

Asymmetric Reactions of N-Phosphonyl/Phosphoryl Imines


{kind=link}
{kind=link}
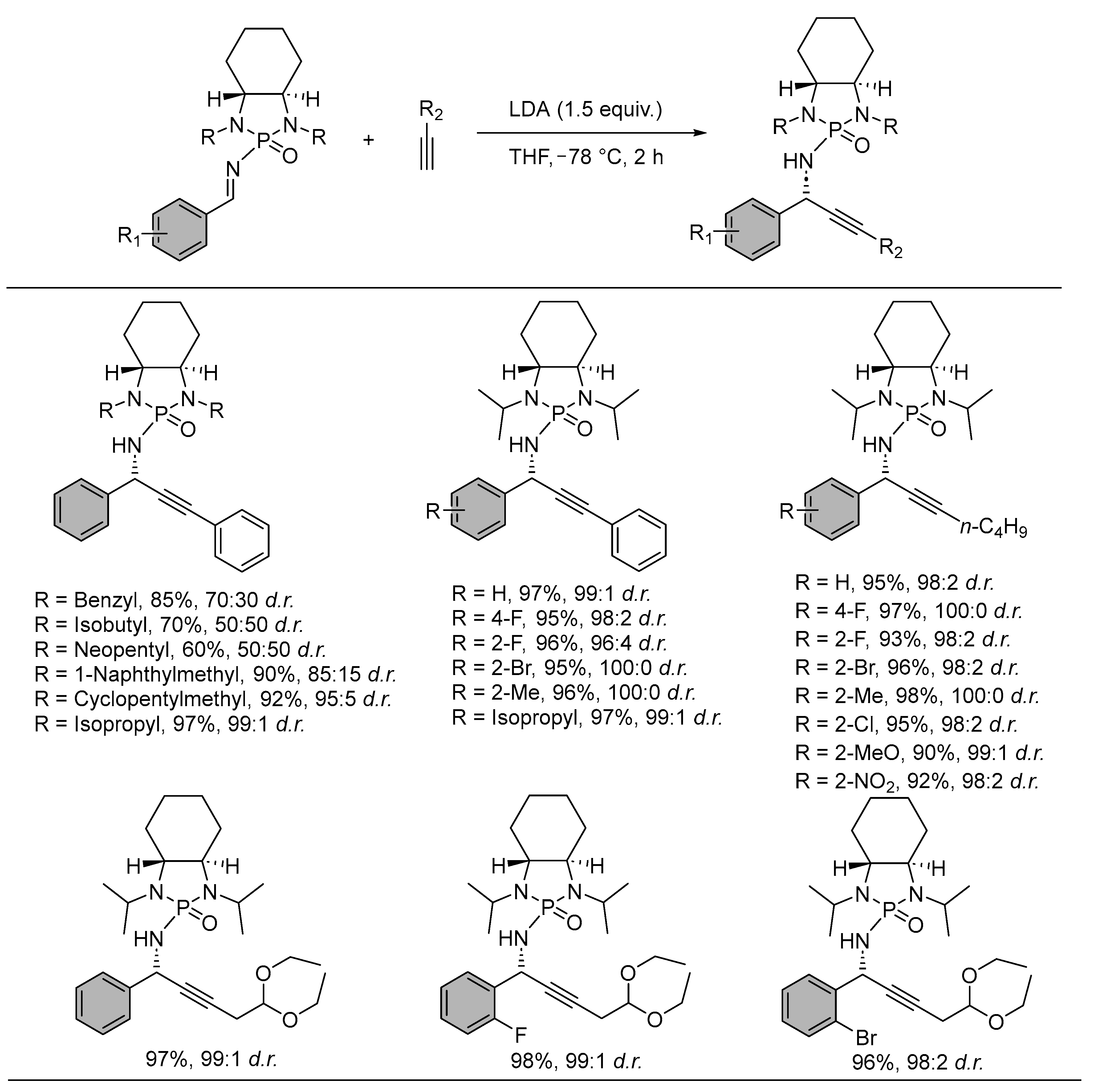{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
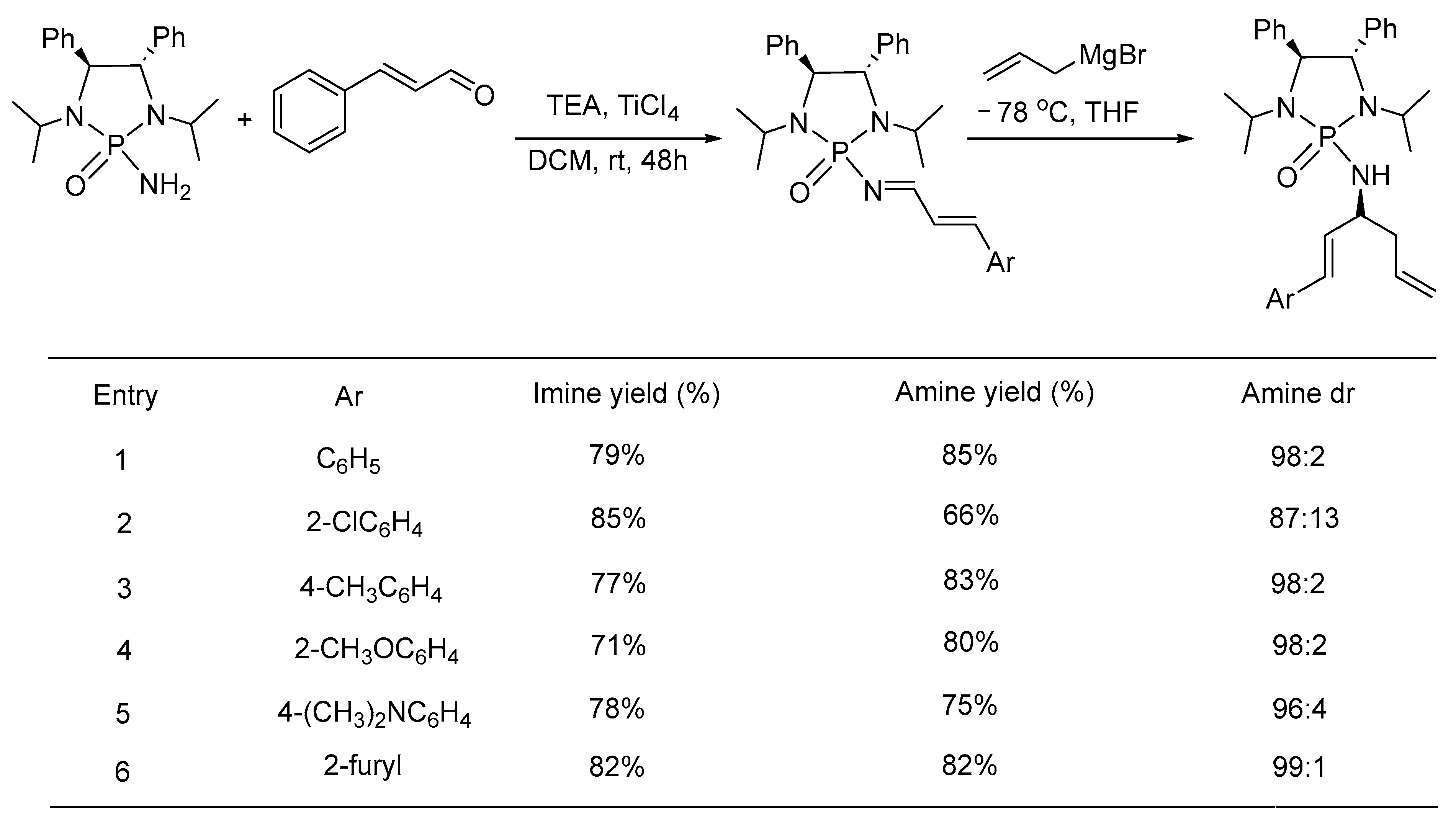{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
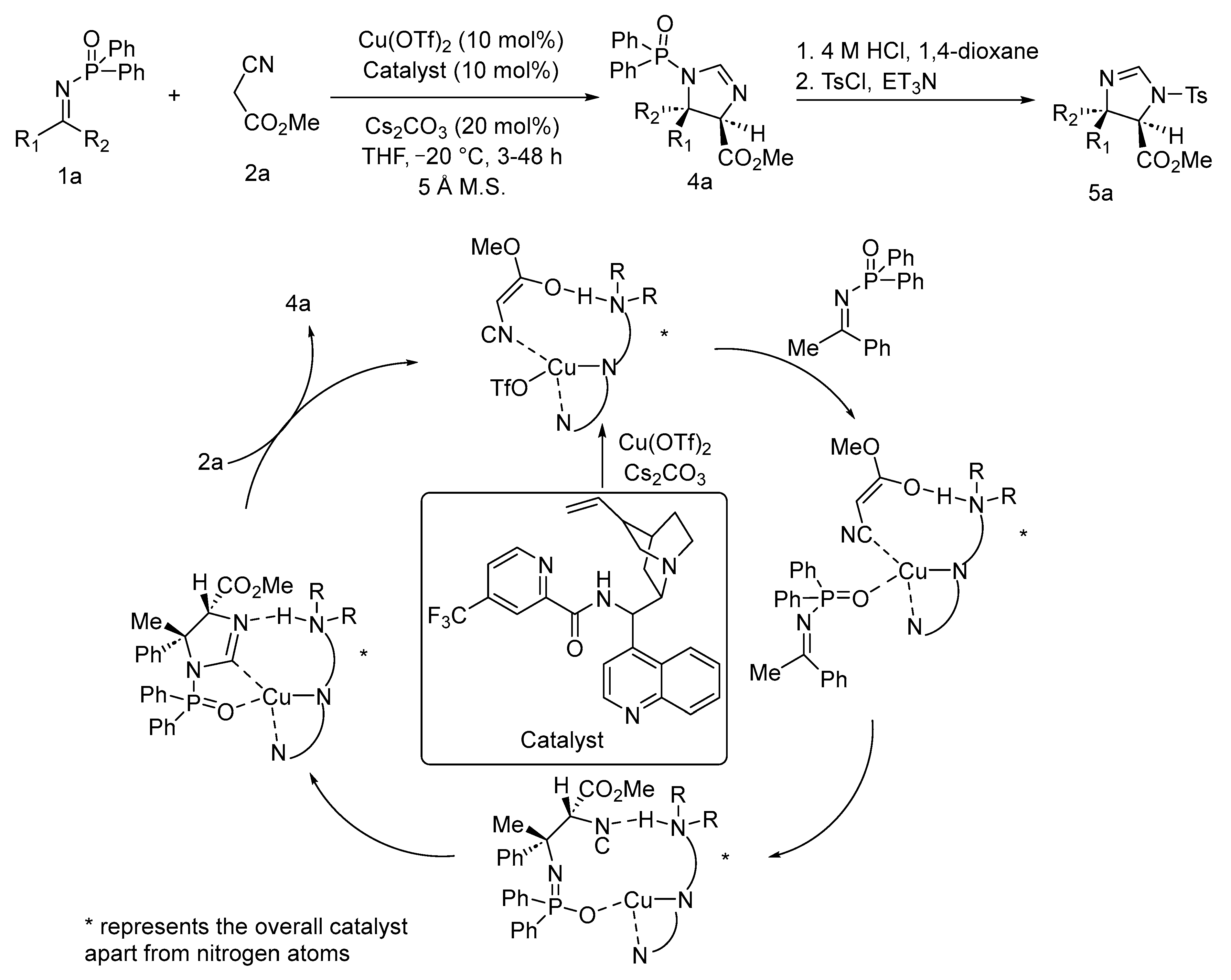{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Asymmetric Reactions in Chiral N-Phosphonyl Imine Auxiliary
3. Asymmetric Reactions in Achiral Phosphonyl Imine Using Chiral Catalyst/Ligand
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hamilton, G.L.; Kang, E.J.; Mba, M.; Toste, F.D. A Powerful Chiral Counterion Strategy for Asymmetric Transition Metal Catalysis. Science 2007, 317, 496–499. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.-H.; Zhu, S.-F.; Zhou, Q.-L. Transition Metal-Catalyzed Enantioselective Hydrogenation of Enamines and Imines. Chem. Rev. 2011, 111, 1713–1760. [Google Scholar] [CrossRef]
- Kobayashi, S.; Mori, Y.; Fossey, J.S.; Salter, M.M. Catalytic Enantioselective Formation of C–C Bonds by Addition to Imines and Hydrazones: A Ten-Year Update. Chem. Rev. 2011, 111, 2626–2704. [Google Scholar] [CrossRef]
- Kang, Q.; Zhao, Z.-A.; You, S.-L. Highly Enantioselective Friedel−Crafts Reaction of Indoles with Imines by a Chiral Phosphoric Acid. J. Am. Chem. Soc. 2007, 129, 1484–1485. [Google Scholar] [CrossRef] [PubMed]
- Xie, D.; Yang, L.; Lin, Y.; Zhang, Z.; Chen, D.; Zeng, X.; Zhong, G. Rapid Access to Spirocylic Oxindoles: Application of Asymmetric N-Heterocyclic Carbene-Catalyzed [3 + 3] Cycloaddition of Imines to Oxindole-Derived Enals. Org. Lett. 2015, 17, 2318–2321. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; An, G.; Lin, S.; Xie, J.; Zhou, W.; Sun, H.; Pan, Y.; Li, G. Solution-Phase-Peptide Synthesis via the Group-Assisted Purification (GAP) Chemistry without Using Chromatography and Recrystallization. Chem. Commun. 2014, 50, 1259–1261. [Google Scholar] [CrossRef]
- Ellman, J.A.; Owens, T.D.; Tang, T.P. N-Tert-Butanesulfinyl Imines: Versatile Intermediates for the Asymmetric Synthesis of Amines. Acc. Chem. Res. 2002, 35, 984–995. [Google Scholar] [CrossRef]
