

The Role of H-Bonds in the Excited-State Properties of Multichromophoric Systems: Static and Dynamic Aspects

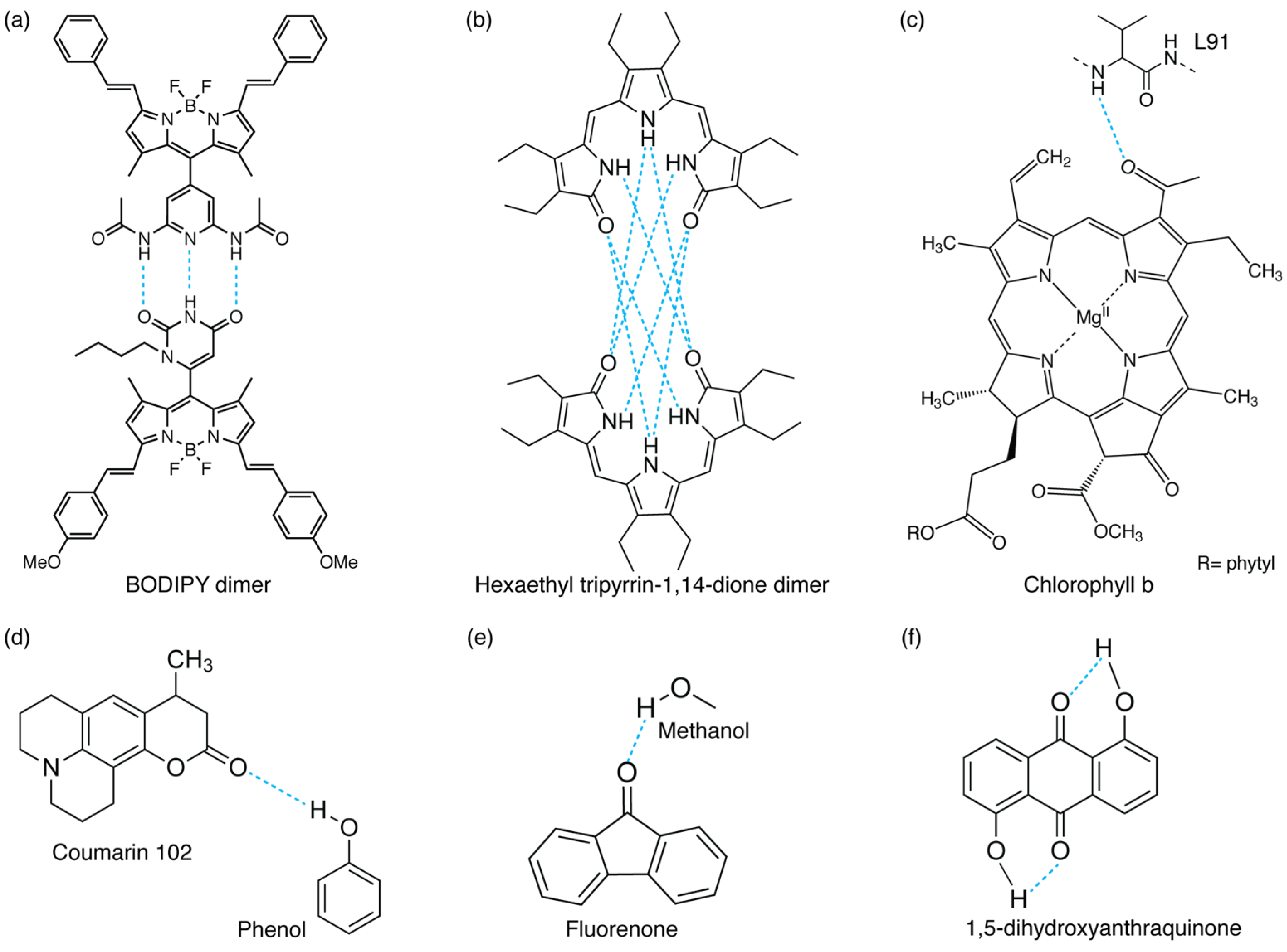{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction

2. Experimental Techniques
2.1. Resonance Raman Spectroscopy
2.2. Time-Resolved Raman Spectroscopy
2.3. Time-Resolved Fluorescence Spectroscopy
2.4. Transient Absorption Spectroscopy
2.5. Multidimensional Optical Spectroscopies

3. Role of H-Bonds in Modulating Electronic Properties
3.1. Shift of Electronic Spectral Peaks in Protic Solvents
3.2. Shift of Electronic Spectral Peaks in a Protein Environment
3.3. Intra- and Intermolecular H-Bonding Interactions
3.4. Excited-State Proton and Hydrogen Transfer
4. Role of H-Bonds in Excited-State Dynamics
4.1. Early Proofs That H-Bonds Affect the Excited-State Relaxation Dynamics: The Example of Anthraquinones
4.2. Femtosecond Dynamics of H-Bonds in the Excited State
4.3. Dynamics of H-Bonded Dimers
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zhao, G.J.; Han, K.L. Hydrogen bonding in the electronic excited state. Acc. Chem. Res. 2012, 45, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Hoober, J.K.; Eggink, L.L.; Chen, M. Chlorophylls, ligands and assembly of light-harvesting complexes in chloroplasts. Photosynth. Res. 2007, 94, 387–400. [Google Scholar] [CrossRef]
- Wang, D.; Hao, C.; Wang, S.; Dong, H.; Qiu, J. A theoretical forecast of the hydrogen bond changes in the electronic excited state for BN and its derivatives. Cent. Eur. J. Phys. 2012, 10, 116–123. [Google Scholar] [CrossRef]
- Fresch, E.; Meneghin, E.; Agostini, A.; Paulsen, H.; Carbonera, D.; Collini, E. How the Protein Environment Can Tune the Energy, the Coupling, and the Ultrafast Dynamics of Interacting Chlorophylls: The Example of the Water-Soluble Chlorophyll Protein. J. Phys. Chem. Lett. 2020, 11, 1059–1067. [Google Scholar] [CrossRef] [PubMed]
- Fresch, E.; Peruffo, N.; Trapani, M.; Cordaro, M.; Bella, G.; Castriciano, M.A.; Collini, E. The effect of hydrogen bonds on the ultrafast relaxation dynamics of a BODIPY dimer. J. Chem. Phys. 2021, 154, 084201. [Google Scholar] [CrossRef] [PubMed]
- Flom, S.R.; Barbara, P.F. Proton transfer and hydrogen bonding in the internal conversion of S1 anthraquinones. J. Phys. Chem. 1985, 89, 4489–4494. [Google Scholar] [CrossRef]
- Palm, D.M.; Agostini, A.; Averesch, V.; Girr, P.; Werwie, M.; Takahashi, S.; Satoh, H.; Jaenicke, E.; Paulsen, H. Chlorophyll a/b binding-specificity in water-soluble chlorophyll protein. Nat. Plants 2018, 4, 920–929. [Google Scholar] [CrossRef]
- Swain, A.; Cho, B.; Gautam, R.; Curtis, C.J.; Tomat, E.; Huxter, V. Ultrafast Dynamics of Tripyrrindiones in Solution Mediated by Hydrogen-Bonding Interactions. J. Phys. Chem. B 2019, 123, 5524–5535. [Google Scholar] [CrossRef]
- Hubbard, R.E.; Kamran Haider, M. Hydrogen Bonds in Proteins: Role and Strength. In Encyclopedia of Life Sciences; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2010. [Google Scholar] [CrossRef]
- Gao, J.; Bosco, D.A.; Powers, E.T.; Kelly, J.W. Localized thermodynamic coupling between hydrogen bonding and microenvironment polarity substantially stabilizes proteins. Nat. Struct. Mol. Biol. 2009, 16, 684–690. [Google Scholar] [CrossRef]
- Horn, R.; Paulsen, H. Early steps in the assembly of light-harvesting chlorophyll a/b complex. J. Biol. Chem. 2004, 279, 44400–44406. [Google Scholar] [CrossRef]
- Natarajan, A.; Schwans, J.P.; Herschlag, D. Using unnatural amino acids to probe the energetics of oxyanion hole hydrogen bonds in the ketosteroid isomerase active site. J. Am. Chem. Soc. 2014, 136, 7643–7654. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.S.; Jacobsen, E.N. Asymmetric catalysis by chiral hydrogen-bond donors. Angew. Chem.-Int. Ed. 2006, 45, 1520–1543. [Google Scholar] [CrossRef] [PubMed]
