
Rhodium-Catalyzed Trans-Bis-Silylation Reactions of 2-Ethynyl-3-pentamethyldisilanylpyridines


Abstract
1. Introduction
2. Results and Discussion
2.1. Synthesis and Reactions
2.2. Theoretical Study
3. Conclusions
4. Materials and Methods
4.1. General Procedure
4.2. Procedures
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Hiyama, T.; Oestreich, M. Organosilicon Chemistry: Novel Approaches and Reactions; Wiley-VCH Press: Weinheim, Germany, 2019. [Google Scholar]
- Yamaguchi, S.; Tamao, K. The Chemistry of Organic Silicon Compounds; Chapter 1; Rappoport, Z., Apeloig, Y., Eds.; Wiley: Chichester, UK, 2001; Volume 3. [Google Scholar]
- Liu, J.; Lam, J.W.Y.; Tang, B.Z. Aggregation-Induced Emission of Silole Molecules and Polymers: Fundamental and Applications. J. Inorg. Organomet. Polym. Mater. 2009, 19, 249–285. [Google Scholar] [CrossRef]
- Sołoducho, J.; Zając, D.; Spychalska, K.; Baluta, S.; Cabaj, J. Conducting Silicone-Based Polymers and Their Application. Molecules 2021, 26, 2012. [Google Scholar] [CrossRef] [PubMed]
- Dang, T.T.; Nguyen, H.M.T.; Nguyen, H.; Dung, T.N.; Nguyen, M.T.; Dehaen, W. Advances in Synthesis of π-Extended Benzosilole Derivatives and Their Analogs. Molecules 2020, 25, 548. [Google Scholar] [CrossRef]
- Chen, J.W.; Cao, Y. Silole-Containing Polymers: Chemistry and Optoelectronic Properties. Macromol. Rapid Commun. 2007, 28, 1714–1742. [Google Scholar] [CrossRef]
- Lu, G.; Usta, H.; Risko, C.; Wang, L.; Facchetti, A.; Ratner, M.A.; Marks, T.J. Synthesis, Characterization, and Transistor Response of Semiconducting Silole Polymers with Substantial Hole Mobility and Air Stability. Experiment and Theory. J. Am. Chem. Soc. 2008, 130, 7670–7685. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X.C.; Ju, C.W.; Zhao, D. Bis-Silylation of Internal Alkynes Enabled by Ni(0) Catalysis. Nat. Commun. 2021, 12, 68. [Google Scholar] [CrossRef]
- Suginome, M.; Ito, Y. Activation of Silicon–Silicon σ Bonds by Transition-Metal Complexes: Synthesis and Catalysis of New Organosilyl Transition-Metal Complexes. J. Chem. Soc. Dalton Trans. 1998, 1925–1934. [Google Scholar] [CrossRef]
- Suginome, M.; Ito, Y. Activation of Si–Si Bonds by Transition-Metal Complexes. Organomet. Chem. 1999, 3, 131–159. [Google Scholar]
- Beletskaya, I.; Moberg, C. Element-Element Addition to Alkynes Catalyzed by the Group 10 Metals. Chem. Rev. 1999, 99, 3435–3462. [Google Scholar] [CrossRef]