- Ellman, J.A. Applications of Tert-Butanesulfinamide in the Asymmetric Synthesis of Amines. Pure Appl. Chem. 2003, 75, 39–46. [Google Scholar] [CrossRef]
- Reddy, N.S.S.; Reddy, B.J.M.; Reddy, B.V.S. A Convergent and Stereoselective Total Synthesis of (−)-Crispine A, (−)-Benzo[a]Quinolizidine and (−)-Salsolidine. Tetrahedron Lett. 2013, 54, 4228–4231. [Google Scholar] [CrossRef]
- Reddy, N.; Babu, R.; Reddy, B. Asymmetric Synthesis of Tetrahydro-β-Carboline Alkaloids Employing Ellman’s Chiral Auxiliary. Synthesis 2016, 48, 1079–1086. [Google Scholar] [CrossRef]
- Huguenot, F.; Brigaud, T. Concise Synthesis of Enantiopure α-Trifluoromethyl Alanines, Diamines, and Amino Alcohols via the Strecker-Type Reaction. J. Org. Chem. 2006, 71, 7075–7078. [Google Scholar] [CrossRef]
- Vilaivan, T.; Winotapan, C.; Banphavichit, V.; Shinada, T.; Ohfune, Y. Indium-Mediated Asymmetric Barbier-Type Allylation of Aldimines in Alcoholic Solvents: Synthesis of Optically Active Homoallylic Amines. J. Org. Chem. 2005, 70, 3464–3471. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.-W.; Liu, M.; Xu, M.-H.; Lin, G.-Q. Remarkable Salt Effect on In-Mediated Allylation of N—Tert-Butanesulfinyl Imines in Aqueous Media: Highly Practical Asymmetric Synthesis of Chiral Homoallylic Amines and Isoindolinones. Org. Lett. 2008, 10, 1259–1262. [Google Scholar] [CrossRef]
- Roy, A.; Gosselin, F.; O’Shea, P.D.; Chen, C. Diastereoselective Arylithium Addition to an α-Trifluoromethyl Imine. Practical Synthesis of a Potent Cathepsin K Inhibitor. J. Org. Chem. 2006, 71, 4320–4323. [Google Scholar] [CrossRef] [PubMed]
- Samanta, D.; Kargbo, R.B.; Cook, G.R. Asymmetric Synthesis of Aminochromanes via Intramolecular Indium-Mediated Allylation of Chiral Hydrazones. J. Org. Chem. 2009, 74, 7183–7186. [Google Scholar] [CrossRef]
- Fustero, S.; Jiménez, D.; Sanz-Cervera, J.F.; Sánchez-Roselló, M.; Esteban, E.; Simón-Fuentes, A. Highly Enantioselective Synthesis of Fluorinated γ-Amino Alcohols through Proline-Catalyzed Cross-Mannich Reaction. Org. Lett. 2005, 7, 3433–3436. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Park, Y.; Yoon, W.; Yun, H.; Yun, J. Asymmetric Synthesis of 1-Benzazepine Derivatives via Copper-Catalyzed Intramolecular Reductive Cyclization. Org. Lett. 2019, 21, 9699–9703. [Google Scholar] [CrossRef]
