
Mechanistic Insights into Peptide Binding and Deactivation of an Adhesion G Protein-Coupled Receptor


Abstract
:
1. Introduction
2. Results
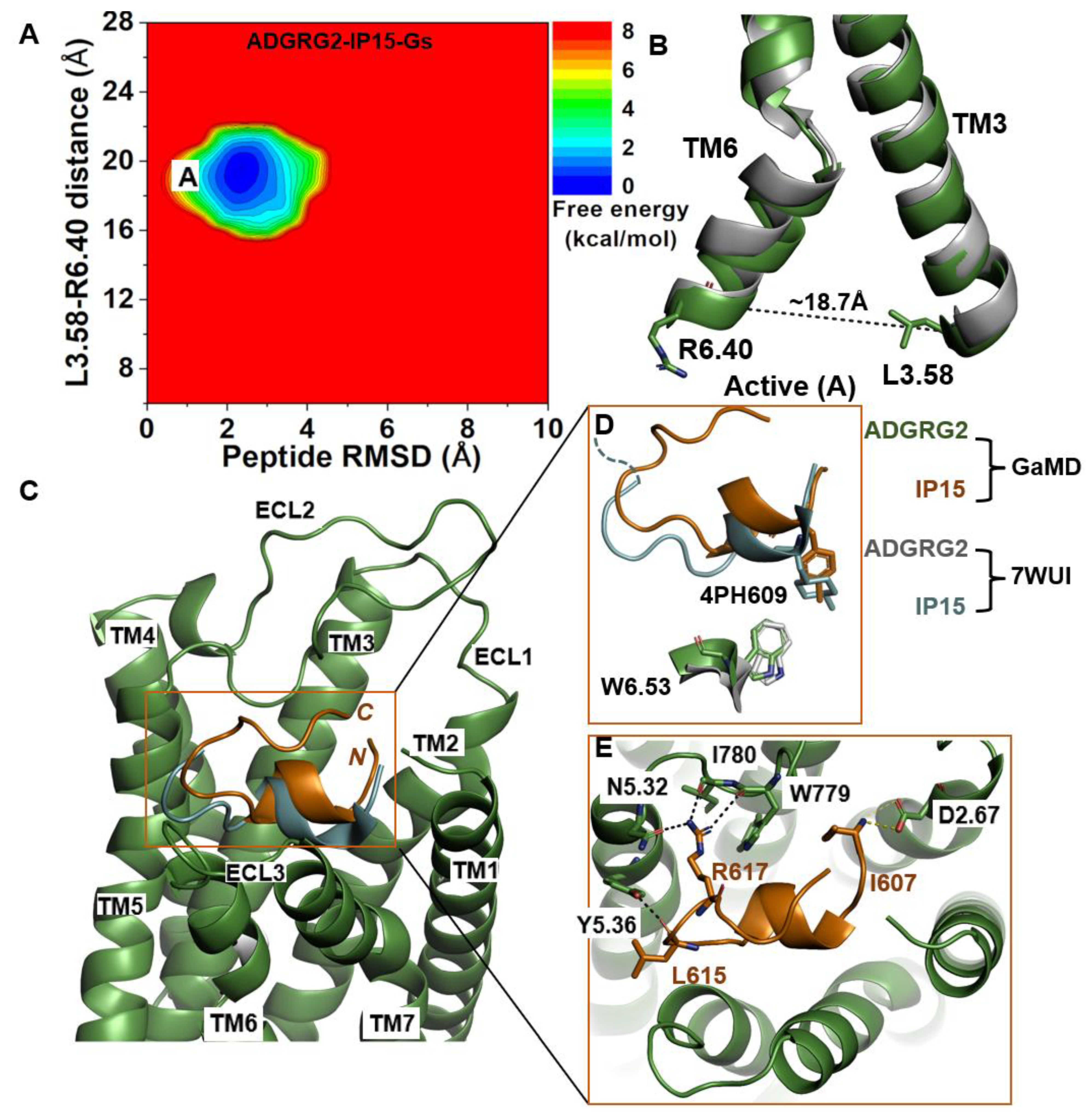
2.1. Agonist IP15-Gs-Bound ADGRG2 Sampled Only the “Active” State
2.2. Agonist IP15-Bound ADGRG2 without Gs Sampled the “Active” and “Intermediate” (“I1”) States
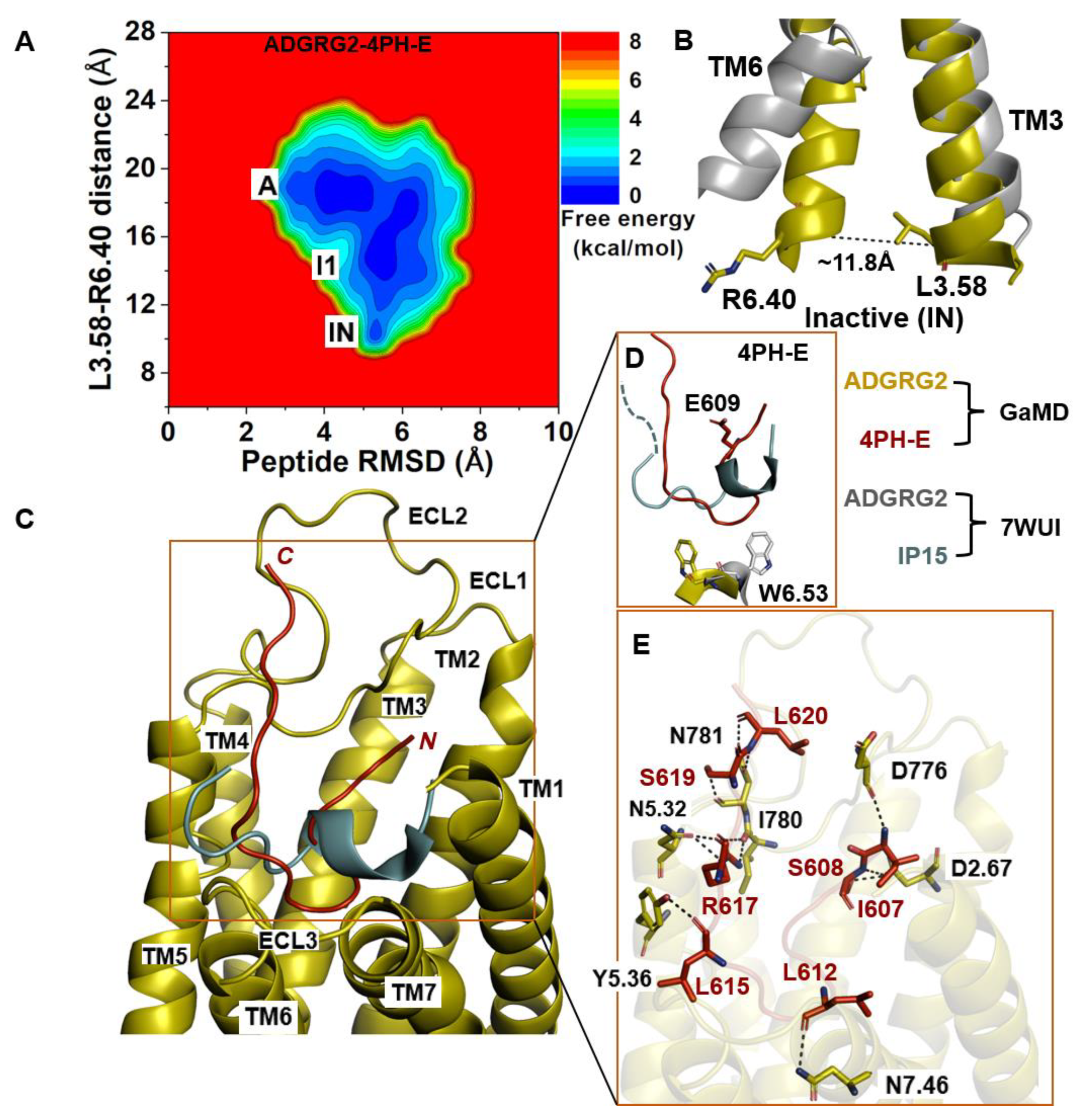
2.3. Antagonist 4PH-E-Bound ADGRG2 System Sampled the “A”, “I1”, and “Inactive” (“IN”) States
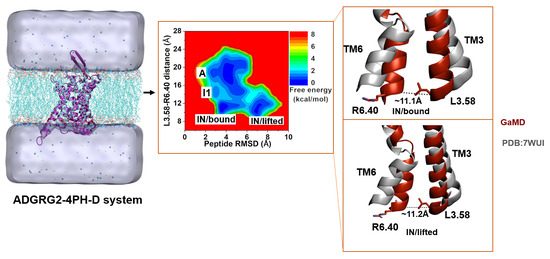
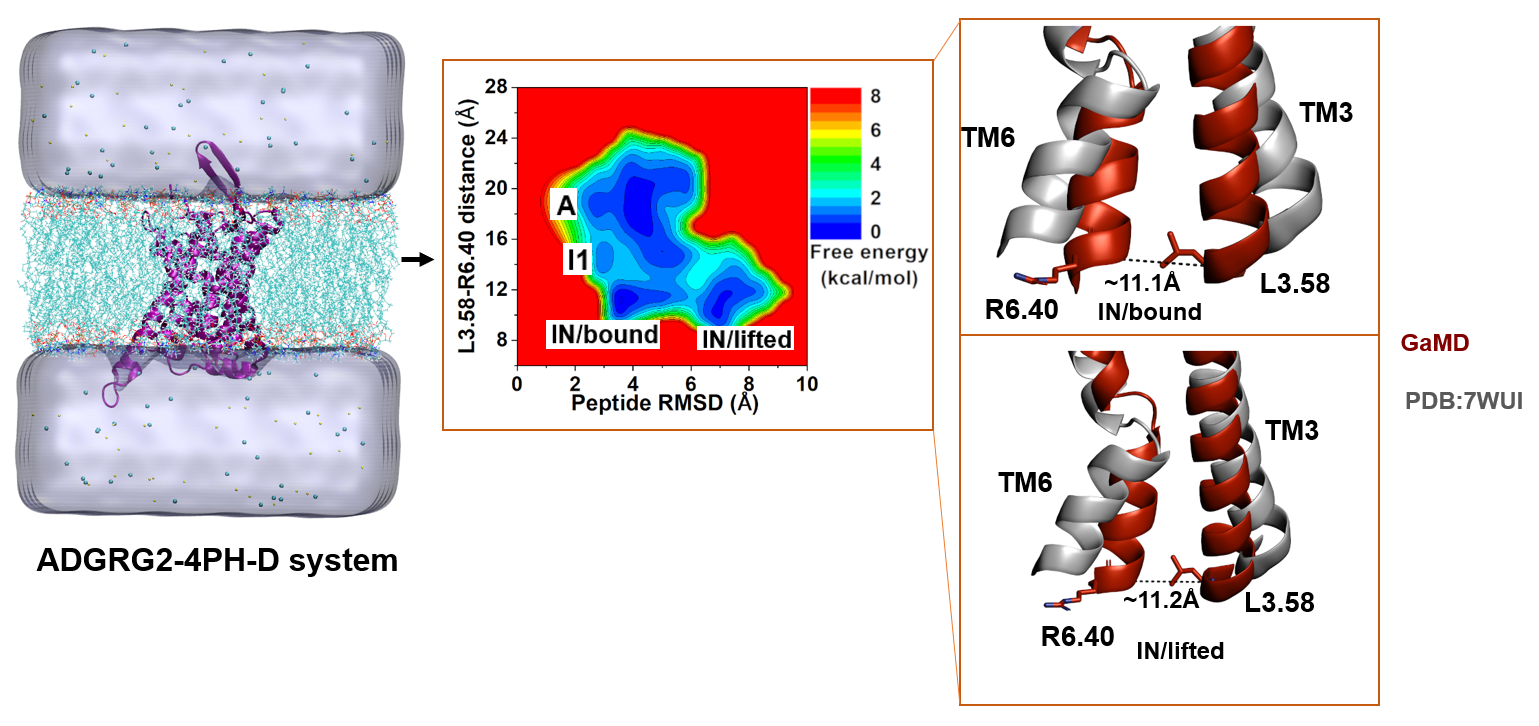
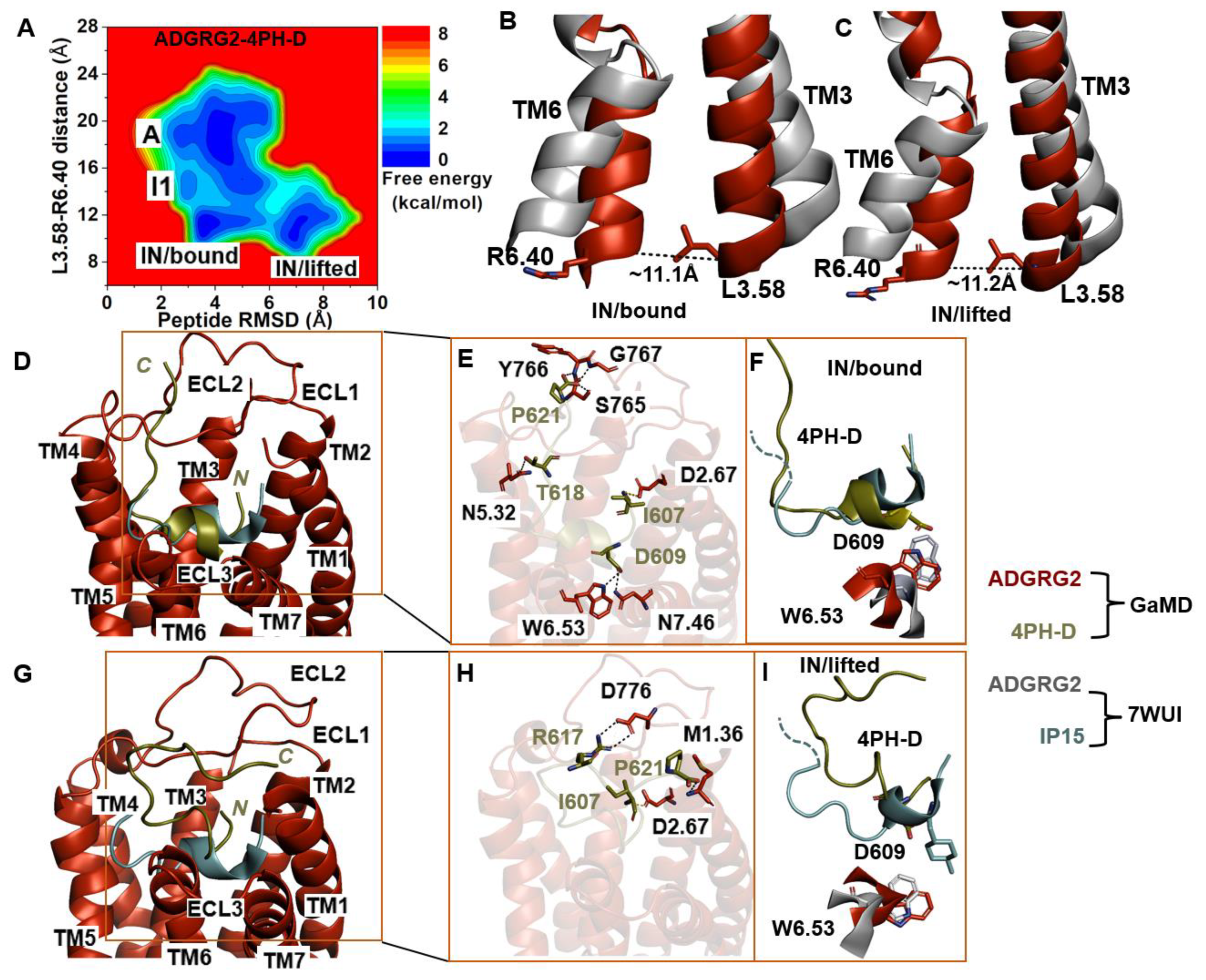
2.4. Antagonist 4PH-D-Bound ADGRG2 Sampled the “A”, “I1”, and “IN/Bound” “IN/Lifted” States
3. Discussion
4. Materials and Methods
4.1. Gaussian Accelerated Molecular Dynamics (GaMD)
4.2. Simulation Protocol
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Irannejad, R.; Tsvetanova, N.G.; Lobingier, B.T.; von Zastrow, M. Effects of endocytosis on receptor-mediated signaling. Curr. Opin. Cell Biol. 2015, 35, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Inoue, A.; Raimondi, F.; Kadji, F.M.N.; Singh, G.; Kishi, T.; Uwamizu, A.; Ono, Y.; Shinjo, Y.; Ishida, S.; Arang, N.; et al. Illuminating G-protein-coupling selectivity of GPCRs. Cell 2019, 177, 1933–1947. [Google Scholar] [CrossRef] [PubMed]
- Ellaithy, A.; Gonzalez-Maeso, J.; Logothetis, D.A.; Levitz, J. Structural and Biophysical Mechanisms of Class C G Protein-Coupled Receptor Function. Trends Biochem. Sci. 2020, 45, 1049–1064. [Google Scholar] [CrossRef] [PubMed]
- Salmaso, V.; Jacobson, K.A. Purinergic Signaling: Impact of GPCR Structures on Rational Drug Design. ChemMedChem 2020, 4, 1958–1973. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.K.; Anderson, G.R.; Araç, D.; Aust, G.; Balenga, N.; Boucard, A.; Bridges, J.P.; Engel, F.B.; Formstone, C.J.; Glitsch, M.D.; et al. The expanding functional roles and signaling mechanisms of adhesion G protein-coupled receptors. Ann. N. Y. Acad. Sci. 2019, 1456, 5–25. [Google Scholar] [CrossRef] [PubMed]
- Bassilana, F.; Nash, M.; Ludwig, M.-G. Adhesion G protein-coupled receptors: Opportunities for drug discovery. Nat. Rev. Drug Discov. 2019, 18, 869–884. [Google Scholar] [CrossRef] [PubMed]
- Bondarev, A.D.; Attwood, M.M.; Jonsson, J.; Chubarev, V.N.; Tarasov, V.V.; Schiöth, H.B. Opportunities and challenges for drug discovery in modulating adhesion G protein-coupled receptor (GPCR) functions. Expert Opin. Drug Discov. 2020, 15, 1291–1307. [Google Scholar] [CrossRef]