- Suginome, M.; Ito, Y. Transition-Metal-Catalyzed Additions of Silicon–Silicon and Silicon-Heteroatom Bonds to Unsaturated Organic Molecules. Chem. Rev. 2000, 100, 3221–3256. [Google Scholar] [CrossRef]
- Beletskaya, I.; Moberg, C. Element-Element Additions to Unsaturated Carbon–Carbon Bonds Catalyzed by Transition Metal Complexes. Chem. Rev. 2006, 106, 2320–2354. [Google Scholar] [CrossRef]
- Suginome, M.; Matsuda, T.; Ohmura, T.; Seki, A.; Murakami, M. Comprehensive Organometallic Chemistry, 10th ed.; Crabtree, R.H., Mingos, D.M.P., Eds.; Elsevier: London, UK, 2007; Volume III, pp. 725–787. [Google Scholar]
- Ansell, M.B.; Navarro, O.; Spencer, J. Transition Metal Catalyzed Element–Element’ Additions to Alkynes. Coord. Chem. Rev. 2017, 336, 54–77. [Google Scholar] [CrossRef]
- Ozawa, F.; Sugawara, M.; Hayashi, T. A New Reactive System for Catalytic Bis-Silylation of Acetylenes and Olefins. Organometallics 1994, 13, 3237–3243. [Google Scholar] [CrossRef]
- Ansell, M.B.; Roberts, D.E.; Cloke, F.G.N.; Navarro, O.; Spencer, J. Synthesis of an [(NHC)2Pd(SiMe3)2] Complex and Catalytic cis-Bis(Silyl)ations of Alkynes with Unactivated Disilanes. Angew. Chem. Int. Ed. 2015, 54, 5578–5582. [Google Scholar] [CrossRef]
- Matsuda, T.; Ichioka, Y. Rhodium-Catalysed Intramolecular Trans-Bis-Silylation of Alkynes to Synthesise 3-Silyl-1-Benzosiloles. Org. Biomol. Chem. 2012, 10, 3175–3177. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Matsuura, T.; Murakami, M. Palladium-catalyzed regular insertion of isonitriles into silicon-silicon linkage of polysilane. J. Am. Chem. Soc. 1988, 110, 3692–3693. [Google Scholar] [CrossRef]
- Hayashi, T.; Kobayashi, T.; Kawamoto, A.; Yamashita, H.; Tanaka, M. Platinum complex catalyzed double silylation of ethylene and norbornene with disilanes. Organometallics 1990, 9, 280–281. [Google Scholar] [CrossRef]
- Tamao, K.; Hayashi, T.; Kumada, M. Fluorinated polysilanes. palladium-catalyzed disilane metathesis, double silylation of acetylenes, and the stereo chemical course. J. Organomet. Chem. 1976, 114, C19–C22. [Google Scholar] [CrossRef]
- Sakurai, H.; Kamiyama, Y.; Nakadaira, Y. Chemistry of organosilicon compounds. 79. Novel [s + p] reactions of hexaorganodisilanes with acetylenes catalyzed by palladium complexes. J. Am. Chem. Soc. 1975, 97, 931–932. [Google Scholar] [CrossRef]