- Chogii, I.; Njardarson, J.T. Asymmetric [3 + 2] Annulation Approach to 3-Pyrrolines: Concise Total Syntheses of (−)-Supinidine, (−)-Isoretronecanol, and (+)-Elacomine. Angew. Chem. Int. Ed. 2015, 54, 13706–13710. [Google Scholar] [CrossRef]
- Si, C.-M.; Shao, L.-P.; Mao, Z.-Y.; Zhou, W.; Wei, B.-G. An Efficient Approach to Trans-4-Hydroxy-5-Substituted 2-Pyrrolidinones through a Stereoselective Tandem Barbier Process: Divergent Syntheses of (3R,4S)-Statines, (+)-Preussin and (−)-Hapalosin. Org. Biomol. Chem. 2017, 15, 649–661. [Google Scholar] [CrossRef]
- Si, C.-M.; Mao, Z.-Y.; Liu, Y.-W.; Du, Z.-T.; Wei, B.-G.; Lin, G.-Q. Stereoselective Formation of Chiral Trans-4-Hydroxy-5-Substituted 2-Pyrrolidinones: Syntheses of Streptopyrrolidine and 3-Epi-Epohelmin A. Org. Chem. Front. 2015, 2, 1485–1499. [Google Scholar] [CrossRef]
- Fustero, S.; Herrera, L.; Lázaro, R.; Rodríguez, E.; Maestro, M.A.; Mateu, N.; Barrio, P. Base-Dependent Stereodivergent Intramolecular Aza-Michael Reaction: Asymmetric Synthesis of 1,3-Disubstituted Isoindolines. Chem. A Eur. J. 2013, 19, 11776–11785. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Yao, L.; Luo, Y.; Liu, T.; Xu, J.; Sun, Y.; Wang, N.; Yan, J.; Tang, B.; Yang, G.; et al. Synthesis of Aziridines with Multiple Chiral Substitutions by Copper-Catalyzed Diastereoselective Radical Aminotrifluoromethylation of Alkenes. Org. Chem. Front. 2020, 7, 3132–3136. [Google Scholar] [CrossRef]
- Reddy, L.R.; Waman, Y.; Kallure, P.; Nalivela, K.S.; Begum, Z.; Divya, T.; Kotturi, S. Asymmetric Synthesis of 1-Substituted 2-Azaspiro[3.3]Heptanes: Important Motifs for Modern Drug Discovery. Chem. Commun. 2019, 55, 5068–5070. [Google Scholar] [CrossRef] [PubMed]
- Mendes, J.A.; Merino, P.; Soler, T.; Salustiano, E.J.; Costa, P.R.R.; Yus, M.; Foubelo, F.; Buarque, C.D. Enantioselective Synthesis, DFT Calculations, and Preliminary Antineoplastic Activity of Dibenzo 1-Azaspiro[4.5]Decanes on Drug-Resistant Leukemias. J. Org. Chem. 2019, 84, 2219–2233. [Google Scholar] [CrossRef] [PubMed]