- Balenga, N.; Azimzadeh, P.; Hogue, J.; Staats, P.N.; Shi, Y.; Koh, J.; Dressman, H.; Olson, J.A. Orphan adhesion GPCR GPR64/ADGRG2 Is overexpressed in parathyroid tumors and attenuates calcium-sensing receptor-mediated signaling. J. Bone Miner. Res. 2017, 32, 654–666. [Google Scholar] [CrossRef]
- Hamann, J.; Aust, G.; Araç, D.; Engel, F.B.; Formstone, C.; Fredriksson, R.; Hall, R.A.; Harty, B.L.; Kirchhoff, C.; Knapp, B.; et al. International Union of Basic and Clinical Pharmacology. XCIV. Adhesion G protein-coupled receptors. Pharmacol. Rev. 2015, 67, 338–367. [Google Scholar] [CrossRef]
- Purcell, R.H.; Hall, R.A. Adhesion G protein-coupled receptors as drug targets. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 429–449. [Google Scholar] [CrossRef] [PubMed]
- Paavola, K.J.; Hall, R.A. Adhesion G protein-coupled receptors: Signaling, pharmacology, and mechanisms of activation. Mol. Pharmacol. 2012, 82, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Patat, O.; Pagin, A.; Siegfried, A.; Mitchell, V.; Chassaing, N.; Faguer, S.; Monteil, L.; Gaston, V.; Bujan, L.; Courtade-Saïdi, M.; et al. Truncating mutations in the adhesion g protein-coupled receptor G2 gene ADGRG2 cause an X-linked congenital bilateral absence of vas deferens. Am. J. Hum. Genet. 2016, 99, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Wang, J.; Zhang, W.; Pan, T. Pathogenic role of ADGRG2 in CBAVD patients replicated in Chinese population. Andrology 2017, 5, 954–957. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Gao, Y.; Ma, C.; Shen, Q.; Wang, J.; Lv, M.; Liu, C.; Cheng, H.; Zhu, F.; Tian, S.; et al. A novel hemizygous loss-of-function mutation in ADGRG2 causes male infertility with congenital bilateral absence of the vas deferens. J. Assist. Reprod. Genet. 2020, 6, 1421–1429. [Google Scholar] [CrossRef] [PubMed]
- Richter, G.H.; Fasan, A.; Hauer, K.; Grunewald, T.G.P.; Berns, C.; Rössler, S.; Naumann, I.; Staege, M.S.; Fulda, S.; Esposito, I.; et al. G-Protein coupled receptor 64 promotes invasiveness and metastasis in Ewing sarcomas through PGF and MMP1. J. Pathol. 2013, 230, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Whittier, K.L.; Boese, E.A.; Gibson-Corley, K.N.; Kirby, P.A.; Darbro, B.W.; Qian, Q.; Ingram, W.J.; Robertson, T.; Remke, M.; Taylor, M.D.; et al. G-protein coupled receptor expression patterns delineate medulloblastoma subgroups. Acta Neuropathol. Commun. 2013, 1, 66. [Google Scholar] [CrossRef] [PubMed]
- Peeters, M.C.; Fokkelman, M.; Boogaard, B.; Egerod, K.L.; van de Water, B.; Ijzerman, A.P.; Schwartz, T.W. The adhesion G protein-coupled receptor G2 (ADGRG2/GPR64) constitutively activates SRE and NFkappaB and is involved in cell adhesion and migration. Cell. Signal. 2015, 27, 2579–2588. [Google Scholar] [CrossRef]
