Abstract
A radionuclide generator of the short-lived alpha emitter 226Th was proposed. An original scheme consisting of two in-series chromatographic columns was developed for rapidly producing a neutral citric-buffered eluate of high purity 226Th. The first column filled with TEVA resin retained the parent 230U, while 226Th was eluted with 7 M HCl solution to be immediately adsorbed on the second column containing DGA resin or UTEVA resin. Having substituted the strongly acidic medium of second column with neutral salt solution, 226Th was desorbed with diluted citric buffer solution. One cycle of generator milking took 5–7 min and produced >90% of 226Th in 1.5 mL of eluate (pH 4.5–5.0) appropriate for direct use in radiopharmaceutical synthesis. The 230U impurity in 226Th eluate was less than 0.01%. The proposed two-column 230U/226Th generator was tested over 2 months including a second loading of 230U additionally accumulated from 230Pa.
1. Introduction
A new fast-paced branch of nuclear medicine called targeted alpha therapy (TAT) is effective for the treatment of various oncological diseases due to the property of α-particles to release a large amount of energy in a limited area of living tissue (~10 cell diameters). One of the promising radionuclides for TAT is 230U (T1/2 = 20.2 d) [1]. The decay of 230U generates a chain of short-lived products that emit five α-particles with a total energy of 33.5 MeV (Figure 1), resulting in effective cell damage [2]. The short-lived daughter alpha emitter 226Th (T1/2 = 30.6 min) is also an attractive radionuclide for using in TAT [3]. In terms of nuclear properties, the 230U/226Th pair is similar to the well-researched 225Ac/213Bi generator pair [4].
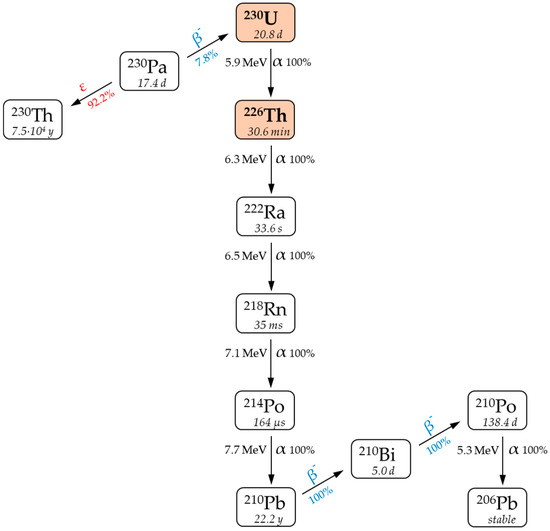
Figure 1.
Decay chain of 230Pa [2].
The expected therapeutic efficacy of 230U/226Th has not been demonstrated yet; nevertheless, research is underway to find optimal chelating agents that can stably bind in vivo 230U [5] and 226Th [6]. Daughter nuclide released from a radiopharmaceutical molecule due to recoil effect can potentially migrate from the target cancer cell, which might cause considerable toxic effects to heathy tissues. If the half-life of a formed nucleus is short, its diffusion is time-limited. Therefore, a major advantage of 226Th-TAT compared with 230U-TAT is that the uncontrolled redistribution of a recoiled daughter nuclide (for 230U, this nuclide is 226Th itself) is significantly diminished (Figure 1).
The effective and reliable production of 230U and 226Th is required in order to accelerate biological studies and implement these radionuclides in TAT. 230U can be produced directly via nuclear reactions 231Pa(p,2n)230U [7] and 231Pa(d,3n)230U [8]. The initial 231Pa (T1/2 = 3.3 × 104 y) is a decay product of 235U, it must be isolated from aged uranium samples. The raw material is hardly accessible, which restricts the method implementation. Another approach uses reactions of thorium nuclei with accelerated protons and deuterons, leading to the formation of the 230Pa precursor decaying into 230U with a branching ratio of 7.8%: 232Th(p,3n)230Pa→230U and 232Th(d,4n)230Pa→230U.
Scientific organizations worldwide are actively developing the production of 230U through irradiation of 232Th with protons [9,10,11,12,13]. This method has proven to be the most effective in terms of product yield compared with the reactions with deuterons [14,15]. Moreover, the maximum of the 232Th(p,3n)230Pa reaction excitation function is around 20 MeV [15,16], which enables up-scaled production of 230U on accessible commercial cyclotrons. At higher proton energies (>100 MeV), 230U can be obtained as a byproduct in 225Ac production [17,18] along with 223Ra [19].
230U from proton-irradiated thorium contains a chemically inseparable impurity of long-lived uranium isotopes. This impurity was evaluated in our previous paper [15] as up to 0.02% 232U (T1/2 = 68.9 y) and 0.001% 233U (T1/2 = 1.6 × 105 y). Long-lived admixtures make the medical use of 230U as a source in a radionuclide generator of 226Th more prospective than direct applications.
226Th is considered to be an alternative to another promising generator-produced short-lived radionuclide 213Bi (T1/2 = 45.6 min) [20]. 226Th presumably provides a greater impact on cancer cells compared with 213Bi. A rapid cascade of four α-particles initiated by 226Th decay deposits totally 27.7 MeV, while 213Bi emits only one α-particle with an energy of 8.4 MeV. 226Th-radiopharmaceuticals can be effective for therapy of epithelial or easily accessible tumors [21]. Radioimmunoconjugates Nimotuzumab-p-SCN-Bn-DTPA(DOTA) were synthesized in our previous paper and their specificity toward EGFR overexpressing epidermoid carcinoma A431 cells were demonstrated [22].
Due to the relatively short half-life of 226Th, time economy becomes a major requirement throughout the entire process from obtaining 226Th to radiopharmaceutical administration. For this reason, the generator system must ensure the rapid and efficient separation of the accumulated thorium radionuclide. Various methods of liquid–liquid extraction [23,24], extraction chromatography [11,25,26,27], and ion exchange chromatography [28,29,30] have been developed for the separation of thorium and uranium. For example, the methods employing extraction chromatographic sorbents TEVATM resin and UTEVA/TRUTM resin were recommended for the selective isolation and determination of U, Th, and a number of other radionuclides in water (sample volume up to 1 L) [31,32]. The chromatographic methods are more appropriate for a 230U/226Th generator since they usually provide 226Th in a small volume of eluate containing a reduced amount of long-lived impurities.
