
Evaluating Software Tools for Lipid Identification from Ion Mobility Spectrometry–Mass Spectrometry Lipidomics Data

 , ,
, ,  , and
, and
Abstract
1. Introduction
2. Lipidomics Data Analysis
3. Survey of Existing Lipidomics Data Analysis Software
4. Evaluation of Lipidomics Data Analysis Software
4.1. Selection of Evaluation Data
4.2. Selection of Software Tools for Evaluation
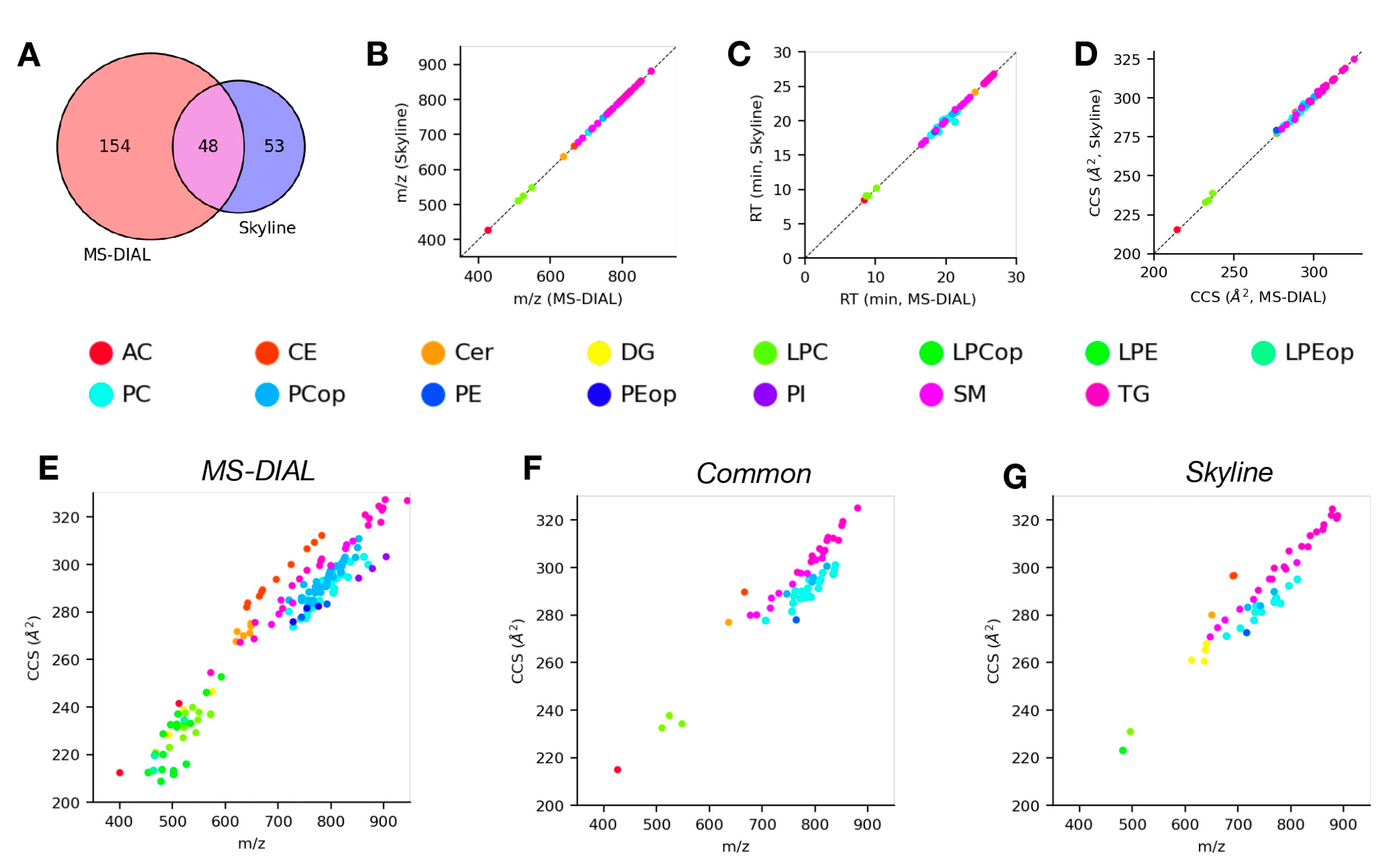
4.3. Analysis of LC-DTIMS-MS/MS Data Using Skyline and MS-DIAL
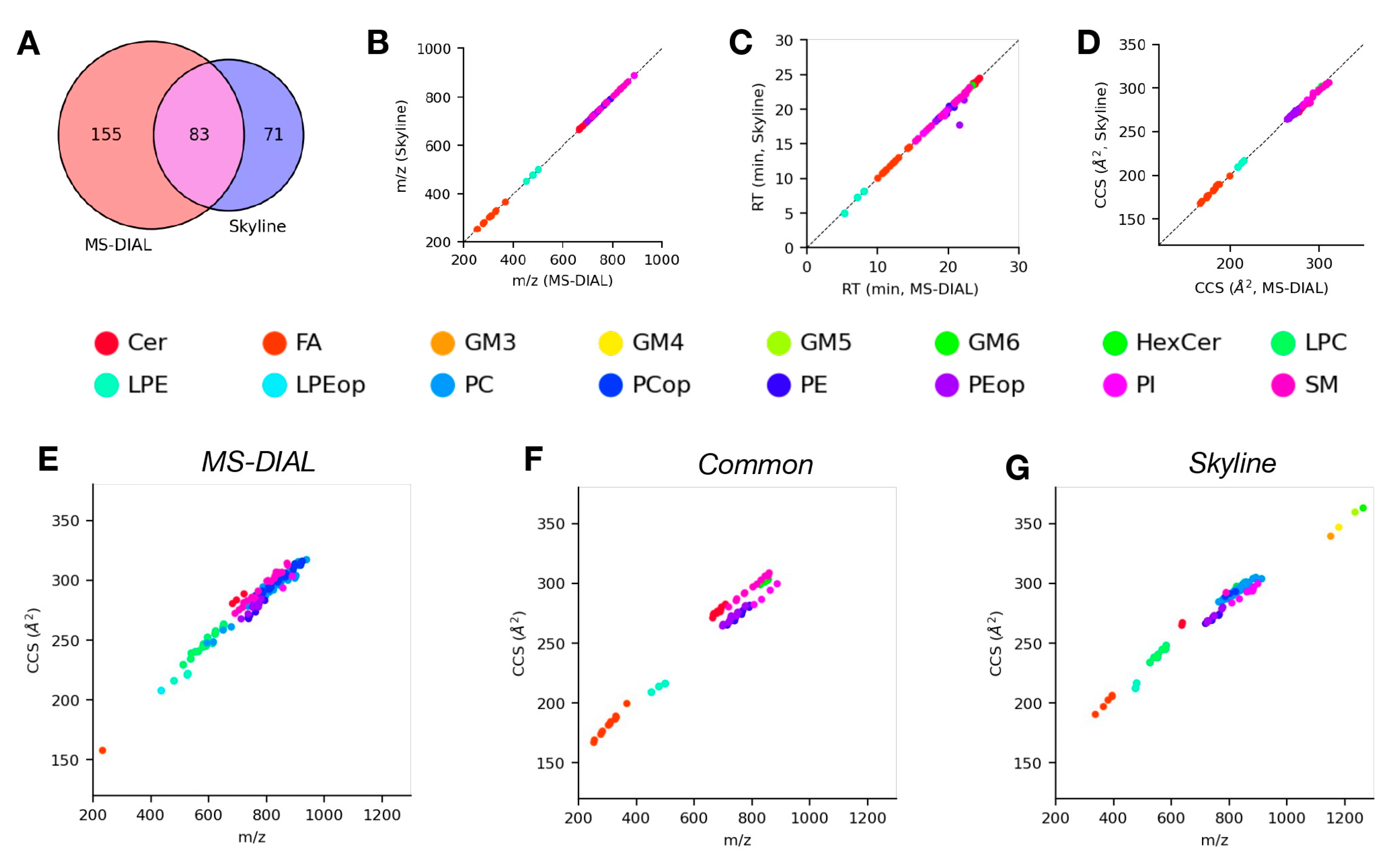
4.4. Analysis of LC-TIMS-MS/MS Data Using Skyline and MS-DIAL
5. Discussion and Future Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harayama, T.; Riezman, H. Understanding the diversity of membrane lipid composition. Nat. Rev. Mol. Cell Biol. 2018, 19, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Meikle, T.G.; Huynh, K.; Giles, C.; Meikle, P.J. Clinical lipidomics: Realizing the potential of lipid profiling. J. Lipid Res. 2021, 62, 100127. [Google Scholar] [CrossRef] [PubMed]
- Butler, L.M.; Perone, Y.; Dehairs, J.; Lupien, L.E.; de Laat, V.; Talebi, A.; Loda, M.; Kinlaw, W.B.; Swinnen, J.V. Lipids and cancer: Emerging roles in pathogenesis, diagnosis and therapeutic intervention. Adv. Drug Deliv. Rev. 2020, 159, 245–293. [Google Scholar] [CrossRef]
- Zheng, X.; Smith, R.D.; Baker, E.S. Recent advances in lipid separations and structural elucidation using mass spectrometry combined with ion mobility spectrometry, ion-molecule reactions and fragmentation approaches. Curr. Opin. Chem. Biol. 2018, 42, 111–118. [Google Scholar] [CrossRef]
- Revercomb, H.E.; Mason, E.A. Theory of plasma chromatography/gaseous electrophoresis. Review. Anal. Chem. 1975, 47, 970–983. [Google Scholar] [CrossRef]
- May, J.C.; McLean, J.A. Ion Mobility-Mass Spectrometry: Time-Dispersive Instrumentation. Anal. Chem. 2015, 87, 1422–1436. [Google Scholar] [CrossRef] [PubMed]
- Dodds, J.N.; Baker, E.S. Ion Mobility Spectrometry: Fundamental Concepts, Instrumentation, Applications, and the Road Ahead. J. Am. Soc. Mass Spectrom. 2019, 30, 2185–2195. [Google Scholar] [CrossRef]