- Nakamura, K.; Asanuma, K.; Okamoto, T.; Yoshida, K.; Matsuyama, Y.; Kita, K.; Hagi, T.; Nakamura, T.; Sudo, A. GPR64, Screened from Ewing Sarcoma Cells, Is a Potential Target for Antibody-Based Therapy for Various Sarcomas. Cancers 2022, 14, 814. [Google Scholar] [CrossRef]
- Kaur, B.; Brat, D.J.; Devi, N.S.; Van Meir, E.G. Vasculostatin, a proteolytic fragment of brain angiogenesis inhibitor 1, is an antiangiogenic and antitumorigenic factor. Oncogene 2005, 24, 3632–3642. [Google Scholar] [CrossRef]
- Eubelen, M.; Bostaille, N.; Cabochette, P.; Gauquier, A.; Tebabi, P.; Dumitru, A.C.; Koehler, M.; Gut, P.; Alsteens, D.; Stainier, D.Y.R.; et al. A molecular mechanism for Wnt ligand-specific signaling. Science 2018, 361, eaat1178. [Google Scholar] [CrossRef] [PubMed]
- Little, K.D.; Hemler, M.E.; Stipp, C.S. Dynamic regulation of a GPCR–tetraspanin–G protein complex on intact cells: Central role of CD81 in facilitating GPR56–Gαq/11 association. Mol. Biol. Cell 2004, 15, 2375–2387. [Google Scholar] [CrossRef] [PubMed]
- Ward, Y.; Lake, R.; Yin, J.J.; Heger, C.D.; Raffeld, M.; Goldsmith, P.K.; Merino, M.; Kelly, K. LPA receptor heterodimerizes with CD97 to amplify LPA-initiated RHO-dependent signaling and invasion in prostate cancer cells. Cancer Res. 2011, 71, 7301–7311. [Google Scholar] [CrossRef] [PubMed]
- Kuffer, A.; Lakkaraju, A.K.K.; Mogha, A.; Petersen, S.C.; Airich, K.; Doucerain, C.; Marpakwar, R.; Bakirci, P.; Senatore, A.; Monnard, A.; et al. The prion protein is an agonistic ligand of the G protein-coupled receptor Adgrg6. Nature 2016, 536, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Jeong, S.-J.; Jin, Z.; Strokes, N.; Li, S.; Piao, X. G protein-coupled receptor 56 and collagen III, a receptor-ligand pair, regulates cortical development and lamination. Proc. Natl Acad. Sci. USA 2011, 108, 12925–12930. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.; Liu, J.; Shi, Q.; Tan, Q.; Wu, D.; Skinner, J.J.; Walker, A.L.; Zhao, L.; Gu, X.; Chen, N.; et al. In vitro expression and analysis of the 826 human G protein-coupled receptors. Protein Cell 2016, 7, 325–337. [Google Scholar] [CrossRef]
- Liebscher, I.; Schoneberg, T. Tethered agonism: A common activation mechanism of adhesion GPCRs. In Handbook of Experimental Pharmacology; Springer: Cham, Switzerland, 2016; Volume 234, pp. 111–125. [Google Scholar]