- Peralta-Hernández, E.; Cordero-Vargas, A. Model Studies Toward the Enantioselective Synthesis of Perhydrohistrionicotoxin: A Free-Radical Approach to the Azaspirocycle Core. Synthesis 2016, 48, 4237–4245. [Google Scholar] [CrossRef]
- de Freitas, G.B.L. Chiral N-Phosphonyl Imine Chemistry: A New Asymmetric Synthesis Strategy. Aust. J. Chem. 2010, 63, 1594. [Google Scholar] [CrossRef]
- Kattuboina, A.; Li, G. Chiral N-Phosphonyl Imine Chemistry: New Reagents and Their Applications for Asymmetric Reactions. Tetrahedron Lett. 2008, 49, 1573–1577. [Google Scholar] [CrossRef]
- Denmark, S.E. Catalysts Break Symmetry. Nature 2006, 443, 40–41. [Google Scholar] [CrossRef]
- Li, G.; Wei, H.-X.; Whittlesey, B.R.; Batrice, N.N. Novel Asymmetric C−C Bond Formation Process Promoted by Et 2 AlCl and Its Application to the Stereoselective Synthesis of Unusual β-Branched Baylis−Hillman Adducts. J. Org. Chem. 1999, 64, 1061–1064. [Google Scholar] [CrossRef]
- Ai, T.; Han, J.; Chen, Z.-X.; Li, G. Chiral N-Phosphonyl Imine Chemistry: Asymmetric Synthesis of α-Alkyl β-Amino Ketones by Reacting Phosphonyl Imines with Ketone-Derived Enolates. Chem. Biol. Drug Des. 2009, 73, 203–208. [Google Scholar] [CrossRef]
- Kattuboina, A.; Kaur, P.; Ai, T.; Li, G. Chiral N-Phosphonyl Imine Chemistry: Asymmetric Aza-Henry Reaction. Chem. Biol. Drug Des. 2008, 71, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Kattuboina, A.; Kaur, P.; Nguyen, T.; Li, G. Chiral N-Phosphonyl Imine Chemistry: Asymmetric 1,2-Additions of Allylmagnesium Bromides. Tetrahedron Lett. 2008, 49, 3722–3724. [Google Scholar] [CrossRef]
- Han, J.; Ai, T.; Nguyen, T.; Li, G. Chiral N-Phosphonyl Imine Chemistry: Asymmetric Additions of Ester Enolates for the Synthesis of β-Amino Acids. Chem. Biol. Drug Des. 2008, 72, 120–126. [Google Scholar] [CrossRef] [PubMed]
- An, G.; Seifert, C.; Li, G. N-Phosphonyl/Phosphinyl Imines and Group-Assisted Purification (GAP) Chemistry/Technology. Org. Biomol. Chem. 2015, 13, 1600–1617. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Xu, L.-W.; Shimizu, Y.; Oisaki, K.; Kanai, M.; Shibasaki, M. Asymmetric Reductive Mannich Reaction to Ketimines Catalyzed by a Cu(I) Complex. J. Am. Chem. Soc. 2008, 130, 16146–16147. [Google Scholar] [CrossRef]