- Shvartsburg, A.A.; Isaac, G.; Leveque, N.; Smith, R.D.; Metz, T.O. Separation and Classification of Lipids Using Differential Ion Mobility Spectrometry. J. Am. Soc. Mass Spectrom. 2011, 22, 1146. [Google Scholar] [CrossRef]
- Kliman, M.; May, J.C.; McLean, J.A. Lipid analysis and lipidomics by structurally selective ion mobility-mass spectrometry. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2011, 1811, 935–945. [Google Scholar] [CrossRef]
- Castro-Perez, J.; Roddy, T.P.; Nibbering, N.M.; Shah, V.; McLaren, D.G.; Previs, S.; Attygalle, A.B.; Herath, K.; Chen, Z.; Wang, S.P.; et al. Localization of fatty acyl and double bond positions in phosphatidylcholines using a dual stage CID fragmentation coupled with ion mobility mass spectrometry. J. Am. Soc. Mass Spectrom. 2011, 22, 1552–1567. [Google Scholar] [CrossRef]
- Lintonen, T.P.I.; Baker, P.R.S.; Suoniemi, M.; Ubhi, B.K.; Koistinen, K.M.; Duchoslav, E.; Campbell, J.L.; Ekroos, K. Differential Mobility Spectrometry-Driven Shotgun Lipidomics. Anal. Chem. 2014, 86, 9662–9669. [Google Scholar] [CrossRef] [PubMed]
- Groessl, M.; Graf, S.; Knochenmuss, R. High resolution ion mobility-mass spectrometry for separation and identification of isomeric lipids. Analyst 2015, 140, 6904–6911. [Google Scholar] [CrossRef] [PubMed]
- Kyle, J.E.; Zhang, X.; Weitz, K.K.; Monroe, M.E.; Ibrahim, Y.M.; Moore, R.J.; Cha, J.; Sun, X.; Lovelace, E.S.; Wagoner, J.; et al. Uncovering biologically significant lipid isomers with liquid chromatography, ion mobility spectrometry and mass spectrometry. Analyst 2016, 141, 1649–1659. [Google Scholar] [CrossRef] [PubMed]
- Bowman, A.P.; Abzalimov, R.R.; Shvartsburg, A.A. Broad Separation of Isomeric Lipids by High-Resolution Differential Ion Mobility Spectrometry with Tandem Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2017, 28, 1552–1561. [Google Scholar] [CrossRef] [PubMed]
- Vasilopoulou, C.G.; Sulek, K.; Brunner, A.D.; Meitei, N.S.; Schweiger-Hufnagel, U.; Meyer, S.W.; Barsch, A.; Mann, M.; Meier, F. Trapped ion mobility spectrometry and PASEF enable in-depth lipidomics from minimal sample amounts. Nat. Commun. 2020, 11, 331. [Google Scholar] [CrossRef]