- Beliu, G.; Altrichter, S.; Guixà-González, R.; Hemberger, M.; Brauer, I.; Dahse, A.-K.; Scholz, N.; Wieduwild, R.; Kuhlemann, A.; Batebi, H.; et al. Tethered agonist exposure in intact adhesion/class B2 GPCRs through intrinsic structural flexibility of the GAIN domain. Mol. Cell 2021, 81, 905–921. [Google Scholar] [CrossRef]
- Bianchi, E.; Sun, Y.; Almansa-Ordonez, A.; Woods, M.; Goulding, D.; Martinez-Martin, N.; Wright, G.J. Control of oviductal fluid flow by the G-protein coupled receptor Adgrd1 is essential for murine embryo transit. Nat. Commun. 2021, 12, 1251. [Google Scholar] [CrossRef]
- Wilde, C.; Fischer, L.; Lede, V.; Kirchberger, J.; Rothemund, S.; Schöneberg, T.; Liebscher, I. The constitutive activity of the adhesion GPCR GPR114/ADGRG5 is mediated by its tethered agonist. FASEB J. 2016, 30, 666–673. [Google Scholar] [CrossRef]
- Frenster, J.D.; Stephan, G.; Ravn-Boess, N.; Bready, D.; Wilcox, J.; Kieslich, B.; Wilde, C.; Sträter, N.; Wiggin, G.R.; Liebscher, I.; et al. Functional impact of intramolecular cleavage and dissociation of adhesion G protein-coupled receptor GPR133 (ADGRD1) on canonical signaling. J. Biol. Chem. 2021, 296, 100798. [Google Scholar] [CrossRef]
- Petersen, S.C.; Luo, R.; Liebscher, I.; Giera, S.; Jeong, S.-J.; Mogha, A.; Ghidinelli, M.; Feltri, M.L.; Schöneberg, T.; Piao, X.; et al. The adhesion GPCR GPR126 has distinct, domain-dependent functions in Schwann cell development mediated by interaction with laminin-211. Neuron 2015, 85, 755–769. [Google Scholar] [CrossRef] [PubMed]
- Scholz, N.; Guan, C.; Nieberler, M.; Grotemeyer, A.; Maiellaro, I.; Gao, S.; Beck, S.; Pawlak, M.; Sauer, M.; Asan, E.; et al. Mechano-dependent signaling by Latrophilin/CIRL quenches cAMP in proprioceptive neurons. eLife 2017, 6, e28360. [Google Scholar] [CrossRef] [PubMed]
- Yeung, J.; Adili, R.; Stringham, E.N.; Luo, R.; Vizurraga, A.; Rosselli-Murai, L.K.; Stoveken, H.M.; Yu, M.; Piao, X.; Holinstat, M.; et al. GPR56/ADGRG1 is a platelet collagen-responsive GPCR and hemostatic sensor of shear force. Proc. Natl. Acad. Sci. USA 2020, 117, 28275–28286. [Google Scholar] [CrossRef] [PubMed]
- Liebscher, I.; Schön, J.; Petersen, S.C.; Fischer, L.; Auerbach, N.; Demberg, L.M.; Mogha, A.; Cöster, M.; Simon, K.-U.; Rothemund, S.; et al. A tethered agonist within the ectodomain activates the adhesion G protein-coupled receptors GPR126 and GPR133. Cell Rep. 2014, 9, 2018–2026. [Google Scholar] [CrossRef] [PubMed]