- Watanabe, H.; Kobayashi, M.; Higuchi, K.; Nagai, Y. Reaction of disilanes with acetylenes: I. Stereoselective addition of methoxymethyldisilanes to phenylacetylene catalyzed by group-VIII metal phosphine complexes. J. Organomet. Chem. 1980, 186, 51–62. [Google Scholar] [CrossRef]
- Naka, A.; Shimomura, N.; Kobayashi, H. Synthesis of Pyridine-Fused Siloles by Palladium-Catalyzed Intramolecular Bis-Silylation. ACS Omega 2022, 7, 30369–30375. [Google Scholar] [CrossRef] [PubMed]
- Kishbaugh, T.L.S. Six-membered ring systems: Pyridine and benzo derivatives. Prog. Heterocycl. Chem. 2012, 24, 343–391. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennuci, B.; Petersson, G.A.; et al. Gaussian09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for the Transition Metal Atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Dunning, T.H., Jr.; Hay, P.J. Modern Theoretical Chemistry; Schaefer, H.F., III, Ed.; Plenum Press: New York, NY, USA, 1976; pp. 1–28. [Google Scholar]
- Fukui, K.; Kato, S.; Fujimoto, H. Constituent analysis of the potential gradient along a reaction coordinate. Method and an application to CH4 + T reaction. J. Am. Chem. Soc. 1975, 97, 1–7. [Google Scholar]







{kind=link}
{kind=link}
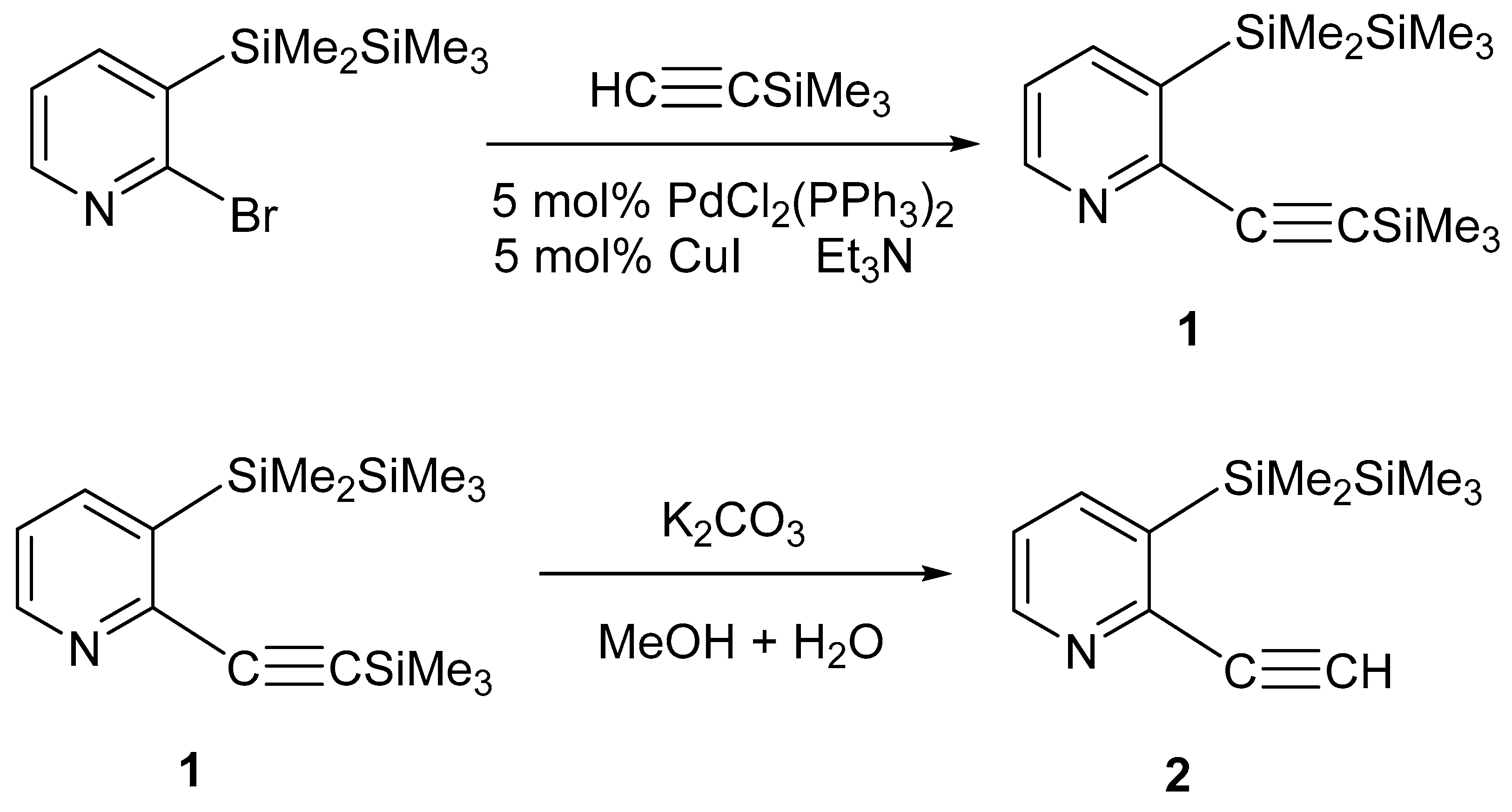{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Starting | Isolated Yield with | ||
|---|---|---|---|
| Compound | [RhCl(CO)2]2 | [RhCl(nbd)]2 | [RhCl(PPh)3]3 |
| 1 | 46% | 6% | 5% |
| 2 | 48% | 5% | 0% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naka, A.; Kobayashi, H. Rhodium-Catalyzed Trans-Bis-Silylation Reactions of 2-Ethynyl-3-pentamethyldisilanylpyridines. Molecules 2023, 28, 3284. https://doi.org/10.3390/molecules28083284
Naka A, Kobayashi H. Rhodium-Catalyzed Trans-Bis-Silylation Reactions of 2-Ethynyl-3-pentamethyldisilanylpyridines. Molecules. 2023; 28(8):3284. https://doi.org/10.3390/molecules28083284
Chicago/Turabian StyleNaka, Akinobu, and Hisayoshi Kobayashi. 2023. "Rhodium-Catalyzed Trans-Bis-Silylation Reactions of 2-Ethynyl-3-pentamethyldisilanylpyridines" Molecules 28, no. 8: 3284. https://doi.org/10.3390/molecules28083284
APA StyleNaka, A., & Kobayashi, H. (2023). Rhodium-Catalyzed Trans-Bis-Silylation Reactions of 2-Ethynyl-3-pentamethyldisilanylpyridines. Molecules, 28(8), 3284. https://doi.org/10.3390/molecules28083284