- Valentine, S.J.; Kulchania, M.; Barnes, C.A.; Clemmer, D.E. Multidimensional separations of complex peptide mixtures: A combined high-performance liquid chromatography/ion mobility/time-of-flight mass spectrometry approach. Int. J. Mass Spectrom. 2001, 212, 97–109. [Google Scholar] [CrossRef]
- Zheng, X.; Wojcik, R.; Zhang, X.; Ibrahim, Y.M.; Burnum-Johnson, K.E.; Orton, D.J.; Monroe, M.E.; Moore, R.J.; Smith, R.D.; Baker, E.S. Coupling Front-End Separations, Ion Mobility Spectrometry, and Mass Spectrometry For Enhanced Multidimensional Biological and Environmental Analyses. Annu. Rev. Anal. Chem. 2017, 10, 71–92. [Google Scholar] [CrossRef]
- Burnum-Johnson, K.E.; Zheng, X.; Dodds, J.N.; Ash, J.; Fourches, D.; Nicora, C.D.; Wendler, J.P.; Metz, T.O.; Waters, K.M.; Jansson, J.K.; et al. Ion mobility spectrometry and the omics: Distinguishing isomers, molecular classes and contaminant ions in complex samples. TrAC Trends Anal. Chem. 2019, 116, 292–299. [Google Scholar] [CrossRef]
- Ibrahim, Y.M.; Baker, E.S.; Danielson, W.F.; Norheim, R.V.; Prior, D.C.; Anderson, G.A.; Belov, M.E.; Smith, R.D. Development of a new ion mobility time-of-flight mass spectrometer. Int. J. Mass Spectrom. 2015, 377, 655–662. [Google Scholar] [CrossRef]
- Pringle, S.D.; Giles, K.; Wildgoose, J.L.; Williams, J.P.; Slade, S.E.; Thalassinos, K.; Bateman, R.H.; Bowers, M.T.; Scrivens, J.H. An investigation of the mobility separation of some peptide and protein ions using a new hybrid quadrupole/travelling wave IMS/oa-ToF instrument. Int. J. Mass Spectrom. 2007, 261, 1–12. [Google Scholar] [CrossRef]
- Moran-Garrido, M.; Camunas-Alberca, S.M.; Gil-de-la Fuente, A.; Mariscal, A.; Gradillas, A.; Barbas, C.; Sáiz, J. Recent developments in data acquisition, treatment and analysis with ion mobility-mass spectrometry for lipidomics. Proteomics 2022, 22, 2100328. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, K.I.; Pratt, B.S.; Shulman, N.; Tamura, K.; MacCoss, M.J.; MacLean, B.X.; Baker, E.S. Utilizing Skyline to analyze lipidomics data containing liquid chromatography, ion mobility spectrometry and mass spectrometry dimensions. Nat. Protoc. 2022, 17, 2415–2430. [Google Scholar] [CrossRef] [PubMed]