- Oña Burgos, P.; Fernández, I.; Iglesias, M.J.; García-Granda, S.; López Ortiz, F. Phosphinamide-Directed Benzylic Lithiation. Application to the Synthesis of Peptide Building Blocks. Org. Lett. 2008, 10, 537–540. [Google Scholar] [CrossRef]
- Boezio, A.A.; Charette, A.B. Catalytic Enantioselective Addition of Dialkylzinc to N-Diphenylphosphinoylimines. A Practical Synthesis of α-Chiral Amines. J. Am. Chem. Soc. 2003, 125, 1692–1693. [Google Scholar] [CrossRef]
- Zhang, X.-M.; Zhang, H.-L.; Lin, W.-Q.; Gong, L.-Z.; Mi, A.-Q.; Cui, X.; Jiang, Y.-Z.; Yu, K.-B. Evaluation of Chiral Oxazolines for the Highly Enantioselective Diethylzinc Addition to N-(Diphenylphosphinoyl) Imines. J. Org. Chem. 2003, 68, 4322–4329. [Google Scholar] [CrossRef]
- Wieland, L.C.; Vieira, E.M.; Snapper, M.L.; Hoveyda, A.H. Ag-Catalyzed Diastereo- and Enantioselective Vinylogous Mannich Reactions of α-Ketoimine Esters. Development of a Method and Investigation of Its Mechanism. J. Am. Chem. Soc. 2009, 131, 570–576. [Google Scholar] [CrossRef]
- Fu, P.; Snapper, M.L.; Hoveyda, A.H. Catalytic Asymmetric Alkylations of Ketoimines. Enantioselective Synthesis of N-Substituted Quaternary Carbon Stereogenic Centers by Zr-Catalyzed Additions of Dialkylzinc Reagents to Aryl-, Alkyl-, and Trifluoroalkyl-Substituted Ketoimines. J. Am. Chem. Soc. 2008, 130, 5530–5541. [Google Scholar] [CrossRef]
- Josephsohn, N.S.; Snapper, M.L.; Hoveyda, A.H. Ag-Catalyzed Asymmetric Mannich Reactions of Enol Ethers with Aryl, Alkyl, Alkenyl, and Alkynyl Imines. J. Am. Chem. Soc. 2004, 126, 3734–3735. [Google Scholar] [CrossRef] [PubMed]
- Viso, A.; Fernández de la Pradilla, R.; Tortosa, M.; García, A.; Flores, A. Update 1 of: α,β-Diamino Acids: Biological Significance and Synthetic Approaches. Chem. Rev. 2011, 111, PR1–PR42. [Google Scholar] [CrossRef]
- Sun, H.; Rajale, T.; Pan, Y.; Li, G. Chiral N-Phosphoryl Imines: Design, Synthesis and Direct Asymmetric Addition Reactions with Diketones and Diesters. Tetrahedron Lett. 2010, 51, 4403–4407. [Google Scholar] [CrossRef]