Effective separation of U(VI) and Th(IV) can be achieved on sorbents displaying anion-exchange properties in strong hydrochloric acid solutions. As it can be seen in Figure 2, U(VI) exhibits high affinity to a strong base anion-exchange resin Dowex 1 (or AG 1) and to an extraction chromatographic resin TEVA at c(HCl) > 6 M, whereas Th(IV) is not retained. Both resins were tested as sorbents for a 230U/226Th «direct» generator [22]. TEVA resin proved to be more preferable, it provided high 226Th yield in less volume of eluate (1–2 mL). Furthermore, the maximal mass distribution ratio Dm of U(VI) adsorbed on TEVA resin is located around 7 M HCl, whereas the largest adsorption of U(VI) on AG 1 corresponds to HCl concentration greater than 9 M (Figure 2). The high acidity of the final 226Th eluate was found to be the main disadvantage assuming the extra-time needed to convert the eluate into neutral solution prior to labeling.
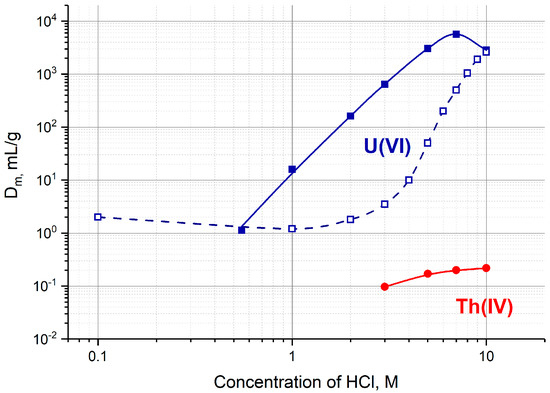
Figure 2.
The mass distribution ratios Dm, mg/mL, of U(VI) and Th(IV) on TEVA resin (solid lines) [31] and Dowex 1 × 10 (dashed line) [33] as a function of HCl concentration.
Another approach is based on a reverse scheme of a 230U/226Th generator, i.e., the parent 230U is not fixed on the column filled with sorbent while the daughter 226Th remains adsorbed. An extraction chromatographic DGA resin containing a diglycolamide derivative was reported to be appropriate for obtaining 226Th in citric buffer solution that was amenable to direct labeling with minimal losses of time [13]. However, the 226Th eluate recovered from the reported reverse 230U/226Th generator contained at least 0.2% of 230U [13], which was unacceptable for clinical trials to date.
In the presented article, we investigated different schemes of two-column 230U/226Th generators pursuing two goals: (i) Obtaining 226Th of high purity in a solution amenable to further labeling; (ii) reducing the time of 226Th production. The first column served for fixing the parent 230U and elution of 226Th. The second column was intended to adsorb 226Th from a strongly acidic solution, and then to desorb it with diluted or neutral solution. This concept was proposed earlier and tested for a 225Ac/213Bi generator [34,35,36]. In the first column, 225Ac formed an extremely strong complex with bis-(2-ethylhexyl)methanediphosphonic acid (H2DEH[MDP]) immobilized on a silica support (Ac resin). Products of the 225Ac decay, 221Fr, and 213Bi were eluted with 1 M HCl and concentrated on the second column filled with the ion exchanger AG-MP 50 from 0.2 M HCl. Then, 213Bi was eluted from the second column with 0.1 M HI. The high efficiency of this approach makes it promising for the 230U/226Th pair, as well.
2. Results and Discussion
A two-column 230U/226Th generator was proposed and investigated for fast 226Th production in diluted or neutral citric solution. The parent 230U was adsorbed onto the first column filled with TEVA resin (Triskem Int.). On reaching the transient equilibrium, the daughter 226Th was separated and eluted with strong HCl solution. The role of the second column was to reduce quickly the acidity of 226Th solution, i.e., a sorbent for second column was to retain 226Th from the strong HCl solution and to desorb it into a diluted solution. Three extraction chromatographic resins eligible for this purpose were considered: TRU resin, UTEVA resin, and DGA resin (all Triskem Int.). According to the reported data [37,38,39] obtained in static conditions and shown in Figure 3a, the resins can be arranged in a row with respect to Th(IV) retention from <5 M HCl solution:
DGA resin > TRU resin > UTEVA resin
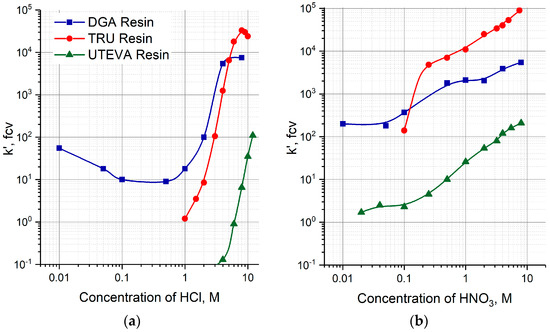
Figure 3.
Capacity factors k′, fcv, of Th(IV) on DGA, TRU and UTEVA resins as a function of HCl (a) and HNO3 (b) concentrations [37,38,39].
In order to evaluate the feasibility of a two-column 230U/226Th generator, column experiments on 226Th sorption from 7 M HCl and its desorption with diluted HCl solutions were carried out.
2.1. Elution of 226Th from the Second Column with HCl Solutions
First, the transfer of 226Th to a second column was investigated (Figure 4a, Step 1). 226Th was easily stripped off the parent column with 7 M HCl solution, the concentration corresponding to the maximum of U(VI) sorption on TEVA resin (Figure 2). The integral 226Th elution curve (blue line in Figure 5a) indicates that the solution volume of 1.5 mL was sufficient to wash out ≥99% of 226Th. DGA resin and TRU resin display high adsorption of 226Th from 7 M HCl solution (Figure 3a), the values of k′ Th(IV) attain 104. When the second column was filled with 0.1 mL of these resins and connected directly to the exit of the parent column, 226Th was completely adsorbed onto the second column. In contrast, the values of k′ Th(IV) on UTEVA resin are below 10 under the same conditions. Therefore, the quantity of UTEVA resin in the second column was increased up to 1 mL to ensure a tolerable breakthrough of 226Th of less than 3% (red line in Figure 5a).
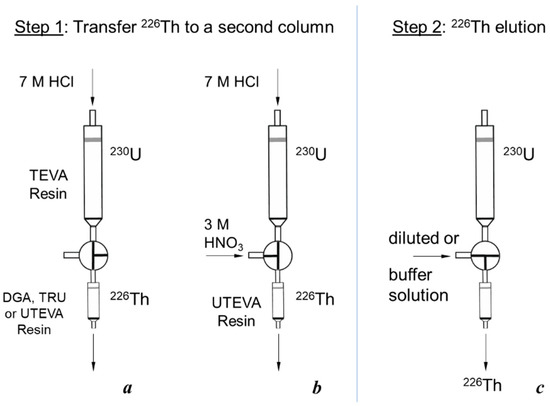
Figure 4.
Scheme of 226Th production by a two-column 230U/226Th generator: (a,b) Two modifications of transferring 226Th from the parent 230U column to a second one; (c) elution of 226Th from the second column.