- Tsugawa, H.; Ikeda, K.; Takahashi, M.; Satoh, A.; Mori, Y.; Uchino, H.; Okahashi, N.; Yamada, Y.; Tada, I.; Bonini, P.; et al. A lipidome atlas in MS-DIAL 4. Nat. Biotechnol. 2020, 38, 1159–1163. [Google Scholar] [CrossRef] [PubMed]
- Ross, D.H.; Cho, J.H.; Zhang, R.; Hines, K.M.; Xu, L. LiPydomics: A Python Package for Comprehensive Prediction of Lipid Collision Cross Sections and Retention Times and Analysis of Ion Mobility-Mass Spectrometry-Based Lipidomics Data. Anal. Chem. 2020, 92, 14967–14975. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Shen, X.; Chen, X.; Tu, J.; Xiong, X.; Zhu, Z.J. LipidIMMS Analyzer: Integrating multi-dimensional information to support lipid identification in ion mobility-mass spectrometry based lipidomics. Bioinformatics 2019, 35, 698–700. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Luo, M.; Chen, X.; Yin, Y.; Xiong, X.; Wang, R.; Zhu, Z.-J. Ion mobility collision cross-section atlas for known and unknown metabolite annotation in untargeted metabolomics. Nat. Commun. 2020, 11, 4334. [Google Scholar] [CrossRef]
- Schmid, R.; Heuckeroth, S.; Korf, A.; Smirnov, A.; Myers, O.; Dyrlund, T.S.; Bushuiev, R.; Murray, K.J.; Hoffmann, N.; Lu, M.; et al. Integrative analysis of multimodal mass spectrometry data in MZmine 3. Nat. Biotechnol. 2023. [Google Scholar] [CrossRef]
- Colby, S.M.; Chang, C.H.; Bade, J.L.; Nunez, J.R.; Blumer, M.R.; Orton, D.J.; Bloodsworth, K.J.; Nakayasu, E.S.; Smith, R.D.; Ibrahim, Y.M.; et al. DEIMoS: An Open-Source Tool for Processing High-Dimensional Mass Spectrometry Data. Anal. Chem. 2022, 94, 6130–6138. [Google Scholar] [CrossRef]
- Bilbao, A.; Gibbons, B.C.; Stow, S.M.; Kyle, J.E.; Bloodsworth, K.J.; Payne, S.H.; Smith, R.D.; Ibrahim, Y.M.; Baker, E.S.; Fjeldsted, J.C. A Preprocessing Tool for Enhanced Ion Mobility-Mass Spectrometry-Based Omics Workflows. J. Proteome Res. 2022, 21, 798–807. [Google Scholar] [CrossRef]