- Ai, T.; Pindi, S.; Kattamuri, P.V.; Li, G. Chiral N-Phosphonyl Imine Chemistry: Asymmetric Additions of Glycine Enolate to Diphenyl Diamine-Based Phosphonyl Imines. Sci. China Chem. 2010, 53, 125–129. [Google Scholar] [CrossRef]
- Kaur, P.; Shakya, G.; Sun, H.; Pan, Y.; Li, G. Chiral N-Phosphonyl Imine Chemistry: An Efficient Asymmetric Synthesis of Chiral N-Phosphonyl Propargylamines. Org. Biomol. Chem. 2010, 8, 1091–1096. [Google Scholar] [CrossRef] [PubMed]
- Kattamuri, P.V.; Xiong, Y.; Pan, Y.; Li, G. N,N-Diisopropyl-N-Phosphonyl Imines Lead to Efficient Asymmetric Synthesis of Aziridine-2-Carboxylic Esters. Org. Biomol. Chem. 2013, 11, 3400. [Google Scholar] [CrossRef] [PubMed]
- Seifert, C.W.; Pindi, S.; Li, G. Asymmetric Carbamoyl Anion Additions to Chiral N-Phosphonyl Imines via the GAP Chemistry Process and Stereoselectivity Enrichments. J. Org. Chem. 2015, 80, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Qiao, S.; Wilcox, C.B.; Unruh, D.K.; Jiang, B.; Li, G. Asymmetric [3 + 2] Cycloaddition of Chiral N-Phosphonyl Imines with Methyl Isocyanoacetate for Accessing 2-Imidazolines with Switchable Stereoselectivity. J. Org. Chem. 2017, 82, 2992–2999. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Shen, M.; Ji, X.; Xu, Z.; Sun, H.; Jiang, B.; Li, G. Chiral N-Phosphonyl Imines for an Aza-Morita-Baylis-Hillman Reaction via Group-Assisted Purification (GAP) Chemistry. J. Org. Chem. 2016, 81, 2488–2493. [Google Scholar] [CrossRef]
- Zhang, H.; Yang, Z.; Zhao, B.N.; Li, G. Group-Assisted Purification Chemistry for Asymmetric Mannich-Type Reaction of Chiral N-Phosphonyl Imines with Azlactones Leading to Syntheses of α-Quaternary α,β-Diamino Acid Derivatives. J. Org. Chem. 2018, 83, 644–655. [Google Scholar] [CrossRef]
- Kaur, P.; Wever, W.; Rajale, T.; Li, G. Asymmetric Hydrophosphylation of Chiral N-Phosphonyl Imines Provides an Efficient Approach to Chiral α-Amino Phosphonates. Chem. Biol. Drug Des. 2010, 76, 314–319. [Google Scholar] [CrossRef]