- Klebe, G.; Böhm, H.-J. Energetic and Entropic Factors Determining Binding Affinity in Protein-Ligand Complexes. J. Recept. Signal Transduct. 1997, 17, 459–473. [Google Scholar] [CrossRef] [PubMed]
- Agostini, A.; Meneghin, E.; Gewehr, L.; Pedron, D.; Palm, D.M.; Carbonera, D.; Paulsen, H.; Jaenicke, E.; Collini, E. How water-mediated hydrogen bonds affect chlorophyll a/b selectivity in Water-Soluble Chlorophyll Protein. Sci. Rep. 2019, 9, 18255. [Google Scholar] [CrossRef]
- van der Lubbe, S.C.C.; Fonseca Guerra, C. The Nature of Hydrogen Bonds: A Delineation of the Role of Different Energy Components on Hydrogen Bond Strengths and Lengths. Chem.-Asian J. 2019, 14, 2760–2769. [Google Scholar]
- International Union of Pure and Applied Chemistry (IUPAC). Hydrogen Bond. In The IUPAC Compendium of Chemical Terminology; International Union of Pure and Applied Chemistry (IUPAC): Zurich, Switzerland, 2008; Volume 1077, p. 2899. [Google Scholar]
- Weinhold, F.; Klein, R.A. What is a hydrogen bond? Mutually consistent theoretical and experimental criteria for characterizing H-bonding interactions. Mol. Phys. 2012, 110, 565–579. [Google Scholar] [CrossRef]
- Rozas, I. On the nature of hydrogen bonds: An overview on computational studies and a word about patterns. Phys. Chem. Chem. Phys. 2007, 9, 2782–2790. [Google Scholar] [CrossRef]
- Phipps, M.J.S.; Fox, T.; Tautermann, C.S.; Skylaris, C.K. Energy decomposition analysis approaches and their evaluation on prototypical protein-drug interaction patterns. Chem. Soc. Rev. 2015, 44, 3177–3211. [Google Scholar] [CrossRef]
- You, X.; Baiz, C.R. Importance of Hydrogen Bonding in Crowded Environments: A Physical Chemistry Perspective. J. Phys. Chem. A 2022, 126, 5881–5889. [Google Scholar] [CrossRef]
- Raffy, G.; Ray, D.; Chu, C.C.; Del Guerzo, A.; Bassani, D.M. Effect of hydrogen-bonding on the excited-state reactivity of fullerene derivatives and its impact on the control of the emission polarisation from photopolic single crystals. Phys. Chem. Chem. Phys. 2012, 14, 8859–8865. [Google Scholar] [CrossRef]
- Kenfack, C.A.; Klymchenko, A.S.; Duportail, G.; Burger, A.; Mély, Y. Ab initio study of the solvent H-bonding effect on ESIPT reaction and electronic transitions of 3-hydroxychromone derivatives. Phys. Chem. Chem. Phys. 2012, 14, 8910–8918. [Google Scholar] [CrossRef] [PubMed]
- Scrutton, N.S.; Louise Groot, M.; Heyes, D.J. Excited state dynamics and catalytic mechanism of the light-driven enzyme protochlorophyllide oxidoreductase. Phys. Chem. Chem. Phys. 2012, 14, 8818–8824. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Ma, F.C. Intermolecular hydrogen-bonding effects on photophysics and photochemistry. Int. Rev. Phys. Chem. 2013, 32, 589–609. [Google Scholar] [CrossRef]
- Krasilnikov, P.M.; Mamonov, P.A.; Knox, P.P.; Paschenko, V.Z.; Rubin, A.B. The influence of hydrogen bonds on electron transfer rate in photosynthetic RCs. Biochim. Biophys. Acta-Bioenerg. 2007, 1767, 541–549. [Google Scholar] [CrossRef]
- Sessler, J.L.; Sathiosatham, M.; Brown, C.T.; Rhodes, T.A.; Wiederrecht, G. Hydrogen-bond-mediated photoinduced electron-transfer: Novel dimethylaniline-anthracene ensembles formed via Watson-Crick base-pairing. J. Am. Chem. Soc. 2001, 123, 3655–3660. [Google Scholar] [CrossRef]
- Sánchez, L.; Sierra, M.; Martín, N.; Myles, A.J.; Dale, T.J.; Rebek, J., Jr.; Seitz, W.; Guldi, D.M. Exceptionally Strong Electronic Communication through Hydrogen Bonds in Porphyrin–C60 Pairs. Angew. Chem. Int. Ed. 2006, 45, 4637–4641. [Google Scholar] [CrossRef]