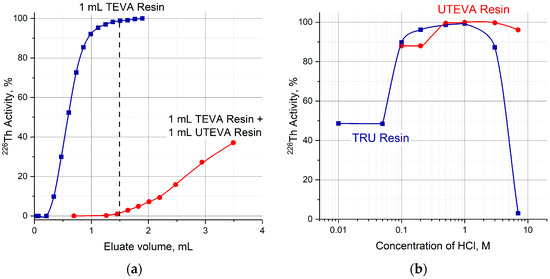
Figure 5.
(a) Optimization of the UTEVA resin volume in the second column; (b) efficiency of 226Th desorption from TRU and UTEVA columns as a function of HCl concentration.
The transferred 226Th was eluted with different HCl solutions as shown in Figure 4c (Step 2). The DGA resin exhibits the greatest retention of 226Th among the studied resins from diluted hydrochloric solutions (Figure 3a). Our results of column experiments were in a good agreement with the k′ data. The elution of 226Th with 0.3 M HCl solution, which is the most favorable for 226Th desorption, resulted in 40% of 226Th yield in 6 mL of eluate. For other HCl concentrations, the 226Th yield was even lower.
The efficiency of 226Th desorption from the second columns filled with TRU and UTEVA resins versus the concentration of hydrochloric solution is presented in Figure 5b. The optimal HCl concentration range of 226Th desorption was 0.4–1 M for TRU resin and 0.5–2 M for UTEVA resin. Typical 226Th elution curves (Figure 6) display that 226Th was completely eluted in ~1 mL of eluate. It is interesting to note that the width of 226Th chromatographic peaks from the TRU resin and UTEVA resin columns was almost the same, although the bed volume of UTEVA resin was 10 times larger than the TRU resin.
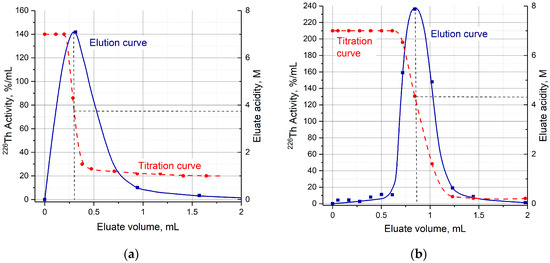
Figure 6.
Typical 226Th elution and titration curves. (a) Elution of 226Th from TRU resin with 1 M HCl; (b) elution of 226Th from UTEVA resin with 0.2 M HCl.
Despite the fact that the HCl concentration of the solution entering the second column for 226Th desorption (Step 2) was relatively low, the eluate acidity from both TRU resin and UTEVA resin columns was 3–4 M [H+]. The detailed titration curves of eluate collected by portions (Figure 6) demonstrate that 226Th is eluted on the drastic HCl concentration gradient when one solution is replaced by another. The maximum of 226Th chromatographic peak corresponds to H+ concentration around 4 M. Therefore, the use of dilute HCl solutions allowed us to decrease eluate acidity by only a factor of two.
2.2. Elution of 226Th from the Second Column with Citric Buffer Solutions
After transferring 226Th from the parent column to a second one containing TRU, UTEVA or DGA resin, 226Th can be desorbed with a neutral citric buffer solution. The efficiency of 226Th desorption was studied as a function of H3Cit (pH 5.0) concentration in the range of 10−4 M–10−1 M. The dependencies plotted on the graph (Figure 7) are arranged in the order reflecting the above-mentioned sequence of resin affinity for thorium (IV). For the studied resins, 0.1 M citric buffer solution fully recovered 226Th.
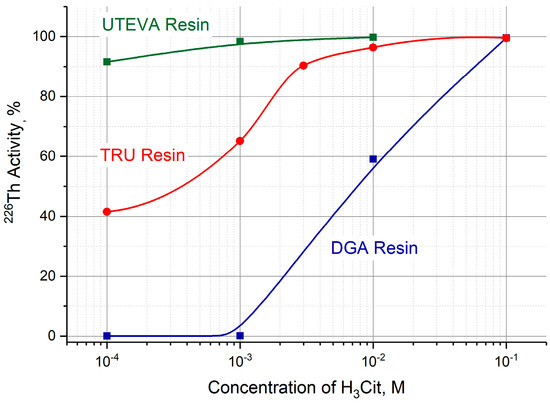
Figure 7.
Efficiency of 226Th desorption from DGA, TRU, and UTEVA resin columns as a function of citric acid concentration (pH 5.0).
Typical curves of 226Th elution from the UTEVA resin and DGA resin columns consisted of the only chromatographic peak within the given range of citric acid concentration (Figure 8a). Similar to the 226Th elution with dilute hydrochloric solutions, the peak maximum followed the acidity gradient. Otherwise, two chromatographic peaks were observed when 226Th was eluted from the TRU column with citric buffer solutions (Figure 8b). The first peak was in the same way related to the HCl concentration gradient. The position of maximum Vmax of the second peak, as well as the capacity factor k′ defined as [37] (Vc is free volume of sorbent in a column), depended on the citric acid concentration.
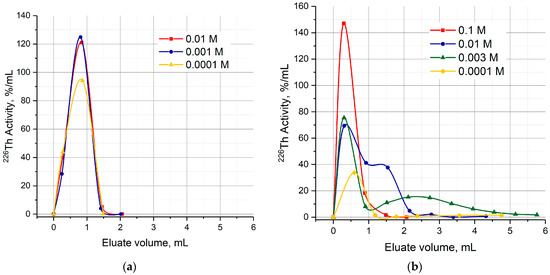
Figure 8.
Curves of 226Th elution with citric buffer solutions of different concentrations. (a) Elution from UTEVA resin; (b) elution from TRU resin.
According to the literature data, various complexes of Th(IV) are coexisting in citric acid media depending on pH values and salinity [40,41]. At pH 5–6, the predominant species are ThCit35− and ThCit2(OH)24− [40] with cumulative formation constants (logβ) of 28 and 15, respectively. In more acidic solution (pH < 1), the speciation shifts toward cation forms of Th(IV), e.g., Th4+ and ThCit+ (logβ = 14). It is evident that all these species may exhibit different affinities to the resins and the detailed analysis is complicated. However, in the case of TRU resin, the dependence of k′ Th(IV) on citric acid concentration may be expressed by a simple correlation helpful for practice use:
where and are empirical constants.
Satisfactory values of 226Th yield (>90%) in a small amount of eluate (1–1.5 mL) were obtained for all the studied resins, the eluate characteristics are listed in Table 1.

Table 1.
Characteristics of 226Th eluted from the second column of 230U/226Th generator with citric buffer solution.
