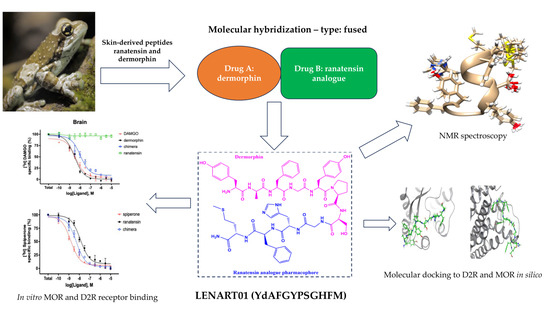
1. Introduction
2. Results and Discussion
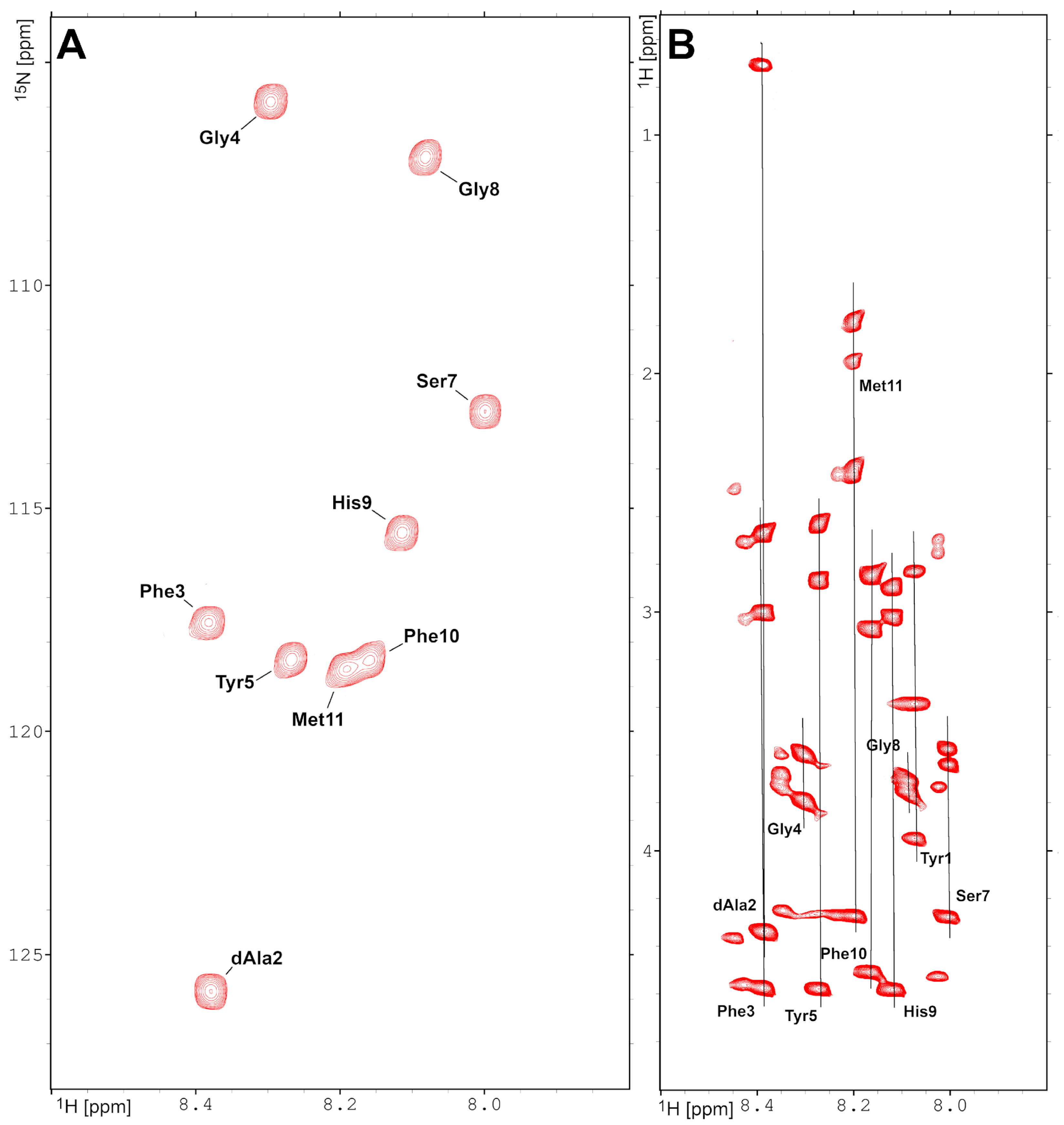
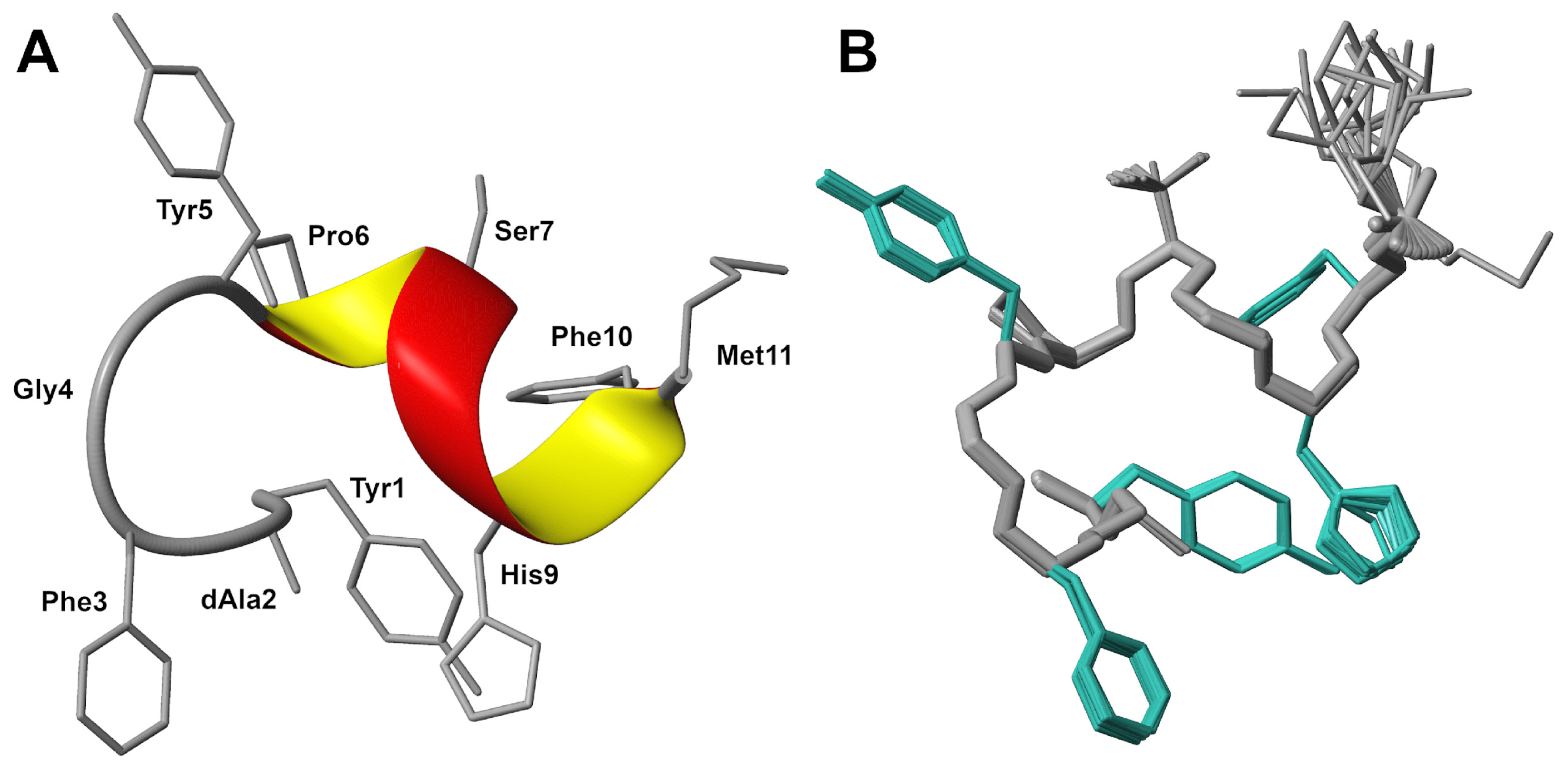
2.1. Multidimensional NMR Spectroscopy
2.2. Receptor Binding Affinities and Activation Efficacy of LENART01 towards Opioid and Dopamine Receptors
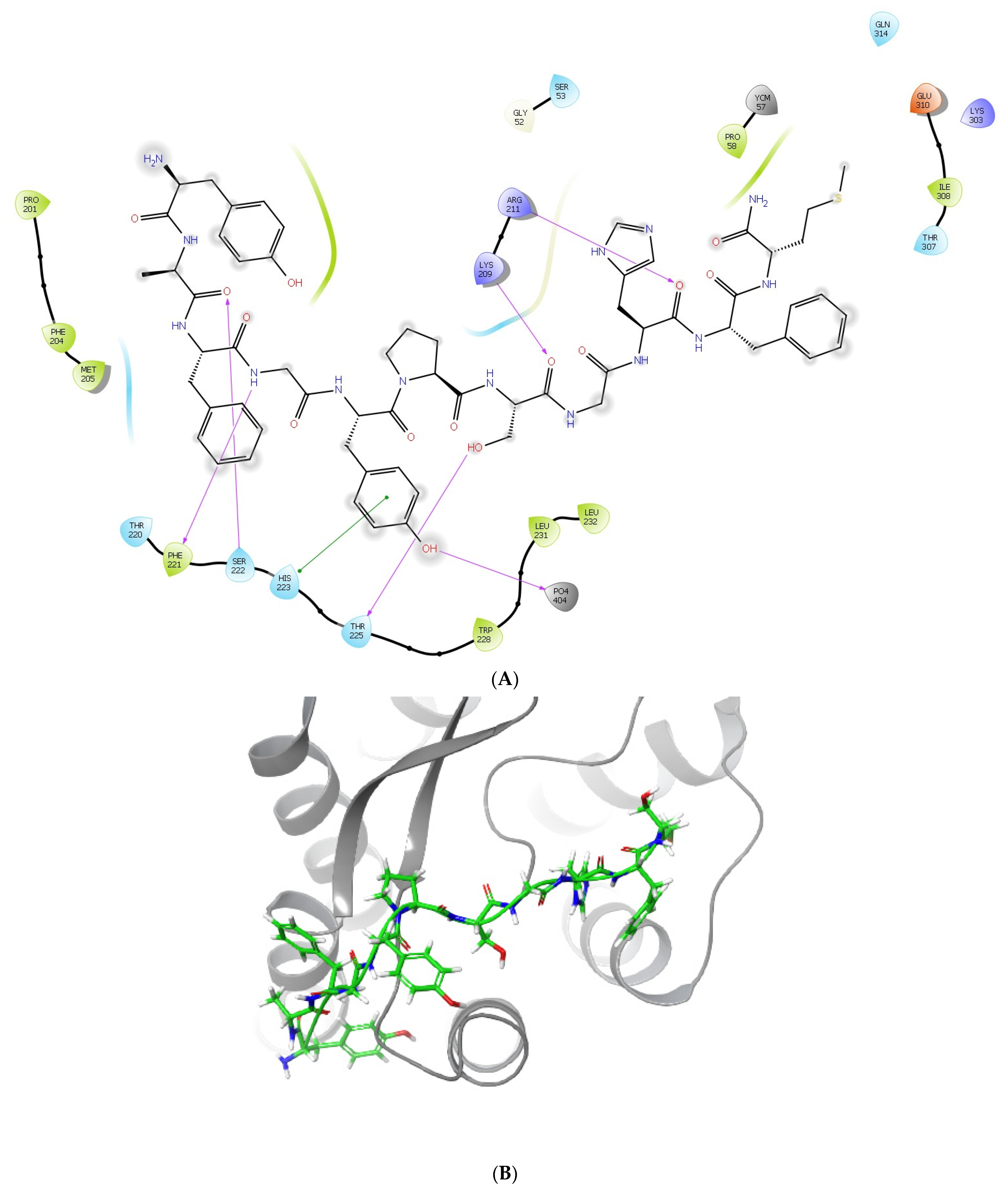
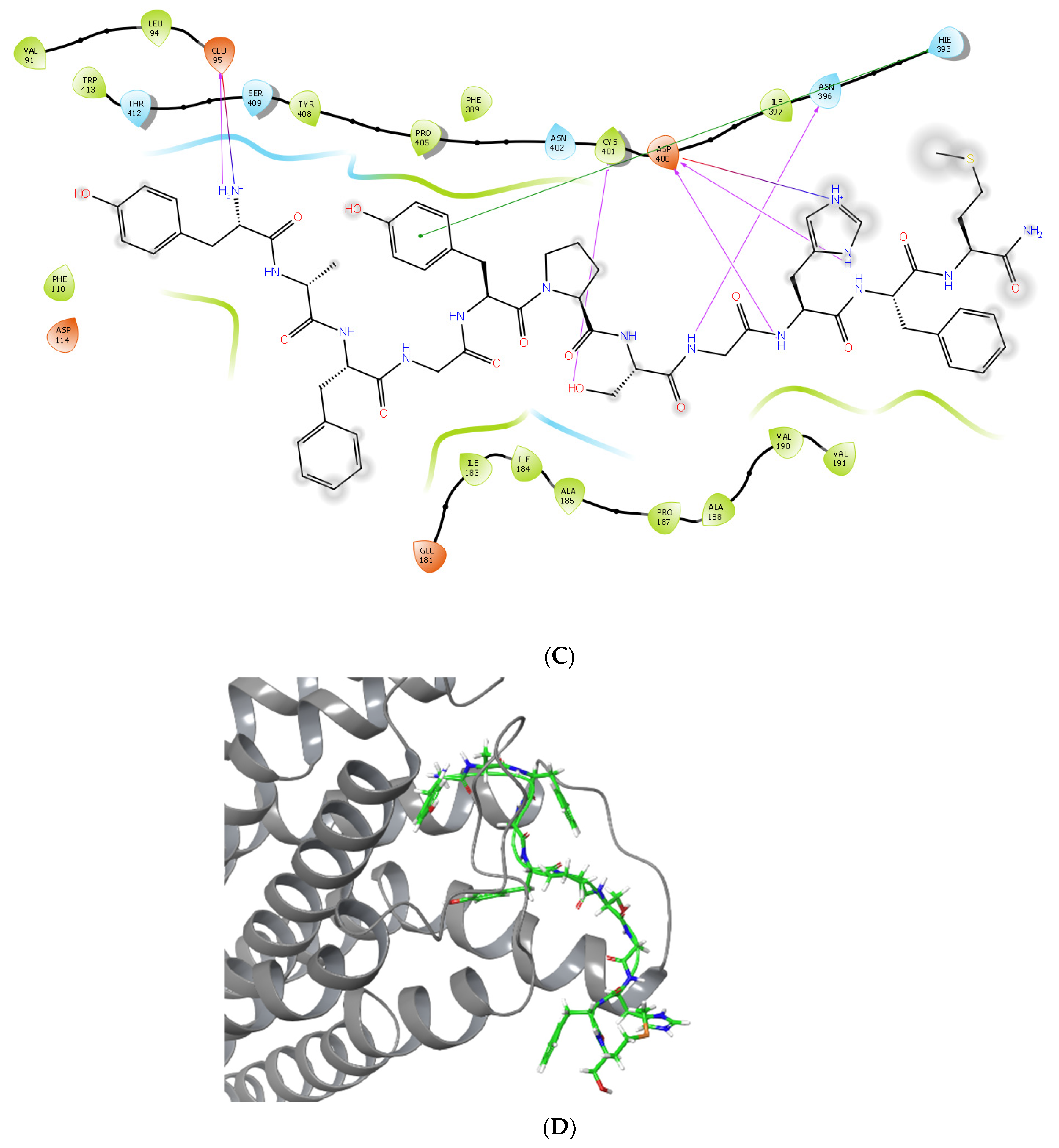
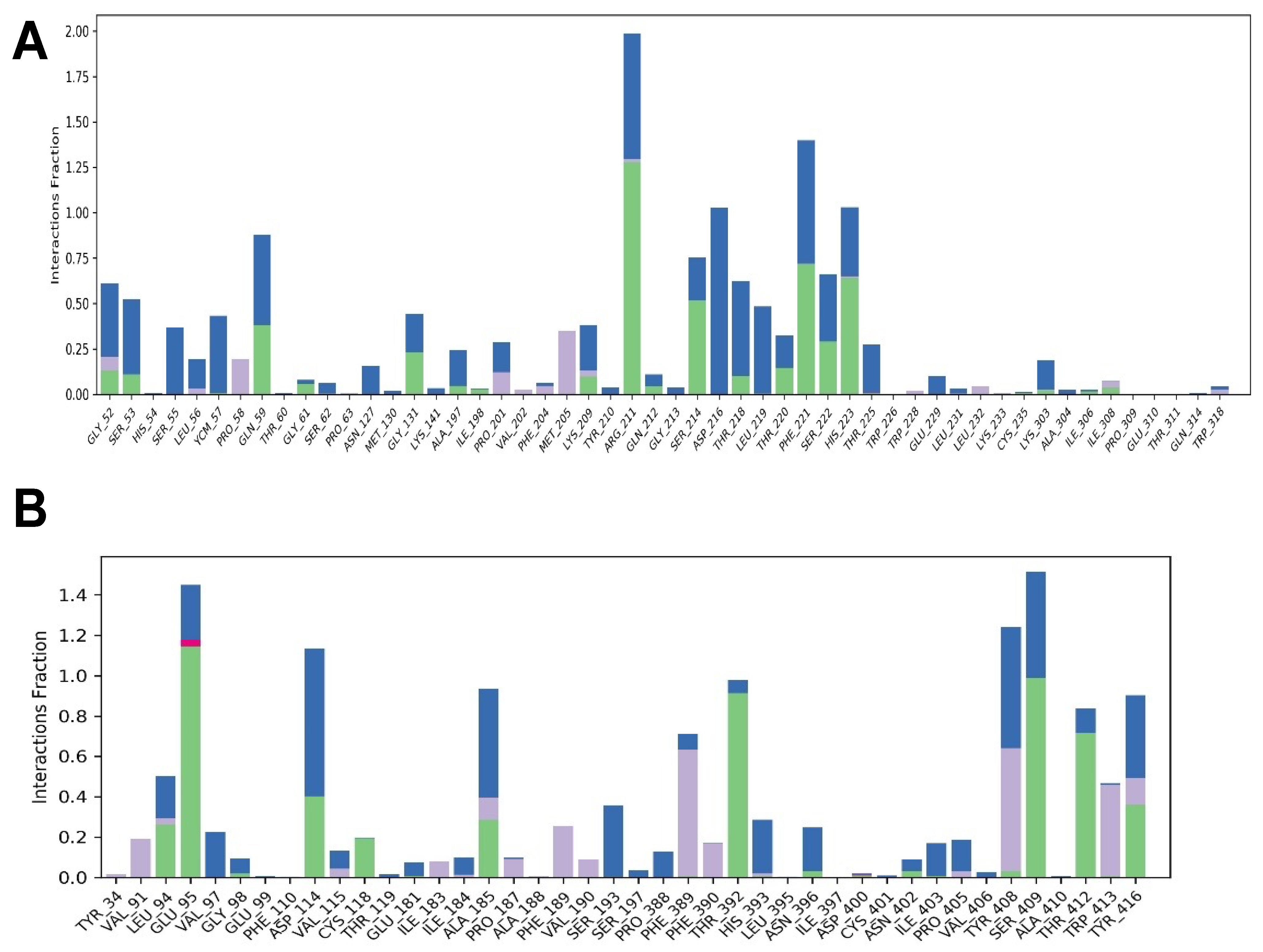
2.3. Molecular Docking and MM/GBSA Calculations of the Complexes Formed between LENART01 and D2R or MOR Receptors
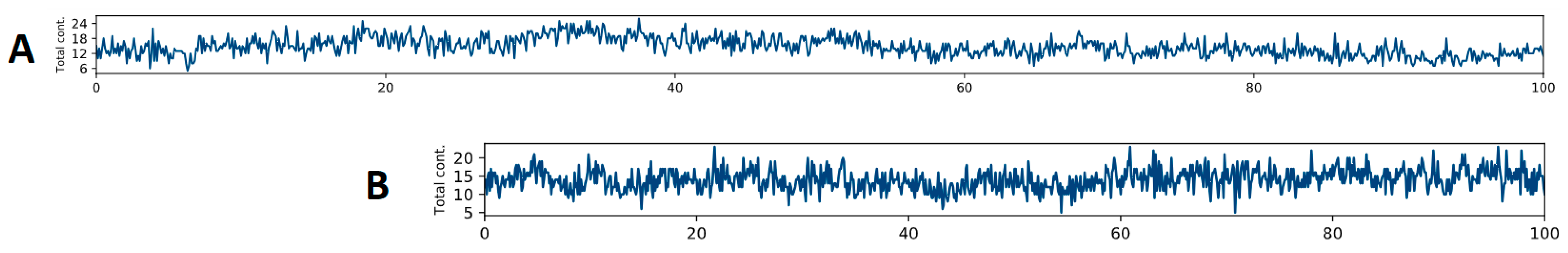
2.4. Molecular Dynamics Simulations of the Complexes Formed between LENART01 and D2R or MOR Receptors
3. Materials and Methods
3.1. Drugs and Reagents
3.2. NMR
3.2.1. Sample Preparation
3.2.2. NMR Measurements
3.2.3. Structure Calculations
3.3. Receptor Binding Assay Ex Vivo
3.3.1. Animals
3.3.2. Competitive Binding Experiments
3.3.3. Functional [35S]GTPγS Binding Experiments
3.4. Molecular Modeling
3.4.1. Structures Preparation
3.4.2. Protein–Peptide Docking
Active Site Identification and Grid Generation
Ligand Preparation
Glide SP–Peptide Docking