- Koelmel, J.P.; Li, X.; Stow, S.M.; Sartain, M.J.; Murali, A.; Kemperman, R.; Tsugawa, H.; Takahashi, M.; Vasiliou, V.; Bowden, J.A.; et al. Lipid Annotator: Towards Accurate Annotation in Non-Targeted Liquid Chromatography High-Resolution Tandem Mass Spectrometry (LC-HRMS/MS) Lipidomics Using a Rapid and User-Friendly Software. Metabolites 2020, 10, 101. [Google Scholar] [CrossRef]
- Goracci, L.; Tortorella, S.; Tiberi, P.; Pellegrino, R.M.; Di Veroli, A.; Valeri, A.; Cruciani, G. Lipostar, a Comprehensive Platform-Neutral Cheminformatics Tool for Lipidomics. Anal. Chem. 2017, 89, 6258–6265. [Google Scholar] [CrossRef] [PubMed]
- Quehenberger, O.; Armando, A.M.; Brown, A.H.; Milne, S.B.; Myers, D.S.; Merrill, A.H.; Bandyopadhyay, S.; Jones, K.N.; Kelly, S.; Shaner, R.L.; et al. Lipidomics reveals a remarkable diversity of lipids in human plasma. J. Lipid Res. 2010, 51, 3299–3305. [Google Scholar] [CrossRef]
- Bowden, J.A.; Heckert, A.; Ulmer, C.Z.; Jones, C.M.; Koelmel, J.P.; Abdullah, L.; Ahonen, L.; Alnouti, Y.; Armando, A.M.; Asara, J.M.; et al. Harmonizing lipidomics: NIST interlaboratory comparison exercise for lipidomics using SRM 1950-Metabolites in Frozen Human Plasma. J. Lipid Res. 2017, 58, 2275–2288. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, K.I.; Christopher, M.W.; Burgess, J.L.; Littau, S.R.; Foster, K.; Richey, K.; Pratt, B.S.; Shulman, N.; Tamura, K.; MacCoss, M.J.; et al. Development and Application of Multidimensional Lipid Libraries to Investigate Lipidomic Dysregulation Related to Smoke Inhalation Injury Severity. J. Proteome Res. 2022, 21, 232–242. [Google Scholar] [CrossRef]
- Hoffmann, N.; Mayer, G.; Has, C.; Kopczynski, D.; Al Machot, F.; Schwudke, D.; Ahrends, R.; Marcus, K.; Eisenacher, M.; Turewicz, M. A Current Encyclopedia of Bioinformatics Tools, Data Formats and Resources for Mass Spectrometry Lipidomics. Metabolites 2022, 12, 584. [Google Scholar] [CrossRef]