- Monk, K.R.; Hamann, J.; Langenhan, T.; Nijmeijer, S.; Schöneberg, T.; Liebscher, I. Adhesion G protein-coupled receptors: From in vitro pharmacology to in vivo mechanisms. Mol. Pharmacol. 2015, 88, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Demberg, L.M.; Winkler, J.; Wilde, C.; Simon, K.-U.; Schön, J.; Rothemund, S.; Schöneberg, T.; Prömel, S.; Liebscher, I. Activation of adhesion G protein-coupled receptors: Agonist specificity of Stachel sequence-derived peptides. J. Biol. Chem. 2017, 292, 4383–4394. [Google Scholar] [CrossRef] [PubMed]
- Salzman, G.S.; Zhang, S.; Gupta, A.; Koide, A.; Koide, S.; Araç, D. Stachel-independent modulation of GPR56/ADGRG1 signaling by synthetic ligands directed to its extracellular region. Proc. Natl. Acad. Sci. USA. 2017, 114, 10095–10100. [Google Scholar] [CrossRef] [PubMed]
- Azimzadeh, P.; Talamantez-Lyburn, S.C.; Chang, K.T.; Inoue, A.; Balenga, N. Spatial regulation of GPR64/ADGRG2 signaling by beta-arrestins and GPCR kinases. Ann. N. Y. Acad. Sci. 2019, 1456, 26–43. [Google Scholar] [CrossRef]
- Xiao, P.; Guo, S.; Wen, X.; He, Q.-T.; Lin, H.; Huang, S.-M.; Gou, L.; Zhang, C.; Yang, Z.; Zhong, Y.-N.; et al. Tethered peptide activation mechanism of the adhesion GPCRs ADGRG2 and ADGRG4. Nature 2022, 7907, 771–778. [Google Scholar] [CrossRef]
- Zhang, D.L.; Sun, Y.J.; Ma, M.L.; Wang, Y.J.; Lin, H.; Li, R.R.; Liang, Z.L.; Gao, Y.; Yang, Z.; He, D.F.; et al. Gq activity- and beta-arrestin-1 scaffolding-mediated ADGRG2/CFTR coupling are required for male fertility. eLife 2018, 7, e33432. [Google Scholar] [CrossRef]
- Kishore, A.; Purcell, R.H.; Nassiri-Toosi, Z.; Hall, R.A. Stalk-dependent and Stalk-independent Signaling by the Adhesion G Protein-coupled Receptors GPR56 (ADGRG1) and BAI1 (ADGRB1). J. Biol. Chem. 2016, 291, 3385–3394. [Google Scholar] [CrossRef] [PubMed]
- Gad, A.A.; Azimzadeh, P.; Balenga, N. Conserved residues in the extracellular loop 2 regulate Stachel-mediated activation of ADGRG2. Sci. Rep. 2021, 11, 14060. [Google Scholar] [CrossRef] [PubMed]
- Stoveken, H.M.; Hajduczok, A.G.; Xu, L.; Tall, G.G. Adhesion G protein-coupled receptors are activated by exposure of a cryptic tethered agonist. Proc. Natl. Acad. Sci. USA 2015, 112, 6194–6199. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhang, D.; Ma, M.-L.; Lin, H.; Song, Y.; Wang, J.; Ma, C.; Yu, K.; An, W.; Guo, S.; et al. Optimization of a peptide ligand for the adhesion GPCR ADGRG2 provides a potent tool to explore receptor biology. J. Biol. Chem. 2021, 296, 100174. [Google Scholar] [CrossRef] [PubMed]