- Yu, M.L.; Wang, S.M.; Feng, K.; Khoury, T.; Crossley, M.J.; Yang, F.; Zhang, J.P.; Tung, C.H.; Wu, L.Z. Photoinduced electron transfer and charge-recombination in 2-ureido-4[1 H]-pyrimidinone quadruple hydrogen-bonded porphyrin-fullerene assemblies. J. Phys. Chem. C 2011, 115, 23634–23641. [Google Scholar] [CrossRef]
- Otsuki, J. Supramolecular approach towards light-harvesting materials based on porphyrins and chlorophylls. J. Mater. Chem. A 2018, 6, 6710–6753. [Google Scholar] [CrossRef]
- Dey, A.; Dana, J.; Aute, S.; Das, A.; Ghosh, H.N. Hydrogen bond assisted photoinduced intramolecular electron transfer and proton coupled electron transfer in an ultrafast time domain using a ruthenium-anthraquinone dyad. Photochem. Photobiol. Sci. 2019, 18, 2430–2441. [Google Scholar] [CrossRef]
- Banno, M.; Ohta, K.; Yamaguchi, S.; Hirai, S.; Tominaga, K. Vibrational Dynamics of Hydrogen-Bonded Complexes in Solutions Studied with Ultrafast Infrared Pump−Probe Spectroscopy. Acc. Chem. Res. 2009, 42, 1259–1269. [Google Scholar] [CrossRef] [PubMed]
- Nibbering, E.T.J.; Elsaesser, T. Ultrafast vibrational dynamics of hydrogen bonds in the condensed phase. Chem. Rev. 2004, 104, 1887–1914. [Google Scholar] [CrossRef] [PubMed]
- Pines, E.; Pines, D.; Ma, Y.Z.; Fleming, G.R. Femtosecond pump-probe measurements of solvation by hydrogen-bonding interactions. ChemPhysChem 2004, 5, 1315–1327. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.J.; Han, K.L. Role of intramolecular and intermolecular hydrogen bonding in both singlet and triplet excited states of aminofluorenones on internal conversion, intersystem crossing, and twisted intramolecular charge transfer. J. Phys. Chem. A 2009, 113, 14329–14335. [Google Scholar] [CrossRef] [PubMed]
- Nibbering, E.T.J.; Tschirschwitz, F.; Chudoba, C.; Elsaesser, T. Femtochemistry of Hydrogen Bonded Complexes after Electronic Excitation in the Liquid Phase: The Case of Coumarin 102. J. Phys. Chem. A 2000, 104, 4247–4255. [Google Scholar] [CrossRef]
- Zhao, G.J.; Han, K.L. Ultrafast hydrogen bond strengthening of the photoexcited fluorenone in alcohols for facilitating the fluorescence quenching. J. Phys. Chem. A 2007, 111, 9218–9223. [Google Scholar] [CrossRef]
- Meneghin, E.; Pedron, D.; Collini, E. Raman and 2D electronic spectroscopies: A fruitful alliance for the investigation of ground and excited state vibrations in chlorophyll a. Chem. Phys. 2018, 514, 132–140. [Google Scholar] [CrossRef]
- Howard, A.A.; Tschumper, G.S.; Hammer, N.I. Effects of hydrogen bonding on vibrational normal modes of pyrimidine. J. Phys. Chem. A 2010, 114, 6803–6810. [Google Scholar] [CrossRef] [PubMed]
- Llansola-Portoles, M.J.; Li, F.; Xu, P.; Streckaite, S.; Ilioaia, C.; Yang, C.; Gall, A.; Pascal, A.A.; Croce, R.; Robert, B. Tuning antenna function through hydrogen bonds to chlorophyll a. Biochim. Biophys. Acta-Bioenerg. 2020, 1861, 148078. [Google Scholar] [CrossRef]
- Lapouge, K.; Näveke, A.; Sturgis, J.N.; Hartwich, G.; Renaud, D.; Simonin, I.; Lutz, M.; Scheer, H.; Robert, B. Non-Bonding Molecular Factors Influencing the Stretching Wavenumbers of the Conjugated Carbonyl Groups of Bacteriochlorophyll a. J. Raman Spectrosc. 1998, 29, 977–981. [Google Scholar] [CrossRef]
- Robert, B.; Lutz, M. Proteic Events following Charge Separation in the Bacterial Reaction Center: Resonance Raman Spectroscopy. Biochemistry 1988, 27, 5108–5114. [Google Scholar] [CrossRef]
- Sahoo, S.K.; Umapathy, S.; Parker, A.W. Time-resolved resonance raman spectroscopy: Exploring reactive intermediates. Appl. Spectrosc. 2011, 65, 1087–1115. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.E.J. Tutorial review. Time-resolved resonance Raman spectroscopy. Analyst 1996, 121, 107R–120R. [Google Scholar] [CrossRef]
- Hub, W.; Schneider, S.; Dörr, F. Time-Resolved Resonance Raman Spectroscopy. In Advances in Multi-Photon Processes and Spectroscopy; World Scientific: Singapore, 1999; Volume 2, pp. 239–332. [Google Scholar]