3.4.3. Binding Free Energy Calculation by MM/GBSA Rescoring
3.4.4. Molecular Dynamics (MD) Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nepali, K.; Sharma, S.; Sharma, M.; Bedi, P.; Dhar, K. Rational approaches, design strategies, structure activity relationship and mechanistic insights for anticancer hybrids. Eur. J. Med. Chem. 2014, 77, 422–487. [Google Scholar] [CrossRef] [PubMed]
- Kleczkowska, P.; Hermans, E.; Kosson, P.; Kowalczyk, A.; Lesniak, A.; Pawlik, K.; Bojnik, E.; Benyhe, S.; Nowicka, B.; Bujalska-Zadrozny, M.; et al. Antinociceptive effect induced by a combination of opioid and neurotensin moieties vs. their hybrid peptide [Ile(9)]PK20 in an acute pain treatment in rodents. Brain Res. 2016, 1648, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Nygaard, R.; Zou, Y.; Dror, R.O.; Mildorf, T.J.; Arlow, D.H.; Manglik, A.; Pan, A.C.; Liu, C.W.; Fung, J.J.; Bokoch, M.P.; et al. The dynamic process of beta(2)-adrenergic receptor activation. Cell 2013, 152, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.Y.; Zhou, F.; Fryszczyn, B.G.; Barth, P. Naturally evolved G protein-coupled receptors adopt metastable conformations. Proc. Natl. Acad. Sci. USA 2012, 109, 13284–13289. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C. Beware of docking! Trends Pharmacol. Sci. 2015, 36, 78–95. [Google Scholar] [CrossRef] [PubMed]
- Mollica, A.; Pelliccia, S.; Famiglini, V.; Stefanucci, A.; Macedonio, G.; Chiavaroli, A.; Orlando, G.; Brunetti, L.; Ferrante, C.; Pieretti, S.; et al. Exploring the first Rimonabant analog-opioid peptide hybrid compound, as bivalent ligand for CB1 and opioid receptors. J. Enzym. Inhib. Med. Chem. 2017, 32, 444–451. [Google Scholar] [CrossRef]
- Serafin, P.; Kowalczyk, P.; Mollica, A.; Stefanucci, A.; Laskowska, A.K.; Zawadzka, M.; Kramkowski, K.; Kleczkowska, P. Evaluation of antimicrobial activities against various E. coli strains of a novel hybrid peptide—LENART01. Molecules 2023, 28, 4955. [Google Scholar] [CrossRef]
- Laskowska, A.K.; Szudzik, M.; Ścieżyńska, A.; Komorowski, M.; Szucs, E.; Gombos, D.; Bączek, B.; Lipka-Miciuk, J.; Benyhe, S.; Kleczkowska, P. The role of a natural amphibian skin-based peptide, ranatensin, in pancreatic cancer expressing dopamine D2 receptors. Cancers 2022, 14, 5535. [Google Scholar] [CrossRef]