- Harris, R.A.; Leaptrot, K.L.; May, J.C.; McLean, J.A. New frontiers in lipidomics analyses using structurally selective ion mobility-mass spectrometry. TrAC Trends Anal. Chem. 2019, 116, 316–323. [Google Scholar] [CrossRef]
- Köfeler, H.C.; Eichmann, T.O.; Ahrends, R.; Bowden, J.A.; Danne-Rasche, N.; Dennis, E.A.; Fedorova, M.; Griffiths, W.J.; Han, X.; Hartler, J.; et al. Quality control requirements for the correct annotation of lipidomics data. Nat. Commun. 2021, 12, 4771. [Google Scholar] [CrossRef] [PubMed]
- Vasilopoulou, C.G.; Sulek, K.; Brunner, A.-D.; Meitei, N.S.; Schweiger-Hufnagel, U.; Meyer, S.W.; Barsch, A.; Mann, M.; Meier, F. Reply to Quality control requirements for the correct annotation of lipidomics data. Nat. Commun. 2021, 12, 4772. [Google Scholar] [CrossRef]
- Köfeler, H.C.; Ahrends, R.; Baker, E.S.; Ekroos, K.; Han, X.; Hoffmann, N.; Holčapek, M.; Wenk, M.R.; Liebisch, G. Recommendations for good practice in MS-based lipidomics. J. Lipid Res. 2021, 62, 100138. [Google Scholar] [CrossRef]
- Ma, X.X.; Xia, Y. Pinpointing Double Bonds in Lipids by Paterno-Buchi Reactions and Mass Spectrometry. Angew. Chem. Int. Ed. 2014, 53, 2592–2596. [Google Scholar] [CrossRef]
- Zhang, D.; Lin, Q.; Xia, T.; Zhao, J.; Zhang, W.; Ouyang, Z.; Xia, Y. LipidOA: A Machine-Learning and Prior-Knowledge-Based Tool for Structural Annotation of Glycerophospholipids. Anal. Chem. 2022, 94, 16759–16767. [Google Scholar] [CrossRef] [PubMed]
- Poad, B.L.J.; Zheng, X.; Mitchell, T.W.; Smith, R.D.; Baker, E.S.; Blanksby, S.J. Online Ozonolysis Combined with Ion Mobility-Mass Spectrometry Provides a New Platform for Lipid Isomer Analyses. Anal. Chem. 2018, 90, 1292–1300. [Google Scholar] [CrossRef] [PubMed]
- Ross, D.H.; Lee, J.-Y.; Bilbao, A.; Orton, D.J.; Eder, J.G.; Burnet, M.C.; Deatherage Kaiser, B.L.; Kyle, J.E.; Zheng, X. LipidOz enables automated elucidation of lipid carbon–carbon double bond positions from ozone-induced dissociation mass spectrometry data. Commun. Chem. 2023, accepted. Available online: https://github.com/PNNL-m-q/lipidoz (accessed on 13 March 2023).