- Demberg, L.M.; Rothemund, S.; Schoneberg, T.; Liebscher, I. Identification of the tethered peptide agonist of the adhesion G protein coupled receptor GPR64/ADGRG2. Biochem. Biophys. Res. Commun. 2015, 464, 743–747. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Feher, V.A.; Mccammon, J.A. Gaussian accelerated molecular dynamics: Unconstrained enhanced sampling and free energy calculation. J. Chem. Theory Comput. 2015, 11, 3584–3595. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Mccammon, J.A. Graded activation and free energy landscapes of a muscarinic G-protein–coupled receptor. Proc. Natl. Acad. Sci. USA 2016, 113, 12162–12167. [Google Scholar] [CrossRef]
- Miao, Y.; Mccammon, J.A. Gaussian Accelerated Molecular Dynamics: Theory, Implementation and Applications. Annu. Rep. Comput. Chem. 2017, 13, 231–278. [Google Scholar]
- Pang, Y.T.; Miao, Y.; Wang, Y.; Mccammon, J.A. Gaussian Accelerated Molecular Dynamics in NAMD. J. Chem. Theory Comput. 2017, 13, 9–19. [Google Scholar] [CrossRef]
- Wang, Y.T.; Chan, Y.H. Understanding the molecular basis of agonist/antagonist mechanism of human mu opioid receptor through gaussian accelerated molecular dynamics method. Sci. Rep. 2017, 7, 7828. [Google Scholar] [CrossRef]
- Chuang, C.H.; Chiou, S.J.; Cheng, T.L.; Wang, Y.T. A molecular dynamics simulation study decodes the Zika virus NS5 methyltransferase bound to SAH and RNA analogue. Sci. Rep. 2018, 8, 6336. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.M.; Wang, Y.T. In silico studies of conformational dynamics of Mu opioid receptor performed using gaussian accelerated molecular dynamics. J. Biomol. Struct. Dynam. 2018, 37, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Bhattarai, A.; Nguyen, A.T.N.; Christopoulos, A.; May, L.T. Structural Basis for Binding of Allosteric Drug Leads in the Adenosine A1 Receptor. Sci. Rep. 2018, 8, 16836. [Google Scholar] [CrossRef] [PubMed]
- Palermo, G.; Miao, Y.; Walker, R.C.; Jinek, M.; Mccammon, J.A. CRISPR-Cas9 conformational activation as elucidated from enhanced molecular simulations. Proc. Natl. Acad. Sci. USA 2017, 114, 7260–7265. [Google Scholar] [CrossRef] [PubMed]
- Park, J.B.; Kim, Y.H.; Yoo, Y.; Kim, J.; Jun, S.H.; Cho, J.W.; El Qaidi, S.; Walpole, S.; Monaco, S.; Garcia-Garcia, A.A.; et al. Structural basis for arginine glycosylation of host substrates by bacterial effector proteins. Nat. Commun. 2018, 9, 4283. [Google Scholar] [CrossRef] [PubMed]
- Sibener, L.V.; Fernandes, R.A.; Kolawole, E.M.; Carbone, C.B.; Liu, F.; Mcaffee, D.; Birnbaum, M.E.; Yang, X.; Su, L.F.; Yu, W.; et al. Isolation of a Structural Mechanism for Uncoupling T Cell Receptor Signaling from Peptide-MHC Binding. Cell 2018, 174, 672–687. [Google Scholar] [CrossRef]
- Bhattarai, A.; Wang, J.; Miao, Y. G-Protein-Coupled Receptor-Membrane Interactions Depend on the Receptor Activation State. J. Comput. Chem. 2019, 41, 460–471. [Google Scholar] [CrossRef]