- Zhu, X.Z.; Ji, X.Q.; Wu, S.X.; Zou, G. Sulpiride attenuates ranatensin-M-induced antinociception. Zhongguo Yao Li Xue Bao 1991, 12, 291–293. [Google Scholar]
- Bird, M.F.; Cerlesi, M.C.; Brown, M.; Malfacini, D.; Vezzi, V.; Molinari, P.; Micheli, L.; Mannelli, L.D.C.; Ghelardini, C.; Guerrini, R.; et al. Characterisation of the novel mixed Mu-NOP peptide ligand dermorphin-N/OFQ (DeNo). PLoS ONE 2016, 11, e0156897. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Bax, A. Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J. Biomol. NMR 2013, 56, 227–241. [Google Scholar] [CrossRef] [PubMed]
- Piela, L.; Némethy, G.; Scheraga, H.A. Proline-induced constraints in alpha-helices. Biopolymers 1987, 26, 1587–1600. [Google Scholar] [CrossRef] [PubMed]
- Dallavalle, S.; Dobričić, V.; Lazzarato, L.; Gazzano, E.; Machuqueiro, M.; Pajeva, I.; Tsakovska, I.; Zidar, N.; Fruttero, R. Improvement of conventional anti-cancer drugs as new tools against multidrug resistant tumors. Drug Resist. Updat. 2020, 50, 100682. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Kumar, A.; Singh, H.; Sonawane, P.; Paliwal, H.; Thareja, S.; Pathak, P.; Grishina, M.; Jaremko, M.; Emwas, A.H.; et al. Concept of hybrid drugs and recent advancements in anticancer hybrids. Pharmaceuticals 2022, 15, 1071. [Google Scholar] [CrossRef]