- Duyne, R.P.V.; Haynes, C.L. Raman Spectroscopy. In Encyclopedia of Physical Science and Technology, 3rd ed.; Meyers, R.A., Ed.; Academic Press: Cambridge, MA, USA, 2003; pp. 845–866. ISBN 9780122274107. [Google Scholar]
- Balos, V.; Kaliannan, N.K.; Elgabarty, H.; Wolf, M.; Kühne, T.D.; Sajadi, M. Time-resolved terahertz–Raman spectroscopy reveals that cations and anions distinctly modify intermolecular interactions of water. Nat. Chem. 2022, 14, 1031–1037. [Google Scholar] [CrossRef]
- Kneipp, J.; Balakrishnan, G.; Chen, R.; Shen, T.J.; Sahu, S.C.; Ho, N.T.; Giovannelli, J.L.; Simplaceanu, V.; Ho, C.; Spiro, T.G. Dynamics of allostery in hemoglobin: Roles of the penultimate tyrosine H bonds. J. Mol. Biol. 2006, 356, 335–353. [Google Scholar] [CrossRef]
- Han, F.; Liu, W.; Zhu, L.; Wang, Y.; Fang, C. Initial hydrogen-bonding dynamics of photoexcited coumarin in solution with femtosecond stimulated Raman spectroscopy. J. Mater. Chem. C 2016, 4, 2954–2963. [Google Scholar] [CrossRef]
- Iwata, K.; Yamaguchi, S.; Hamaguchi, H.O. Construction of a transform-limited picosecond time-resolved Raman spectrometer. Rev. Sci. Instrum. 1993, 64, 2140–2146. [Google Scholar] [CrossRef]
- Kukura, P.; McCamant, D.W.; Mathies, R.A. Femtosecond Stimulated Raman Spectroscopy. Annu. Rev. Phys. Chem. 2007, 58, 461–488. [Google Scholar] [CrossRef]
- Frontiera, R.R.; Mathies, R.A. Femtosecond stimulated Raman spectroscopy. Laser Photonics Rev. 2011, 5, 102–113. [Google Scholar] [CrossRef]
- Fang, C.; Frontiera, R.R.; Tran, R.; Mathies, R.A. Mapping GFP structure evolution during proton transfer with femtosecond Raman spectroscopy. Nature 2009, 462, 200–204. [Google Scholar] [CrossRef]
- Frontiera, R.R.; Fang, C.; Dasgupta, J.; Mathies, R.A. Probing structural evolution along multidimensional reaction coordinates with femtosecond stimulated Raman spectroscopy. Phys. Chem. Chem. Phys. 2012, 14, 405–414. [Google Scholar] [CrossRef]
- Dasgupta, J.; Frontiera, R.R.; Taylor, K.C.; Lagarias, J.C.; Mathies, R.A. Ultrafast excited-state isomerization in phytochrome revealed by femtosecond stimulated Raman spectroscopy. Proc. Natl. Acad. Sci. USA 2009, 106, 1784–1789. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Tang, L. Mapping structural dynamics of proteins with femtosecond stimulated raman spectroscopy. Annu. Rev. Phys. Chem. 2020, 71, 239–265. [Google Scholar] [CrossRef] [PubMed]
- Sciortino, A.; Messina, F. Ultrafast Optical Spectroscopies. In Spectroscopy for Materials Characterization; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2021; pp. 65–95. ISBN 9781119698029. [Google Scholar]
- Parson, W.W. Modern Optical Spectroscopy, 2nd ed.; Springer: Berlin/Heidelberg, Germany, 2015. [Google Scholar]
- Liu, Y.H.; Zhao, G.J.; Li, G.Y.; Han, K.L. Fluorescence quenching phenomena facilitated by excited-state hydrogen bond strengthening for fluorenone derivatives in alcohols. J. Photochem. Photobiol. A Chem. 2010, 209, 181–185. [Google Scholar] [CrossRef]
- Hossen, T.; Sahu, K. New Insights on Hydrogen-Bond-Induced Fluorescence Quenching Mechanism of C102-Phenol Complex via Proton Coupled Electron Transfer. J. Phys. Chem. A 2018, 122, 2394–2400. [Google Scholar] [CrossRef]
- Barman, N.; Singha, D.; Sahu, K. Fluorescence Quenching of Hydrogen-Bonded Coumarin 102-Phenol Complex: Effect of Excited-State Hydrogen Bonding Strength. J. Phys. Chem. 2013, 117, 3945–3953. [Google Scholar] [CrossRef]
- Biczók, L.; Bérces, T.; Linschitz, H. Quenching processes in hydrogen-bonded pairs: Interactions of excited fluorenone with alcohols and phenols. J. Am. Chem. Soc. 1997, 119, 11071–11077. [Google Scholar] [CrossRef]