- Ricci, C.G.; Chen, J.S.; Miao, Y.; Jinek, M.; Doudna, J.A.; Mccammon, J.A.; Palermo, G. Deciphering Off-Target Effects in CRISPR-Cas9 through Accelerated Molecular Dynamics. ACS Cent. Sci. 2019, 5, 651–662. [Google Scholar] [CrossRef]
- Pawnikar, S.; Miao, Y. Pathway and mechanism of drug binding to chemokine receptors revealed by accelerated molecular simulations. Future Med. Chem. 2020, 12, 1213–1225. [Google Scholar] [CrossRef]
- Vuckovic, Z.; Wang, J.; Pham, V.; Mobbs, J.I.; Belousoff, M.J.; Bhattarai, A.; Burger, W.A.C.; Thompson, G.; Yeasmin, M.; Nawaratne, V.; et al. Pharmacological hallmarks of allostery at the M4 muscarinic receptor elucidated through structure and dynamics. eLife 2023, 12, e83477. [Google Scholar] [CrossRef]
- Li, Y.; Sun, J.; Li, D.; Lin, J. The full activation mechanism of the adenosine A1 receptor revealed by GaMD and Su-GaMD simulations. Proc. Natl. Acad. Sci. USA 2022, 18, e2203702119. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Bai, Q.; Bing, Z.; Liu, H.; Yao, X. Insights into the molecular mechanism of positive cooperativity between partial agonist MK-8666 and full allosteric agonist AP8 of hGPR40 by Gaussian accelerated molecular dynamics (GaMD) simulations. Comput. Struct. Biotechnol. J. 2021, 19, 3978–3989. [Google Scholar] [CrossRef] [PubMed]
- Úsuga-Acevedo, B.; García, Y.; Díaz, C.F.; Jiménez, V.A. Rational Discovery of Microtubule-Stabilizing Peptides. J. Chem. Inf. Model. 2022, 62, 6844–6856. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; Zhang, H.; Gou, R.; Luo, D.; Liu, Z.; Zhu, F.; Xue, W. Structure-Based Discovery of a Novel Allosteric Inhibitor against Human Dopamine Transporter. J. Chem. Inf. Model. 2023, 63, 4458–4467. [Google Scholar] [CrossRef] [PubMed]
- Bao, H.; Wang, W.; Sun, H.; Chen, J. Probing mutation-induced conformational transformation of the GTP/M-RAS complex through Gaussian accelerated molecular dynamics simulations. J. Enzym. Inhib. Med. Chem. 2023, 38, 2195995. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, J.; Zeng, Q.; Wang, W.; Sun, H.; Wei, B. Exploring the deactivation mechanism of human β2 adrenergic receptor by accelerated molecular dynamic simulations. Front. Mol. Biosci. 2022, 9, 972463. [Google Scholar] [CrossRef] [PubMed]
- Ballesteros, J.A.; Weinstein, H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 1995, 25, 366–428. [Google Scholar] [CrossRef]
- ADGRG2 (agrg2_human). Available online: https://gpcrdb.org/protein/agrg2_human/ (accessed on 18 December 2023).