It was found that eluate acidity remained relatively excessive for immediate synthesis of labeled compounds. In order to maintain 226Th on the second column during the substitution of the acidic medium with the neutral one, the influence of nitrate ions was studied.
2.3. Stabilization of 226Th on the Second Column before Elution
The ability of Th(IV) to form stable anionic complexes with nitrate ions is widely used to separate it from other elements. Comparison of k′ values for DGA, TRU, and UTEVA resins reveals higher sorption of Th(IV) from nitric solutions (Figure 3b) than from hydrochloric ones (Figure 3a), especially for the acidity below 1 M. Taking this fact into account, we modified the procedure of 226Th production from the two-column 230U/226Th generator.
The initial part of modification consisted of the pre-treatment of the second column. Before transferring 226Th from the parent column (Step 1), 10 mL of HNO3 solution of a certain concentration was passed through the second column at a flowrate of 1 mL/min. For the DGA and TRU resins, the HNO3 concentration was 0.1 M, which corresponds to moderate values of the capacity factor 150 < k′ Th(IV) < 350 (Figure 3b). In the case of UTEVA resin, the values of k′ Th(IV) for dilute nitric acid solutions are small; they grow with an increasing acid concentration and reach values around 100 in the region of 3–4 M HNO3.
Starting from these data obtained in static conditions, the UTEVA column was pre-treated with a HNO3 solution of various concentrations and 226Th losses during Step 1 were studied. The results presented in Figure 9 display that the 226Th losses when transferring from the TEVA column to UTEVA one with 7 M HCl solution noticeably diminished along with increasing the concentration of HNO3 used for UTEVA pre-treatment. For 3 M HNO3 solution, the breakthrough of 226Th began after passing not less than 2.5 mL of 7 M HCl.
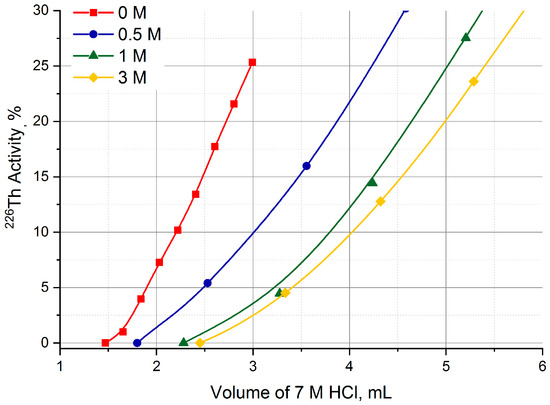
Figure 9.
Choice of HNO3 concentration for the pre-treatment of UTEVA resin column.
In addition, a variation of Step 1 was tested (Figure 4b) that provided the flow of 7 M HCl solution carrying 226Th to be equally mixed with the flow of 3 M HNO3 solution at the entrance of the UTEVA column (“two flows” variation). As a result, 226Th breakthrough was not observed even after passing 20 mL of 7 M HCl (>20 bed volumes).
The other part of the second column modification was carried out after Step 1 and involved substituting the 7 M HCl medium with dilute or neutral one. Solutions of 0.1 M HNO3, 0.15 M NaCl, and 0.15 M NaNO3 were studied. Full change in medium in the DGA column took place after passing 1.0–1.2 mL of each solution, and 226Th losses did not exceed 3%. In the case of TRU column, 226Th was partially washed out on the HCl concentration gradient (similar to Figure 6a) regardless of the substituting solution, its losses ranged from 10% to 40% and were poorly reproducible. The UTEVA resin retained 226Th well when the solutions of 0.1 M HNO3 and 0.15 M NaNO3 were passed through the second column, whereas the 0.15 M NaCl solution washed out up to 28% of 226Th (Figure 10).
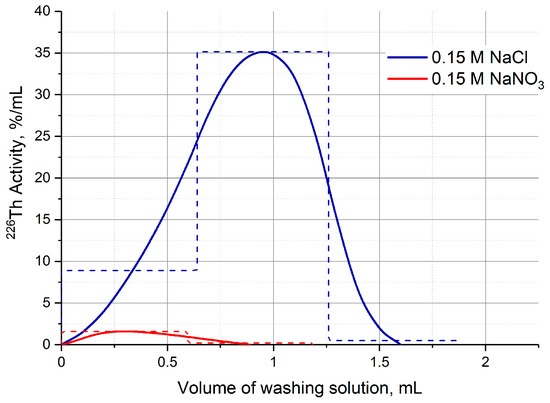
Figure 10.
226Th losses during the substitution of the 7 M HCl medium on the UTEVA resin column with neutral salt solutions.
Having replaced the strongly acidic medium in the second column, 226Th was washed out with a citric buffer solution (pH 5.0) of various concentrations (Figure 11a). The results obtained for the UTEVA column:
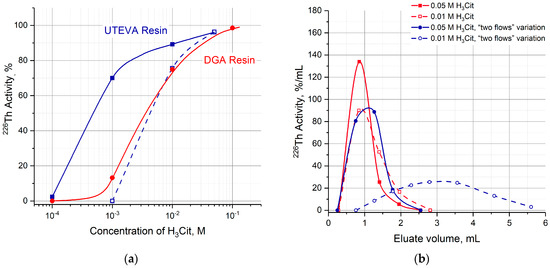
Figure 11.
(a) Efficiency of 226Th desorption from the pre-treated DGA and UTEVA resin columns as a function of citric acid concentration (pH 5.0). DGA resin column pre-treated with 0.1 M HNO3 (red solid curve); UTEVA resin: Pre-treated with 3 M HNO3 (blue solid curve), pre-treated with 3 M HNO3 + “two flows” variation of 226Th transfer (blue dashed curve); (b) curves of 226Th elution depending on the variations of 226Th recovery from UTEVA columns.
allow us to suggest that increasing contact time of the UTEVA resin and nitric solution led to an increasing difficult 226Th recovery. The relative positions of 226Th elution curves (Figure 11b) were also in line with these suggestions.
Moreover, the influence of the contact time was evaluated in parallel experiments that included the 3 M HNO3 pre-treatment of the UTEVA column, the HCl substitution with 0.15 M NaNO3 solution, and the 226Th elution with a 10−3 M citric buffer solution. As the pre-treatment duration increased from 10 min to 10 h, the efficiency of 226Th production decreased from 70% (see in Figure 11a) to zero. The behavior of the UTEVA resin may be explained by the fact that the k′ Th(IV) dependence in nitric solutions is higher than the hydrochloric solutions over the entire concentration range (Figure 3).
Two most effective procedures for obtaining 226Th from the two-column 230U/226Th generator are presented in Table 2.
Table 2.
Effective procedures of 226Th extraction from the two-column 230U/226Th generator (>90% of 226Th in 1.5 mL of eluate, pH 4.5–5.0).