- Starnowska-Sokół, J.; Przewłocka, B. Multifunctional opioid-derived hybrids in neuropathic pain: Preclinical evidence, ideas and challenges. Molecules 2020, 25, 5520. [Google Scholar] [CrossRef] [PubMed]
- Raschka, S.; Wolf, A.; Bemister-Buffington, J.; Kuhn, L. Protein-Ligand interfaces are polarized: Discovery of a strong trend for intermolecular hydrogen bonds to favor donors on the protein side with implications for predicting and designing ligand complexes. J. Comput. Aided Mol. Des. 2018, 32, 511–528. [Google Scholar] [CrossRef]
- Martínez, L. Automatic identification of mobile and rigid substructures in molecular dynamics simulations and fractional structural fluctuation analysis. PLoS ONE 2015, 10, e0119264. [Google Scholar] [CrossRef]
- Liu, K.; Watanabe, E.; Kokubo, H. Exploring the stability of ligand binding modes to proteins by molecular dynamics simulations. J. Comput. Aided Mol. Des. 2017, 31, 201–211. [Google Scholar] [CrossRef]
- Delaglio, F.; Grzesiek, S.; Vuister, G.; Zhu, G.; Pfeifer, J.; Bax, A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar] [CrossRef]
- Lee, W.; Tonelli, M.; Markley, J.L. NMRFAM-SPARKY: Enhanced software for biomolecular NMR spectroscopy. Bioinformatics 2015, 31, 1325. [Google Scholar] [CrossRef] [PubMed]
- Guntert, P. Automated NMR protein structure calculation with CYANA. Methods Mol. Biol. 2004, 278, 353–378. [Google Scholar] [PubMed]
- Shen, Y.; Bax, A. Prediction of Xaa-Pro peptide bond conformation from sequence and chemical shifts. J. Biomol. NMR 2010, 46, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/GBSA methods to estimate ligand-binding affinities. Expert. Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 26. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Emax ± S.E.M. (%) | ||||
|---|---|---|---|---|---|
| Opioid System | Dopaminergic System | ||||
| Ligand + Cyprodime | Ligand + SCH-39166 | Ligand + Risperidone | |||
| Brain membrane | dermorphin | 140.5 ± 2.9 | 100.0 ± 1.1 *** | n.d.a | n.d.a |
| chimera | 140.6 ± 1.0 | 100.4 ± 1.4 *** | 140.8 ± 0.8 ns | 101.1 ± 0.7 *** | |
| ranatensin | 124.1 ± 1.9 | n.d.a | 119.1 ± 0.8 *** | 101.5 ± 1.7 *** | |