- Herbich, J.; Kijak, M.; Zielińska, A.; Thummel, R.P.; Waluk, J. Fluorescence quenching by pyridine and derivatives induced by intermolecular hydrogen bonding to pyrrole-containing heteroaromatics. J. Phys. Chem. A 2002, 106, 2158–2163. [Google Scholar] [CrossRef]
- Biczók, L.; Bérces, T.; Inoue, H. Effects of molecular structure and hydrogen bonding on the radiationless deactivation of singlet excited fluorenone derivatives. J. Phys. Chem. A 1999, 103, 3837–3842. [Google Scholar] [CrossRef]
- Berera, R.; van Grondelle, R.; Kennis, J.T.M. Ultrafast transient absorption spectroscopy: Principles and application to photosynthetic systems. Photosynth. Res. 2009, 101, 105–118. [Google Scholar] [CrossRef]
- Fushitani, M. Applications of pump-probe spectroscopy. Annu. Rep. Prog. Chem.-Sect. C 2008, 104, 272–297. [Google Scholar] [CrossRef]
- van Stokkum, I.H.M.; Kloz, M.; Polli, D.; Viola, D.; Weißenborn, J.; Peerbooms, E.; Cerullo, G.; Kennis, J.T.M. Vibronic dynamics resolved by global and target analysis of ultrafast transient absorption spectra. J. Chem. Phys. 2021, 155, 114113. [Google Scholar] [CrossRef] [PubMed]
- Gelzinis, A.; Augulis, R.; Butkus, V.; Robert, B.; Valkunas, L. Two-dimensional spectroscopy for non-specialists. Biochim. Biophys. Acta-Bioenerg. 2019, 1860, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Collini, E. 2D electronic spectroscopic techniques towards quantum technology applications. In Proceedings of the SPIE OPTO: Ultrafast Phenomena and Nanophotonics XXVI, 2022, San Francisco, CA, USA, 22–28 February 2022; Volume 11999. [Google Scholar]
- Brańczyk, A.M.; Turner, D.B.; Scholes, G.D. Crossing disciplines-A view on two-dimensional optical spectroscopy. Ann. Phys. 2014, 526, 31–49. [Google Scholar] [CrossRef]
- Jonas, D.M. Two-dimensional femtosecond spectroscopy. Annu. Rev. Phys. Chem. 2003, 54, 425–463. [Google Scholar] [CrossRef]
- Hamm, P.; Zanni, M. Concepts and Methods of 2D Infrared Spectroscopy; Cambridge University Press: Cambridge, UK, 2011; ISBN 9781107000056. [Google Scholar]
- Branczyk, A.M.; Turner, D.B.; Scholes, G.D. Two-dimensional electronic spectroscopy for the quantum-optics enthusiast. arXiv 2013, arXiv:1307.5855. [Google Scholar]
- Lima, M.; Candelaresi, M.; Foggi, P. 2D-IR spectroscopy an additional dimension to investigate ultrafast structural. J. Raman Spectrosc. 2013, 44, 1470–1477. [Google Scholar] [CrossRef]
- Cho, M. Two-Dimensional Optical Spectroscopy; CRC Press: Boca Raton, Florida, 2009; ISBN 9781420084306. [Google Scholar]
- Petti, M.K.; Lomont, J.P.; Maj, M.; Zanni, M.T. Two-Dimensional Spectroscopy Is Being Used to Address Core Scientific Questions in Biology and Materials Science. J. Phys. Chem. B 2018, 122, 1771–1780. [Google Scholar] [CrossRef]
- Ghosh, A.; Ostrander, J.S.; Zanni, M.T. Watching Proteins Wiggle: Mapping Structures with Two-Dimensional Infrared Spectroscopy. Chem. Rev. 2017, 117, 10726–10759. [Google Scholar] [CrossRef]
- Roberts, S.T.; Ramasesha, K.; Tokmakoff, A. Structural rearrangements in water viewed through two-dimensional infrared spectroscopy. Acc. Chem. Res. 2009, 42, 1239–1249. [Google Scholar] [CrossRef]
- Elsaesser, T. Two-Dimensional Infrared Spectroscopy of Intermolecular Hydrogen Bonds in the Condensed Phase. Acc. Chem. Res. 2009, 42, 1220–1228. [Google Scholar] [CrossRef] [PubMed]
- Oliver, T.A.A. Recent advances in multidimensional ultrafast spectroscopy. R. Soc. Open Sci. 2018, 5, 171425. [Google Scholar] [CrossRef]
- Kolano, C.; Helbing, J.; Kozinski, M.; Sander, W.; Hamm, P. Watching hydrogen-bond dynamics in a β-turn by transient two-dimensional infrared spectroscopy. Nature 2006, 444, 469–472. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Hochstrasser, R.M. Chemical exchange 2D IR of hydrogen-bond making and breaking. Proc. Natl. Acad. Sci. USA 2005, 102, 11185–11190. [Google Scholar] [CrossRef] [PubMed]