- Dror, R.O.; Arlow, D.H.; Maragakis, P.; Mildorf, T.J.; Pan, A.C.; Xu, H.; Borhani, D.W.; Shaw, D.E. Activation mechanism of the β2-adrenergic receptor. Proc. Natl. Acad. Sci. USA 2011, 108, 18684–18689. [Google Scholar] [CrossRef]
- Nygaard, R.; Zou, Y.; Dror, R.O.; Mildorf, T.J.; Arlow, D.H.; Manglik, A.; Pan, A.C.; Liu, C.W.; Fung, J.J.; Bokoch, M.P.; et al. The dynamic process of β(2)-adrenergic receptor activation. Cell 2013, 152, 532–542. [Google Scholar] [CrossRef]
- Miao, Y.; Nichols, S.E.; Gasper, P.M.; Metzger, V.T.; McCammon, J.A. Activation and dynamic network of the M2 muscarinic receptor. Proc. Natl. Acad. Sci. USA 2013, 110, 10982–10987. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Im, W. Automated builder and database of protein/membrane complexes for molecular dynamics simulations. PLoS ONE 2007, 2, e880. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Brooks, C.L., 3rd; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Lim, J.B.; Klauda, J.B.; Im, W. CHARMM-GUI Membrane Builder for mixed bilayers and its application to yeast membranes. Biophys. J. 2009, 97, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Wu, E.L.; Cheng, X.; Jo, S.; Rui, H.; Song, K.C.; Dávila-Contreras, E.M.; Qi, Y.; Lee, J.; Monje-Galvan, V.; Venable, R.M.; et al. CHARMM-GUI Membrane Builder toward realistic biological membrane simulations. J. Comput. Chem. 2014, 35, 1997–2004. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Mackerell, A.D., Jr. CHARMM additive and polarizable force fields for biophysics and computer-aided drug design. Biochem. Biophys. Acta 2014, 1850, 861–871. [Google Scholar] [CrossRef]
- Case, D.A.; Aktulga, H.M.; Belfon, K.; Ben-Shalom, I.Y.; Berryman, J.T.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cisneros, G.A.; Cruzeiro, V.W.D. Amber 2023; University of California: San Francisco, CA, USA, 2023. [Google Scholar]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., 3rd. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Grant, B.J.; Rodrigues, A.P.; ElSawy, K.M.; McCammon, J.A.; Caves, L.S. Bio3d: An R package for the comparative analysis of protein structures. Bioinformatics 2006, 22, 2695–2696. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Sinko, W.; Pierce, L.; Bucher, D.; Walker, R.C.; Mccammon, J.A. Improved reweighting of accelerated molecular dynamics simulations for free energy calculation. J. Chem. Theory Comput. 2014, 10, 2677–2689. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | State | L3.58-R6.40 Distance (Å) | Peptide-Receptor Interactions | |
|---|---|---|---|---|
| Hydrogen Bonding | Salt-Bridge | |||
| ADGRG2-IP15-Gs | Active | ~18.7 Å | R617-N5.32, R617-I780ECL2, R617-W779ECL2, L615-Y5.36 | I607-D2.67 |
| ADGRG2-IP15 | Intermediate | ~14.7 Å | R617-N5.32, R617-I780ECL2, L612-W779ECL2, L615-Y5.36, I607-D2.67 | I607-D2.67 |
| ADGRG2-4PH-E | Inactive | ~11.8 Å | L620-N781ECL2, S619-N781ECL2, R617- N5.32, R617-I780ECL2, L612-N7.46, S608- D2.67, I607-D776ECL2 | _ |
| ADGRG2-4PH-D | Inactive/bound | ~11.1 Å | P621-Y766ECL2, P621- S765ECL2, P621-G767ECL2, T618-N5.32, D609-W6.53, D609-N7.46 | I607-D2.67 |
| Inactive/lifted | ~11.2 Å | P621-M1.36, R617-D776ECL2 | I607-D2.67 | |
| System (a) | Natoms | Dimension (Å3) | cMD Runs (ns) | GaMD Equilibration (ns) | GaMD Production (ns) | ΔV(b) (Kcal/mol) |
|---|---|---|---|---|---|---|
| ADGRG2-IP15-Gs | 199,976 | 108 × 108 × 180 | 10 | 64 | 1200 ns × 3 | 14.42 ± 4.31 |
| ADGRG2-IP15 | 95,486 | 96 × 96 × 128 | 10 | 40 | 2000 ns × 3 | 14.08 ± 4.23 |
| ADGRG2-4PH-E | 93,332 | 96 × 96 × 128 | 10 | 40 | 2000 ns × 3 | 13.85 ± 4.20 |
| ADGRG2-4PH-D | 93,929 | 96 × 96 × 128 | 10 | 40 | 2000 ns × 3 | 14.26 ± 4.27 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adediwura, V.A.; Miao, Y. Mechanistic Insights into Peptide Binding and Deactivation of an Adhesion G Protein-Coupled Receptor. Molecules 2024, 29, 164. https://doi.org/10.3390/molecules29010164
Adediwura VA, Miao Y. Mechanistic Insights into Peptide Binding and Deactivation of an Adhesion G Protein-Coupled Receptor. Molecules. 2024; 29(1):164. https://doi.org/10.3390/molecules29010164
Chicago/Turabian StyleAdediwura, Victor A., and Yinglong Miao. 2024. "Mechanistic Insights into Peptide Binding and Deactivation of an Adhesion G Protein-Coupled Receptor" Molecules 29, no. 1: 164. https://doi.org/10.3390/molecules29010164
APA StyleAdediwura, V. A., & Miao, Y. (2024). Mechanistic Insights into Peptide Binding and Deactivation of an Adhesion G Protein-Coupled Receptor. Molecules, 29(1), 164. https://doi.org/10.3390/molecules29010164