The overall time of 226Th eluate production was within 5–7 min. The time expenditure was slightly shorter when the DGA column was used, since its size was 10 times smaller than the UTEVA column. Meanwhile, the use of UTEVA resin for the second column resulted in high yield of 226Th in a less concentrated solution (0.01–0.05 M H3Cit, pH 4.5–5.0).
2.4. Long-Term Performance of the Two-Column 230U/226Th Generator
Following the developed procedure, the solution of 7 M HCl was only passed through the parent column containing 230U adsorbed on TEVA resin. The distribution of 230U along with the length of parent column was monitored throughout the generator lifetime (Figure A2). Usually, two cycles of 226Th production per weekday were performed over 2 months. The total volume of 7 M HCl solution that passed through the parent column was about 200 mL including accessory operations and a loading of a second portion of 230U. According to the calculations illustrated by Figure A3, one third of the initial amount of 230U was additionally accumulated from 230Pa in 27–28 days after the first separation, and then loaded onto the parent column.
Due to the two-column scheme of 226Th production, the proposed generator provided deep purification 226Th from 230U. It was found that the 230U impurity in the 226Th eluates did not exceed 0.01%, which was at least one order of magnitude better in comparison with the reported literature data [13].
In this work, we used low 230U activities that do not lead to radiolysis and destruction of the sorbent. Influence of these processes on the generator performance is to be investigated in future studies.
3. Materials and Methods
All chemicals were of p.a. (pro analysis) quality or higher, obtained from Merck (Darmstadt, Germany), and used without additional purifications. All experiments were carried out using de-ionized “Milli-Q” water (18 MΩ∙cm−1). DGA resin (N,N,N′,N′tetroctyldiglicolamide as an extracting agent), TEVA resin (quaternary ammonium salt Aliquate 336 as an extracting agent), TRU resin (octyl(phenyl)-N,N-di-isobutylcarbomoylmethylphosphine oxide dissolved in tributylphosphate), and UTEVA resin (dipentil pentylphosphonate as an extracting agent) with 50–150 μm particle size were obtained from Triskem, France.
Citric buffer solutions (10−4–10−1 M, pH 5.0 ± 0.1) were prepared by dissolving the corresponding solid acid sample and adding small portions of 1 M NaOH to obtain a solution with the required pH value.
Measurements of operational pH values were performed with an Orion 2 Star Benchtop pH meter using an Orion 8103SC combination pH electrode. Commercial pH Titrisol buffer concentrates (Merck p.a.) were used to calibrate the setup at room temperature.
Acid-base titration with indicators methyl orange and phenolphthalein were used to determine the acid content in commercial solutions of concentrated HCl and HNO3, as well as in the 226Th containing eluate.
The experiments were carried out at a temperature of 21 ± 2 °C.
3.1. Gamma-ray Spectroscopy
The measurement of radionuclide activities was performed by γ-ray spectrometry using a high resolution HP Ge detector (ORTEC GEM15P4-70). Samples were counted at different detector-source distances respecting a level of dead-time of less than 10%. The detector efficiency at the used distances were determined with standard calibration sources. Net peak areas in the detected photopeaks were evaluated by means of the GammaVision32 software.
The characteristic γ-ray emission of 226Th (111.1 keV, 3.29%) and 222Ra (324.3 keV, 2.77%) [2] were used for 230U and 226Th activity quantification of various generator testing samples respecting the transient equilibrium of daughter radionuclides.
3.2. Target Preparation and Irradiation, and 230U Isolation
Metallic thorium supplied by Institute for Physics and Power Engineering (IPPE, Russia) was used as target material. Thorium plates of (2.2 × 2.5) cm2 approximate dimensions with thickness of 1.5–2.0 mm were fabricated and packed in copper and aluminum foil envelopes, which served for beam monitoring purposes, as well. Each package was encapsulated in a graphite shell sealed with high-temperature silicone adhesive. Several targets were irradiated at the linear proton accelerator of the Institute for Nuclear Research of the Russian Academy of Sciences (INR RAS, Moscow, Russia) [42] with an initial energy of 120–130 MeV. The beam current and total beam charge were 3–5 µA and 12–18 µA·h, respectively.
The dissolution of the irradiated thorium was performed as described previously [18,43] 4 to 5 days after the end of bombardment (EOB). The protactinium fraction including 230Pa was recovered from the solution according to the procedure reported in [15] and remained in 7 M HCl/0.1 M HF solution for 230U accumulation during 27–28 days. Traces of Nb (mainly 95Nb) and Ru (103,106Ru) radionuclides were impurities of 230Pa/230U.
A chromatographic technique close to the one developed by A.W. Knight [44] was implemented for separation of 230U from 230Pa. The solution with radionuclides was loaded onto a column filled with 2 mL of TEVA resin. Due to the presence of fluoride ions, Pa(V) together with Ru(IV) were eluted, while U(VI) and Nb(V) were retained on the resin. The column was washed with 7 M HCl/0.1 M HF solution to remove Pa(V). The washing was added to the Pa(V) eluate and the combined solution was maintained for the next 230U accumulation. Then, having the column washed with 7 M HCl solution, U(VI) and a part of Nb(V) were desorbed with 0.1 M HCl solution. The desorbate was adjusted to 3 M HCl by adding concentrated hydrochloric acid, and following the known procedure [13,45], this solution was passed through a column filled with 1 mL of DGA resin. The uranium fraction was adsorbed, while the 95Nb was washed out of the column. Finally, 230U was eluted with a small amount of 0.1 M HCl solution.
3.3. Generator Schemes for Producing 226Th
3.3.1. Preparation of a Parent 230U Column
A plastic column of ~5 mm in diameter was filled with 1 mL of TEVA resin equilibrated with 7 M HCl. The resin was fixed inside by two frits at the bottom and at the top of column. The 230U solution was evaporated and reconstituted in 7 M HCl. The resulting solution was passed through the column followed by washing with a solution of 7 M HCl. The uranium was adsorbed under these conditions, the loaded activity of 230U was 300–350 kBq. Approximately a month after the 230U isolation and loading, a second part of 230U was accumulated, separated from 230Pa as described above and added to the column.
The prepared column served as parent one, and it could work as a one-column generator providing 226Th elution in 7 M HCl. A typical differential curve of 226Th elution is shown in Figure A1. Furthermore, the parent column was a part of the two-column generator schemes.