- Stingel, A.M.; Petersen, P.B. Couplings Across the Vibrational Spectrum Caused by Strong Hydrogen Bonds: A Continuum 2D IR Study of the 7-Azaindole-Acetic Acid Heterodimer. J. Phys. Chem. B 2016, 120, 10768–10779. [Google Scholar] [CrossRef] [PubMed]
- Hunt, N.T. Transient 2D-IR spectroscopy of inorganic excited states. Dalt. Trans. 2014, 43, 17578–17589. [Google Scholar] [CrossRef]
- Collini, E. 2D Electronic Spectroscopic Techniques for Quantum Technology Applications. J. Phys. Chem. C 2021, 125, 13096–13108. [Google Scholar] [CrossRef] [PubMed]
- Volpato, A.; Collini, E. Time-frequency methods for coherent spectroscopy. Opt. Express 2015, 23, 20040. [Google Scholar] [CrossRef]
- Volpato, A.; Collini, E. Optimization and selection of time-frequency transforms for wave-packet analysis in ultrafast spectroscopy. Opt. Express 2019, 27, 2975–2987. [Google Scholar] [CrossRef]
- Butkus, V.; Zigmantas, D.; Valkunas, L.; Abramavicius, D. Vibrational vs. electronic coherences in 2D spectrum of molecular systems. Chem. Phys. Lett. 2012, 545, 40–43. [Google Scholar] [CrossRef]
- Meneghin, E.; Pedron, D.; Collini, E. Characterization of the coherent dynamics of bacteriochlorophyll a in solution. Chem. Phys. 2019, 519, 85–91. [Google Scholar] [CrossRef]
- Biswas, S.; Kim, J.W.; Zhang, X.; Scholes, G.D. Coherent Two-Dimensional and Broadband Electronic Spectroscopies. Chem. Rev. 2022, 122, 4257–4321. [Google Scholar] [CrossRef]
- Mukamel, S. Principles of Nonlinear Optical Spectroscopy; Oxford series in optical and imaging sciences; Oxford University Press: Oxford, UK, 1995; ISBN 9780195092783. [Google Scholar]
- Han, K.L.; Zhao, G.J. Hydrogen Bonding and Transfer in the Excited State; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2010; Volume 1–2, ISBN 9780470666777. [Google Scholar]
- Zhao, G.J.; Han, K.L. Effects of hydrogen bonding on tuning photochemistry: Concerted hydrogen-bond strengthening and weakening. ChemPhysChem 2008, 9, 1842–1846. [Google Scholar] [CrossRef]
- Tuna, D.; Sobolewski, A.L.; Domcke, W. Mechanisms of ultrafast excited-state deactivation in adenosine. J. Phys. Chem. A 2014, 118, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.J.; Han, K.L. Early time hydrogen-bonding dynamics of photoexcited coumarin 102 in hydrogen-donating solvents: Theoretical study. J. Phys. Chem. A 2007, 111, 2469–2474. [Google Scholar] [CrossRef] [PubMed]
- Sturgis, J.N.; Robert, B. Pigment Binding-Site and Electronic Properties in Light-Harvesting Proteins of Purple Bacteria. J. Phys. Chem. B 1997, 101, 7227–7231. [Google Scholar] [CrossRef]
- Sturgis, J.N.; Robert, B. The role of chromophore coupling in tuning the spectral properties of peripheral light-harvesting protein of purple bacteria. Photosynth. Res. 1996, 50, 5–10. [Google Scholar] [CrossRef]
- Theiss, C.; Trostmann, I.; Andree, S.; Schmitt, F.J.; Renger, T.; Eichler, H.J.; Paulsen, H.; Renger, G. Pigment-Pigment and pigment-Protein interactions in recombinant Water-Soluble Chlorophyll Proteins (WSCP) from cauliflower. J. Phys. Chem. B 2007, 111, 13325–13335. [Google Scholar] [CrossRef] [PubMed]
- Pieper, J.; Trostmann, I.; Schmitt, F.; Theiss, C.; Paulsen, H.; Eichler, H.J.; Freiberg, A. Excitonic Energy Level Structure and Pigment-Protein Interactions in the Recombinant Water-Soluble Chlorophyll Protein II. Spectral Hole-Burning Experiments. J. Phys. Chem. B 2011, 115, 4053–4065. [Google Scholar] [CrossRef] [PubMed]
- Orlowski, R.; Clark, J.A.; Derr, J.B.; Espinoza, E.M.; Mayther, M.F.; Staszewska-Krajewska, O.; Winkler, J.R.; Jedrzejewska, H.; Szumna, A.; Gray, H.B.; et al. Role of intramolecular hydrogen bonds in promoting electron flow through amino acid and oligopeptide conjugates. Proc. Natl. Acad. Sci. USA 2021, 118, e2026462118. [Google Scholar] [CrossRef] [PubMed]