- Kattamuri, P.V.; Ai, T.; Pindi, S.; Sun, Y.; Gu, P.; Shi, M.; Li, G. Asymmetric Synthesis of α-Amino-1,3-Dithianes via Chiral N-Phosphonyl Imine-Based Umpolung Reaction without Using Chromatography and Recrystallization. J. Org. Chem. 2011, 76, 2792–2797. [Google Scholar] [CrossRef] [PubMed]
- Pindi, S.; Kaur, P.; Shakya, G.; Li, G. N-Phosphinyl Imine Chemistry (I): Design and Synthesis of Novel N-Phosphinyl Imines and Their Application to Asymmetric Aza-Henry Reaction. Chem. Biol. Drug Des. 2011, 77, 20–29. [Google Scholar] [CrossRef]
- Kaur, P.; Wever, W.; Pindi, S.; Milles, R.; Gu, P.; Shi, M.; Li, G. The GAP Chemistry for Chiral N-Phosphonyl Imine-Based Strecker Reaction. Green Chem. 2011, 13, 1288–1292. [Google Scholar] [CrossRef]
- Xie, J.B.; Luo, J.; Winn, T.R.; Cordes, D.B.; Li, G. Group-Assisted Purification (GAP) Chemistry for the Synthesis of Velcade via Asymmetric Borylation of N-Phosphinylimines. Beilstein J. Org. Chem. 2014, 10, 746–751. [Google Scholar] [CrossRef]
- Xiong, Y.; Mei, H.; Xie, C.; Han, J.; Li, G.; Pan, Y. Asymmetric Synthesis of α-Alkenyl Homoallylic Primary Amines via 1,2-Addition of Grignard Reagent to α,β-Unsaturated Phosphonyl Imines. RSC Adv. 2013, 3, 15820–15826. [Google Scholar] [CrossRef] [PubMed]
- Qiao, S.; Pindi, S.; Spigener, P.T.; Jiang, B.; Li, G. Asymmetric Synthesis of Homoallylic Amines via 1,2-Addition of Grignard Reagent to Aliphatic N-Phosphonyl Hemiaminal. Tetrahedron Lett. 2016, 57, 619–622. [Google Scholar] [CrossRef]
- Silverio, D.L.; Torker, S.; Pilyugina, T.; Vieira, E.M.; Snapper, M.L.; Haeffner, F.; Hoveyda, A.H. Simple Organic Molecules as Catalysts for Enantioselective Synthesis of Amines and Alcohols. Nature 2013, 494, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Vieira, E.M.; Snapper, M.L.; Hoveyda, A.H. Enantioselective Synthesis of Homoallylic Amines through Reactions. J. Am. Chem. Soc. 2011, 133, 3332–3335. [Google Scholar] [CrossRef]
- Vieira, E.M.; Haeffner, F.; Snapper, M.L.; Hoveyda, A.H. A Robust, Efficient, and Highly Enantioselective Method for Synthesis of Homopropargyl Amines. Angew. Chem. Int. Ed. 2012, 51, 6618–6621. [Google Scholar] [CrossRef]