3.3.2. Two-Column 230U/226Th Generator Scheme and 226Th Elution Cycle
A general scheme of the 230U/226Th generator comprised two columns connected in series via a three-valve cock as shown in Figure 4. The parent TEVA column containing 230U was the first column and a column filled with TRU, UTEVA or DGA resin served as a second one that could be interchanged when necessary. A milking procedure included two common steps: (1) Transfer 226Th with 7 M HCl solution from the parent column to the second one (Figure 4a,b); (2) 226Th elution with diluted HCl or citric buffer solution (Figure 4c). Eluates were collected for measurement of 226Th and 230U activity and eluate acidity. Flow rates of solutions passing through the columns were maintained and controlled with a peristaltic pump. The values of flow rate were 1 and 0.6 mL/min for Steps 1 and 2, respectively.
The TRU or DGA resin was loaded into a plastic column of ~3 mm in diameter, the height of resin bed was 11–12 mm (resin volume ~0.1 mL). Diameter of the column for the UTEVA resin was ~5 mm, the height of the UTEVA bed was 55–58 mm (~1 mL). Each column was equipped with bottom and top frits.
For some experiments with the UTEVA column, we modified Step 1 as it is shown in Figure 4b. A flow of 7 M HCl solution (0.5 mL/min) after passing the parent 230U column was mixed with a flow of 3 M HNO3 solution (0.5 mL/min) resulting in 1 mL/min flow of 3.5 M HCl/1.5 M HNO3 solution at the entrance of the UTEVA resin column.
3.4. 230U Measurements
The activity of 230U in eluates was usually measured overnight for complete 226Th decay. 230U was assayed by γ-ray spectroscopy via the daughter radionuclides 226Th and 226Ra.
Distribution of 230U along the length of TEVA resin column was monitored by successive scanning of the column through a 4 mm wide slit between lead blocks. The measurements were performed after 230U loading onto the TEVA resin column and regularly after milking.
4. Conclusions
We have proposed and tested the proof-of-concept of a two-column 230U/226Th generator for rapidly producing 226Th amenable to further labeling. The first 230U column with TEVA resin furnished 226Th in 7 M HCl solution. The second column retained 226Th from the strongly acidic solution, and then released it with a diluted hydrochloric or neutral citric buffer solution. Analysis based on the dependence of the capacity factor k′ Th(IV) on the concentration of hydrochloric and nitric acid allowed us to consider DGA, TRU, and UTEVA resins as promising sorbents for the second column.
High yields (>97%) of 226Th elution from TRU and UTEVA resins with a small volume (~1 mL) of diluted HCl solutions were obtained. However, the resulting acidity of the eluate was 3–4 M [H+] regardless of the solution concentration entering the second column. The titration analysis displayed that 226Th was eluted on the HCl concentration gradient when one solution was replaced by another.
Elution of 226Th transferred to the second column containing DGA, TRU or UTEVA resin was studied with citric buffer solutions (pH 5.0) in two modes. Direct 226Th desorption was also influenced by the acidity gradient. While 226Th was stripped off the UTEVA and DGA columns in one chromatographic peak, a typical curve of 226Th elution from the TRU column consisted of two chromatographic peaks within the studied range of citric acid concentration. The first peak followed the HCl concentration gradient, and the second one may be attributed to 226Th complexation with citrate ions in the course of TRU column elution. Satisfactory yields were achieved by 226Th elution from the second column filled with any of the studied resins. For TRU and DGA resins, 1–1.5 mL of 0.1 M H3Cit (pH 5.0) solution extracted more than 90% of 226Th, while the UTEVA resin column demonstrated similar effectiveness with less concentrated citric buffer solutions (down to 10−3 M H3Cit). The acidity of citric eluates was about two times lower than the diluted HCl solution’s eluates but still relatively high for immediate labeling.
Neutral citric-buffered 226Th eluates were obtained when nitrate ions were introduced. The second column was initially put in contact with a nitric acid solution. Then, after 226Th transfer from the parent column, the acidic medium of the second column was substituted with the neutral one maintaining 226Th immobile. Solutions of 0.15 M NaCl and of 0.15 M NaNO3 were used for the DGA and UTEVA column, respectively. Finally, 226Th was extracted with citric buffer solution: 0.1 M H3Cit from the DGA column and 0.01–0.05 M H3Cit from the UTEVA one. Therefore, one cycle of generator milking took 5–7 min and produced >90% of 226Th in 1.5 mL of eluate, pH 4.5–5.0.
The proposed two-column 230U/226Th generator was tested over 2 months including a second loading of 230U additionally accumulated from 230Pa. The 230U impurity in the 226Th eluate was less than 0.01% allowing its use directly in synthesis of radiopharmaceutical compounds.
Author Contributions
Conceptualization, S.V.E.; methodology, S.V.E. and A.K.S.; validation, S.V.E., A.K.S. and A.N.V.; formal analysis, S.V.E. and A.K.S.; investigation, S.V.E., A.K.S. and A.N.V.; writing—original draft preparation, S.V.E. and A.N.V.; writing—review and editing, S.V.E. and A.N.V.; visualization, S.V.E. and A.N.V.; supervision, S.V.E.; funding acquisition, S.V.E. All authors have read and agreed to the published version of the manuscript.
Funding
The reported study was funded by RFBR, project number 20-53-15007.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Not applicable.
Appendix A

Figure A1.
Typical curve of 226Th elution from the first column with TEVA resin (1 mL).
Figure A1.
Typical curve of 226Th elution from the first column with TEVA resin (1 mL).


Figure A2.
Distribution of 230U activity along the length of the first column with TEVA resin depending on the number of elutions.
Figure A2.
Distribution of 230U activity along the length of the first column with TEVA resin depending on the number of elutions.


Figure A3.
Illustration of 230Pa production by a 10-day irradiation of thorium target followed by isolation and periodic extraction of 230U (see irradiation details in [15]).
Figure A3.
Illustration of 230Pa production by a 10-day irradiation of thorium target followed by isolation and periodic extraction of 230U (see irradiation details in [15]).

References
- Alfassi, Z.B.; Bonardi, M.; Groppi, F.; Menapace, E. A new alpha-emitter for nuclear medicine: 230U. J. Radioanal. Nucl. Chem. 2006, 270, 483–487. [Google Scholar] [CrossRef]
- National Nuclear Data Center, Brookhaven National Laboratory, USA. Available online: http://www.nndc.bnl.gov/nudat3/ (accessed on 21 February 2023).
- Makvandi, M.; Dupis, E.; Engle, J.W.; Nortier, F.M.; Fassbender, M.E.; Simon, S.; Birnbaum, E.R.; Atcher, R.W.; John, K.D.; Rixe, O.; et al. Alpha-emitters and targeted alpha therapy in oncology: From basic science to clinical investigations. Target. Oncol. 2018, 13, 189–203. [Google Scholar] [CrossRef] [PubMed]
- Le Du, A. Study of In Vivo Generators Pb-212/Bi–212 and U-230/Th-226 for Alpha Radioimmunotherapy. Ph.D. Thesis, Université de Nantes, Nantes, France, 2011. No. FRNC-TH—8349. Available online: https://tel.archives-ouvertes.fr/tel-00779481 (accessed on 8 April 2023).