| Spinal cord membrane | dermorphin | 140.0 ± 1.8 | 100.4 ± 0.9 *** | n.d.a | n.d.a |
| chimera | 136.3 ± 2.2 | 100.8 ± 1.1 *** | 136.1 ± 1.7 ns | 101.6 ± 1.2 *** | |
| ranatensin | 119.8 ± 1.3 | n.d.a | 117.4 ± 1.1 ns | 100.0 ± 0.7 *** | |
| Glide G-Score (kcal/mol) | Glide Emodel (kcal/mol) | MM/GBSA ΔG Binding (kcal/mol) | Residues Forming Hydrogen Bonds | Residues Forming π Interactions | |
|---|---|---|---|---|---|
| MOR (5C1M) | −10.343 | −147.199 | −23.53 | LYS209, ARG211, PHE221, SER222, THR225 | HIS223 |
| D2R (7JVR) | −13.800 | −205.959 | −48.75 | CYS182, ASP400, CYS401 | HIS393 |
| MM/GBSA ΔG Binding | CE | CB | HB | LE | PP | GB | SA | |
|---|---|---|---|---|---|---|---|---|
| MOR (5C1M) | −23.53 | 8.72 | −11.12 | −2.86 | −5.81 | 0.75 | 36.09 | −49.30 |
| D2R (7JVR) | −48.75 | −97.26 | −2.39 | −1.24 | −34.51 | −0.26 | 164.69 | −77.78 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serafin, P.; Szeleszczuk, Ł.; Zhukov, I.; Szűcs, E.; Gombos, D.; Stefanucci, A.; Mollica, A.; Pisklak, D.M.; Kleczkowska, P. Opioid/Dopamine Receptor Binding Studies, NMR and Molecular Dynamics Simulation of LENART01 Chimera, an Opioid-Bombesin-like Peptide. Molecules 2024, 29, 272. https://doi.org/10.3390/molecules29010272