- Sobolewski, A.L.; Domcke, W. Photophysics of intramolecularly hydrogen-bonded aromatic systems: Ab initio exploration of the excited-state deactivation mechanisms of salicylic acid. Phys. Chem. Chem. Phys. 2006, 8, 3410–3417. [Google Scholar] [CrossRef] [PubMed]
- Shemesh, D.; Domcke, W. Effect of the Chirality of Residues and γ-Turns on the Electronic Excitation Spectra, Excited-State Reaction Paths and Conical Intersections of Capped Phenylalanine–Alanine Dipeptides. ChemPhysChem 2011, 12, 1833–1840. [Google Scholar] [CrossRef]
- Guerrero-Corella, A.; Fraile, A.; Alemán, J. Intramolecular Hydrogen-Bond Activation: Strategies, Benefits, and Influence in Catalysis. ACS Org. Inorg. Au 2022, 2, 197–204. [Google Scholar] [CrossRef]
- Sobolewski, A.L.; Domcke, W.; Dedonder-Lardeux, C.; Jouvet, C. Excited-state hydrogen detachment and hydrogen transfer driven by repulsive 1πσ* states: A new paradigm for nonradiative decay in aromatic biomolecules. Phys. Chem. Chem. Phys. 2002, 4, 1093–1100. [Google Scholar] [CrossRef]
- Jouvet, C.; Miyazaki, M.; Fujii, M. Revealing the role of excited state proton transfer (ESPT) in excited state hydrogen transfer (ESHT): Systematic study in phenol-(NH3)nclusters. Chem. Sci. 2021, 12, 3836–3856. [Google Scholar] [CrossRef]
- Miyazaki, M.; Ohara, R.; Dedonder, C.; Jouvet, C.; Fujii, M. Electron-Proton Transfer Mechanism of Excited-State Hydrogen Transfer in Phenol-(NH3)n (n = 3 and 5). Chem. Eur. J. 2018, 24, 881–890. [Google Scholar] [CrossRef]
- Šolomek, T.; Bochet, C.G.; Bally, T. The primary steps in excited-state hydrogen transfer: The phototautomerization of o-nitrobenzyl derivatives. Chem. Eur. J. 2014, 20, 8062–8067. [Google Scholar] [CrossRef]
- Ehrmaier, J.; Karsili, T.N.V.; Sobolewski, A.L.; Domcke, W. Mechanism of Photocatalytic Water Splitting with Graphitic Carbon Nitride: Photochemistry of the Heptazine-Water Complex. J. Phys. Chem. A 2017, 121, 4754–4764. [Google Scholar] [CrossRef]
- Lan, Z.; Frutos, L.M.; Sobolewski, A.L.; Domcke, W. Photochemistry of hydrogen-bonded aromatic pairs: Quantum dynamical calculations for the pyrrole-pyridine complex. Proc. Natl. Acad. Sci. USA 2008, 105, 12707–12712. [Google Scholar] [CrossRef]
- Schultz, T.; Samoylova, E.; Radloff, W.; Hertel, I.V.; Sobolewski, A.L.; Domcke, W. Efficient deactivation of a model base pair via excited-state hydrogen transfer. Science 2004, 306, 1765–1768. [Google Scholar] [CrossRef] [PubMed]
- Plasser, F.; Barbatti, M.; Aquino, A.J.A.; Lischka, H. Excited-state diproton transfer in [2,2′-bipyridyl]-3,3′-diol: The mechanism is sequential, not concerted. J. Phys. Chem. A 2009, 113, 8490–8499. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sun, M.; Li, Y. How was the proton transfer process in bis-3, 6-(2- benzoxazolyl)-pyrocatechol, single or double proton transfer? Sci. Rep. 2016, 6, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.T.; Wu, P.J.; Peng, C.Y.; Shen, J.Y.; Tsai, C.C.; Hu, W.P.; Chou, P.T. A study of the competitive multiple hydrogen bonding effect and its associated excited-state proton transfer tautomerism. Phys. Chem. Chem. Phys. 2017, 19, 28641–28646. [Google Scholar] [CrossRef]
- Weiß, J.; May, V.; Ernsting, N.P.; Farztdinov, V.; Mühlpfordt, A. Frequency and time-domain analysis of excited-state intramolecular proton transfer. Double-proton transfer in 2,5-bis(2-benzoxazolyl)-hydroquinone? Chem. Phys. Lett. 2001, 346, 503–511. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, J.; Liu, J.; Hoffmann, M.R. Competitive excited-state single or double proton transfer mechanisms for bis-2,5-(2-benzoxazolyl)-hydroquinone and its derivatives. Phys. Chem. Chem. Phys. 2015, 17, 11990–11999. [Google Scholar] [CrossRef]