- Fan, W.; Kong, S.; Cai, Y.; Wu, G.; Miao, Z. Diastereo- and Enantioselective Nitro-Mannich Reaction of α-Substituted Nitroacetates to N-Phosphoryl Imines Catalyzed by Cinchona Alkaloid Thiourea Organocatalysts. Org. Biomol. Chem. 2013, 11, 3223–3229. [Google Scholar] [CrossRef] [PubMed]
- Li, S.S.; Wu, L.; Qin, L.; Zhu, Y.Q.; Su, F.; Xu, Y.J.; Dong, L. Iridium(III)-Catalyzed Tandem [3 + 2] Annulation: Synthesis of Spirocyclic Phosphoramide Derivatives. Org. Lett. 2016, 18, 4214–4217. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Chen, X.Y.; Enders, D. Aldehyde Catalysis: New Options for Asymmetric Organocatalytic Reactions. Chem 2018, 4, 2026–2028. [Google Scholar] [CrossRef]
- Zhang, C.; Yang, J.; Zhou, W.; Tan, Q.; Yang, Z.; He, L.; Zhang, M. Enantioselective Mannich Reaction of Glycine Iminoesters with N-Phosphinoyl Imines: A Bifunctional Approach. Org. Lett. 2019, 21, 8620–8624. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Iwanaga, M.; Shiomi, N.; Nakane, D.; Masuda, H.; Nakamura, S. Direct Asymmetric Mannich-Type Reaction of α-Isocyanoacetates with Ketimines Using Cinchona Alkaloid/Copper(II) Catalysts. Angew. Chem. Int. Ed. 2014, 53, 8411–8415. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Yoshino, T.; Morimoto, H.; Matsunaga, S.; Shibasaki, M. Stereodivergent Direct Catalytic Asymmetric Mannich-Type Reactions of α-Isothiocyanato Ester with Ketimines. Angew. Chem. Int. Ed. 2011, 50, 4382–4385. [Google Scholar] [CrossRef]

























Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ray, D.; Majee, S.; Yadav, R.N.; Banik, B.K. Asymmetric Reactions of N-Phosphonyl/Phosphoryl Imines. Molecules 2023, 28, 3524. https://doi.org/10.3390/molecules28083524
Ray D, Majee S, Yadav RN, Banik BK. Asymmetric Reactions of N-Phosphonyl/Phosphoryl Imines. Molecules. 2023; 28(8):3524. https://doi.org/10.3390/molecules28083524
Chicago/Turabian StyleRay, Devalina, Suman Majee, Ram Naresh Yadav, and Bimal Krishna Banik. 2023. "Asymmetric Reactions of N-Phosphonyl/Phosphoryl Imines" Molecules 28, no. 8: 3524. https://doi.org/10.3390/molecules28083524
APA StyleRay, D., Majee, S., Yadav, R. N., & Banik, B. K. (2023). Asymmetric Reactions of N-Phosphonyl/Phosphoryl Imines. Molecules, 28(8), 3524. https://doi.org/10.3390/molecules28083524