Serafin P, Szeleszczuk Ł, Zhukov I, Szűcs E, Gombos D, Stefanucci A, Mollica A, Pisklak DM, Kleczkowska P. Opioid/Dopamine Receptor Binding Studies, NMR and Molecular Dynamics Simulation of LENART01 Chimera, an Opioid-Bombesin-like Peptide. Molecules. 2024; 29(1):272. https://doi.org/10.3390/molecules29010272
Chicago/Turabian StyleSerafin, Pawel, Łukasz Szeleszczuk, Igor Zhukov, Edina Szűcs, Dávid Gombos, Azzurra Stefanucci, Adriano Mollica, Dariusz Maciej Pisklak, and Patrycja Kleczkowska. 2024. "Opioid/Dopamine Receptor Binding Studies, NMR and Molecular Dynamics Simulation of LENART01 Chimera, an Opioid-Bombesin-like Peptide" Molecules 29, no. 1: 272. https://doi.org/10.3390/molecules29010272
APA StyleSerafin, P., Szeleszczuk, Ł., Zhukov, I., Szűcs, E., Gombos, D., Stefanucci, A., Mollica, A., Pisklak, D. M., & Kleczkowska, P. (2024). Opioid/Dopamine Receptor Binding Studies, NMR and Molecular Dynamics Simulation of LENART01 Chimera, an Opioid-Bombesin-like Peptide. Molecules, 29(1), 272. https://doi.org/10.3390/molecules29010272