- Serdiuk, I.E.; Roshal, A.D. Exploring double proton transfer: A review on photochemical features of compounds with two proton-transfer sites. Dye. Pigment. 2017, 138, 223–244. [Google Scholar] [CrossRef]
- Yatsuhashi, T.; Inoue, H. Molecular mechanism of radiationless deactivation of aminoanthraquinones through intermolecular hydrogen-bonding interaction with alcohols and hydroperoxides. J. Phys. Chem. A 1997, 101, 8166–8173. [Google Scholar] [CrossRef]
- Inoue, H.; Hida, M.; Nakashima, N.; Yoshihara, K. Picosecond fluorescence lifetimes of anthraquinone derivatives. Radiationless deactivation via intra- and intermolecular hydrogen bonds. J. Phys. Chem. 1982, 86, 3184–3188. [Google Scholar] [CrossRef]
- Mondal, J.A.; Samant, V.; Varne, M.; Singh, A.K.; Ghanty, T.K.; Ghosh, H.N.; Palit, D.K. The role of hydrogen-bonding interactions in the ultrafast relaxation dynamics of the excited states of 3- and 4-aminofluoren-9-ones. ChemPhysChem 2009, 10, 2995–3012. [Google Scholar] [CrossRef]
- Ghosh, R.; Mondal, J.A.; Palit, D.K. Ultrafast dynamics of the excited states of curcumin in solution. J. Phys. Chem. B 2010, 114, 12129–12143. [Google Scholar] [CrossRef] [PubMed]
- Mondal, J.A.; Ghosh, H.N.; Mukherjee, T.; Palit, D.K. S2 fluorescence and ultrafast relaxation dynamics of the S2 and S1 states of a ketocyanine dye. J. Phys. Chem. A 2005, 109, 6836–6846. [Google Scholar] [CrossRef]
- Zvereva, E.; Segarra-Martí, J.; Marazzi, M.; Brazard, J.; Nenov, A.; Weingart, O.; Léonard, J.; Garavelli, M.; Rivalta, I.; Dumont, E.; et al. The effect of solvent relaxation in the ultrafast time-resolved spectroscopy of solvated benzophenone. Photochem. Photobiol. Sci. 2018, 17, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Volpato, A.; Bolzonello, L.; Meneghin, E.; Collini, E. Global analysis of coherence and population dynamics in 2D electronic spectroscopy. Opt. Express 2016, 24, 24773–24785. [Google Scholar] [CrossRef] [PubMed]
- Fresch, E.; Collini, E. Relaxation dynamics of chlorophyll b in the sub-ps ultrafast timescale measured by 2d electronic spectroscopy. Int. J. Mol. Sci. 2020, 21, 2836. [Google Scholar] [CrossRef]
- Meneghin, E.; Leonardo, C.; Volpato, A.; Bolzonello, L.; Collini, E. Mechanistic insight into internal conversion process within Q-bands of chlorophyll a. Sci. Rep. 2017, 7, 1–7. [Google Scholar] [CrossRef]
- Meneghin, E.; Pedron, D.; Collini, E. Spectroscopy data for the time and frequency characterization of vibrational coherences in bacteriochlorophyll a. Data Br. 2019, 23, 103707. [Google Scholar] [CrossRef]
- Turner, D.B.; Dinshaw, R.; Lee, K.-K.; Belsley, M.S.; Wilk, K.E.; Curmi, P.M.G.; Scholes, G.D. Quantitative investigations of quantum coherence for a light-harvesting protein at conditions simulating photosynthesis. Phys. Chem. Chem. Phys. 2012, 14, 4857. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fresch, E.; Collini, E. The Role of H-Bonds in the Excited-State Properties of Multichromophoric Systems: Static and Dynamic Aspects. Molecules 2023, 28, 3553. https://doi.org/10.3390/molecules28083553
Fresch E, Collini E. The Role of H-Bonds in the Excited-State Properties of Multichromophoric Systems: Static and Dynamic Aspects. Molecules. 2023; 28(8):3553. https://doi.org/10.3390/molecules28083553
Chicago/Turabian StyleFresch, Elisa, and Elisabetta Collini. 2023. "The Role of H-Bonds in the Excited-State Properties of Multichromophoric Systems: Static and Dynamic Aspects" Molecules 28, no. 8: 3553. https://doi.org/10.3390/molecules28083553
APA StyleFresch, E., & Collini, E. (2023). The Role of H-Bonds in the Excited-State Properties of Multichromophoric Systems: Static and Dynamic Aspects. Molecules, 28(8), 3553. https://doi.org/10.3390/molecules28083553