- Woods, J.J.; Unnerstall, R.; Hasson, A.; Abou, D.S.; Radchenko, V.; Thorek, D.L.J.; Wilson, J.J. Stable Chelation of the Uranyl Ion by Acyclic Hexadentate Ligands: Potential Applications for 230U Targeted α-Therapy. Inorg. Chem. 2022, 61, 3337–3350. [Google Scholar] [CrossRef] [PubMed]
- Ferrier, M.G.; Li, Y.; Chyanand, M.K.; Wong, R.; Li, L.; Spreckelmeyer, S.; Hamlin, D.K.; Mastren, T.; Fassbender, M.E.; Orvig, C.; et al. Thorium chelators for targeted alpha therapy: Rapid chelation of thorium-226. J. Label. Compd. Radiopharm. 2020, 63, 502–516. [Google Scholar] [CrossRef] [PubMed]
- Morgenstern, A.; Lebeda, O.; Stursa, J.; Bruchertseifer, F.; Capote, R.; McGinley, J.; Rasmussen, G.; Sin, M.; Zielinska, B.; Apostolidis, C. Production of 230U/226Th for Targeted Alpha Therapy via Proton Irradiation of 231Pa. Anal. Chem. 2008, 80, 8763–8770. [Google Scholar] [CrossRef]
- Morgenstern, A.; Lebeda, O.; Stursa, J.; Capote, R.; Sin, M.; Bruchertseifer, F.; Zielinska, B.; Apostolidis, C. Cross-Sections of the Reaction Pa231(d, 3n)U230 for the Production of U230/Th226 for Targeted α Therapy. Phys. Rev. C 2009, 80, 054612. [Google Scholar] [CrossRef]
- Radchenko, V.; Engle, J.W.; Wilson, J.J.; Maassen, J.R.; Nortier, M.F.; Birnbaum, E.R.; John, K.D.; Fassbender, M.E. Formation Cross-Sections and Chromatographic Separation of Protactinium Isotopes Formed in Proton-Irradiated Thorium Metal. Radiochim. Acta 2016, 104, 291–304. [Google Scholar] [CrossRef]
- Friend, M.; Mastren, T.; Parker, T.; Vermeulen, C.; Brugh, M.; Birnbaum, E.; Nortier, F.; Fassbender, M. Production of 230Pa by Proton Irradiation of 232Th at the LANL Isotope Production Facility: Precursor of 230U for Targeted Alpha Therapy. Appl. Radiat. Isot. 2019, 156, 108973. [Google Scholar] [CrossRef]
- Mastren, T.; Stein, B.W.; Parker, T.G.; Radchenko, V.; Copping, R.; Owens, A.; Wyant, L.; Brugh, M.; Kozimor, S.; Nortier, F.; et al. Separation of Protactinium Employing Sulfur-Based Extraction Chromatographic Resins. Anal. Chem. 2018, 90, 7012–7017. [Google Scholar] [CrossRef]
- Steyn, G.F.; Motetshwane, M.A.; Szelecsényi, F.; Brümmer, J.W. Pairing of thorium with selected primary target materials in tandem configurations: Co-production of 225Ac/213Bi and 230U/226Th generators with a 70 MeV H− cyclotron. Appl. Radiat. Isot. 2021, 168, 109514. [Google Scholar] [CrossRef]
- Mastren, T.; Akin, A.; Copping, R.; Brugh, M.; Wilbur, D.S.; Birnbaum, E.R.; Nortier, F.M.; John, K.D.; Fassbender, M.E. A reverse 230U/226Th radionuclide generator for targeted alpha therapy applications. Nucl. Med. Biol. 2020, 90, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Duchemin, C.; Guertin, A.; Haddad, F.; Michel, N.; Metivier, V. 232Th(d, 4n)230Pa Cross-Section Measurements at ARRONAX Facility for Production of 230U. Nucl. Med. Biol. 2014, 41, e19–e22. [Google Scholar] [CrossRef] [PubMed]
- Vasiliev, A.N.; Ermolaev, S.V.; Lapshina, E.V.; Bravo, M.G.; Skasyrskaya, A.K. Production of 230Pa as a Source for Medical Radionuclides 230U and 226Th Including Isolation by Liquid–liquid Extraction. Solvent Extr. Ion Exch. 2022, 40, 735–755. [Google Scholar] [CrossRef]
- Morgenstern, A.; Apostolidis, C.; Bruchertseifer, F.; Capote, R.; Gouder, T.; Simonelli, F.; Sin, M.; Abbas, K. Cross-Sections of the Reaction 232Th(p,3n)230Pa for Production of 230U for Targeted Alpha Therapy. Appl. Radiat. Isot. 2008, 66, 1275–1280. [Google Scholar] [CrossRef] [PubMed]
- Ermolaev, S.V.; Zhuikov, B.L.; Kokhanyuk, V.; Matushko, V.L.; Kalmykov, S.N.; Aliev, R.A.; Tananaev, I.G.; Myasoedov, B.F. Production of actinium, thorium and radium isotopes from natural thorium irradiated with protons up to 141 MeV. Radiochim. Acta 2012, 100, 223–229. [Google Scholar] [CrossRef]
- Aliev, R.; Ermolaev, S.V.; Vasiliev, A.; Ostapenko, V.S.; Lapshina, E.V.; Zhuikov, B.L.; Zakharov, N.V.; Pozdeev, V.V.; Kokhanyuk, V.M.; Myasoedov, B.F.; et al. Isolation of medicine-applicable actinium-225 from thorium targets irradiated by medium-energy protons. Solv. Extr. Ion Exch. 2014, 32, 468. [Google Scholar] [CrossRef]
- Vasiliev, A.N.; Ostapenko, V.S.; Lapshina, E.V.; Ermolaev, S.V.; Danilov, S.S.; Zhuikov, B.L.; Kalmykov, S.N. Recovery of Ra-223 from natural thorium irradiated by protons. Radiochim. Acta 2016, 104, 539–547. [Google Scholar] [CrossRef]
- Ermolaev, S.; Skasyrskaya, A.; Vasiliev, A. A Radionuclide Generator of High-purity Bi-213 for Instant Labeling. Pharmaceutics 2021, 13, 914. [Google Scholar] [CrossRef]
- Ahenkorah, S.; Cassells, I.; Deroose, C.M.; Cardinaels, T.; Burgoyne, A.R.; Bormans, G.; Ooms, M.; Cleeren, F. Bismuth-213 for Targeted Radionuclide Therapy: From Atom to Bedside. Pharmaceutics 2021, 13, 599. [Google Scholar] [CrossRef]
- Bravo, M.G.; Egorova, B.V.; Vasiliev, A.N.; Lapshina, E.V.; Ermolaev, S.V.; Durymanov, M.O. DTPA(DOTA)-Nimotuzumab Radiolabeling with Generator-Produced Thorium for Radioimmunotherapy of EGFR-Overexpressing Carcinomas. Cur. Radiopharm. 2023; accepted for publication. [Google Scholar]