- Wojcik, R.; Webb, I.K.; Deng, L.; Garimella, S.V.B.; Prost, S.A.; Ibrahim, Y.M.; Baker, E.S.; Smith, R.D. Lipid and Glycolipid Isomer Analyses Using Ultra-High Resolution Ion Mobility Spectrometry Separations. Int. J. Mol. Sci. 2017, 18, 183. [Google Scholar] [CrossRef] [PubMed]
- Rose, B.S.; May, J.C.; Reardon, A.R.; McLean, J.A. Collision Cross-Section Calibration Strategy for Lipid Measurements in SLIM-Based High-Resolution Ion Mobility. J. Am. Soc. Mass Spectr. 2022, 33, 1229–1237. [Google Scholar] [CrossRef] [PubMed]
- Ellis, S.R.; Michael, J.A.; Mutuku, S.M.; Ucur, B.; Sarretto, T.; Maccarone, A.T.; Niehaus, M.; Trevitt, A.J.; Ellis, S.R. Mass Spectrometry Imaging of Lipids Using MALDI Coupled with Plasma-Based Post-Ionization on a Trapped Ion Mobility Mass Spectrometer. Anal. Chem. 2022, 94, 17494–17503. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Software | Supported File Formats | Workflow | Acquisition Modes | Annotation Method | Ref. |
|---|---|---|---|---|---|
 | Input: raw files (Agilent, Bruker, Sciex, Waters, Thermo and Shimadzu), mzML, mz5, mzXML Output: csv | Targeted | DDA, DIA | MS2 fragmentation, iRT calibrated RT, CCS experimental library containing 516 unique lipids | [22] |
 MS-DIAL | Input: raw files (Agilent, Waters and Bruker), must be converted to IBF Output: mztab-M | Untargeted | DDA, DIA | MS2 fragmentation, RT, CCS experimental and predicted library containing 581,047 unique lipids | [23] |
| LiPydomics | Input: csv (feature table) Output: png, xlsx | Untargeted | DDA, DIA | HILIC RT, CCS experimental and predicted library containing 145,388 unique lipids | [24] |
| LipidIMMS (Lipid4DAnalyzer) | Input: supports Agilent, Bruker, Waters, Sciex MS1 peak table (.csv format), MS2 data files (.mgf/.msp /.cef format), RT calibration table (.csv format, optional) Output: html, pdf, csv | Untargeted | DDA, DIA | MS2 fragmentation, RT, CCS experimental and predicted library containing 267,716 unique lipids | [25] |
 | Input: csv Output: csv | Untargeted | DDA, DIA | Experimental and predicted CCS library | [26] |
 | Input: tdf, tsf (Bruker), mzML Output: csv | Untargeted | MS1 | User-supplied RT and CCS library | [27] |
| DEIMoS | Input: mzML Output: csv, mgf, mzML | Untargeted | DDA, DIA | NA | [28] |
| FA | DTIM (+) | DTIM (−) | TIMS (+) | TIMS (−) | |||||
|---|---|---|---|---|---|---|---|---|---|
| sum | |||||||||
| Skyline | 149 | 212 | 9 | 32 | |||||
| 68 | 11 | 92 | 122 | ||||||
| MS-DIAL | 86 | 36 | 111 | 175 | |||||
| 137 | 13 | 91 | 63 | ||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ross, D.H.; Guo, J.; Bilbao, A.; Huan, T.; Smith, R.D.; Zheng, X. Evaluating Software Tools for Lipid Identification from Ion Mobility Spectrometry–Mass Spectrometry Lipidomics Data. Molecules 2023, 28, 3483. https://doi.org/10.3390/molecules28083483
Ross DH, Guo J, Bilbao A, Huan T, Smith RD, Zheng X. Evaluating Software Tools for Lipid Identification from Ion Mobility Spectrometry–Mass Spectrometry Lipidomics Data. Molecules. 2023; 28(8):3483. https://doi.org/10.3390/molecules28083483
Chicago/Turabian StyleRoss, Dylan H., Jian Guo, Aivett Bilbao, Tao Huan, Richard D. Smith, and Xueyun Zheng. 2023. "Evaluating Software Tools for Lipid Identification from Ion Mobility Spectrometry–Mass Spectrometry Lipidomics Data" Molecules 28, no. 8: 3483. https://doi.org/10.3390/molecules28083483
APA StyleRoss, D. H., Guo, J., Bilbao, A., Huan, T., Smith, R. D., & Zheng, X. (2023). Evaluating Software Tools for Lipid Identification from Ion Mobility Spectrometry–Mass Spectrometry Lipidomics Data. Molecules, 28(8), 3483. https://doi.org/10.3390/molecules28083483