- Sharma, J.N.; Ruhela, R.; Harindaran, K.N.; Mishra, S.L.; Tangri, S.K.; Suri, A.K. Separation studies of uranium and thorium using tetra(2-ethylhexyl) diglycolamide (TEHDGA) as an extractant. J. Radioanal. Nucl. Chem. 2008, 278, 173–177. [Google Scholar] [CrossRef]
- Pathak, P.N.; Veeraraghavan, R.; Prabhu, D.R.; Mahajan, G.R.; Manchanda, V.K. Separation studies of uranium and thorium using di-2-ethylhexyl isobutyramide (D2EHIBA). Separ. Sci. Technol. 1999, 34, 2601–2614. [Google Scholar] [CrossRef]
- Raju, C.S.K.; Subramanian, M.S. Sequential separation of lanthanides, thorium and uranium using novel solid phase extraction method from high acidic nuclear wastes. J. Hazard Mater. 2007, 145, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Carter, H.E.; Warwick, P.; Cobb, J.; Longworth, G. Determination of uranium and thorium in geological materials using extraction chromatography. Analyst 1999, 124, 271–274. [Google Scholar] [CrossRef]
- Yokoyama, T.; Makishima, A.; Nakamura, E. Separation of thorium and uranium from silicate rock samples using two commercial extraction chromatographic resins. Anal. Chem. 1999, 71, 135–141. [Google Scholar] [CrossRef]
- Korkisch, J.; Tera, F. Anion Exchange Separation of Uranium, Thorium and Bismuth. Z. Anal. Chem. 1962, 186, 290–295. [Google Scholar] [CrossRef]
- Alhassanieh, O.; Abdul-Hadi, A.; Ghafar, M.; Aba, A. Separation of Th, U, Pa, Ra and Ac from natural uranium and thorium series. Appl. Radiat. Isot. 1999, 51, 493–498. [Google Scholar] [CrossRef]
- Carswell, D.J. Separation of Thorium and Uranium Nitrates by Anion Exchange. J. Inorg. Nucl. Chem. 1957, 3, 384–387. [Google Scholar] [CrossRef]
- Horwitz, E.P.; Dietz, M.L.; Chiarizia, R.; Diamond, H.; Maxwell, S.L.; Nelson, M.R. Separation and Preconcentration of Actinides by Extraction Chromatography using a supported liquid anion exchanger: Application of the characterization of high-level nuclear waste solutions. Analyt. Chim. Acta 1995, 310, 63–78. [Google Scholar] [CrossRef]
- Maxwell, S.L. Rapid Analysis of Emergency Urine and Water Samples. J. Radioanal. Nucl. Chem. 2008, 275, 497–502. [Google Scholar] [CrossRef]
- Kraus, K.A.; Moore, G.E.; Nelson, F. Anion-exchange Studies. XXI. Th(IV) and U(IV) in Hydrochloric Acid. Separation of Thorium, Protactinium and Uranium. J. Am. Chem. Soc. 1956, 78, 2692–2695. [Google Scholar] [CrossRef]
- Wu, C.; Brechbiel, M.W.; Gansow, O.A. An Improved Generator for the Production of 213Bi from 225Ac. Radiochim. Acta 1997, 79, 141–144. [Google Scholar] [CrossRef]
- Dietz, M.L.; Horwitz, E.P. Applications of Extraction Chromatography in the Development of Radionuclide Generator Systems for Nuclear Medicine. Ind. Eng. Chem. Res. 2000, 39, 3181–3188. [Google Scholar] [CrossRef]
- Vasiliev, A.N.; Zobnin, V.A.; Pavlov, Y.S.; Chudakov, V.M. Radiation Stability of Sorbents in Medical 225Ac/213Bi Generators. Solvent. Extr. Ion Exch. 2021, 39, 353–372. [Google Scholar] [CrossRef]
- Horwitz, E.P.; McAlister, D.R.; Bond, A.H.; Barrans, R.E., Jr. Novel extraction of chromatographic resins based on tetraalkyldiglycolamides: Characterization and potential applications. Solv. Extr. Ion Exch. 2005, 23, 319–344. [Google Scholar] [CrossRef]
- Horwitz, E.P.; Dietz, M.L.; Chiarizia, R.; Diamond, H.; Essling, A.M.; Graczyk, D. Separation and preconcentration of uranium from acidic media by extraction chromatography. Anal. Chim. Acta 1992, 266, 25–37. [Google Scholar] [CrossRef]
- Horwitz, E.P.; Chiarizia, R.; Dietz, M.L.; Diamond, H.; Nelson, D.M. Separation and preconcentration of actinides from acidic media by extraction chromatography. Anal. Chim. Acta 1993, 281, 361–372. [Google Scholar] [CrossRef]
- Raymond, D.P.; Duffield, J.R.; Williams, D.R. Complexation of plutonium and thorium in aqueous environments. Inorg. Chim. Acta 1987, 140, 309–313. [Google Scholar] [CrossRef]
- Choppin, G.R.; Erten, H.N.; Xia, Y.X. Variation of stability constants of thorium citrate complexes with ionic strength. Radiochim. Acta 1996, 74, 123–128. [Google Scholar] [CrossRef]
- Zhuikov, B.L.; Kokhanyuk, V.M.; Konyakhin, N.A.; Vincent, J. Target irradiation facility and targetry development at 160 MeV proton beam of Moscow linac. Nucl. Instrum. Methods Phys. Res. Sect. A 1999, 438, 173. [Google Scholar] [CrossRef]
- Ermolaev, S.V.; Vasilev, A.N.; Lapshina, E.V.; Zhuikov, B.L. Method of Producing Actinium-225. Russian Patent № 2 725 414, 12 December 2019. [Google Scholar]
- Knight, A.W.; Eitrheim, E.S.; Nelson, A.W.; Nelson, S.; Schultz, M.K. A simple-rapid method to separate uranium, thorium, and protactinium for U-series age-dating of materials. J. Environ. Radioact. 2014, 134, 66–74. [Google Scholar] [CrossRef]
- Pourmand, A.; Dauphas, N. Distribution coefficients of 60 elements on TODGA resin: Application to Ca, Lu, Hf, U and Th isotope geochemistry. Talanta 2010, 81, 741–753. [Google Scholar] [CrossRef] [PubMed]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).