

Discovery of Novel EGFR Inhibitor Targeting Wild-Type and Mutant Forms of EGFR: In Silico and In Vitro Study

 , , ,
, , ,  , ,
, ,  , and
, and
Abstract
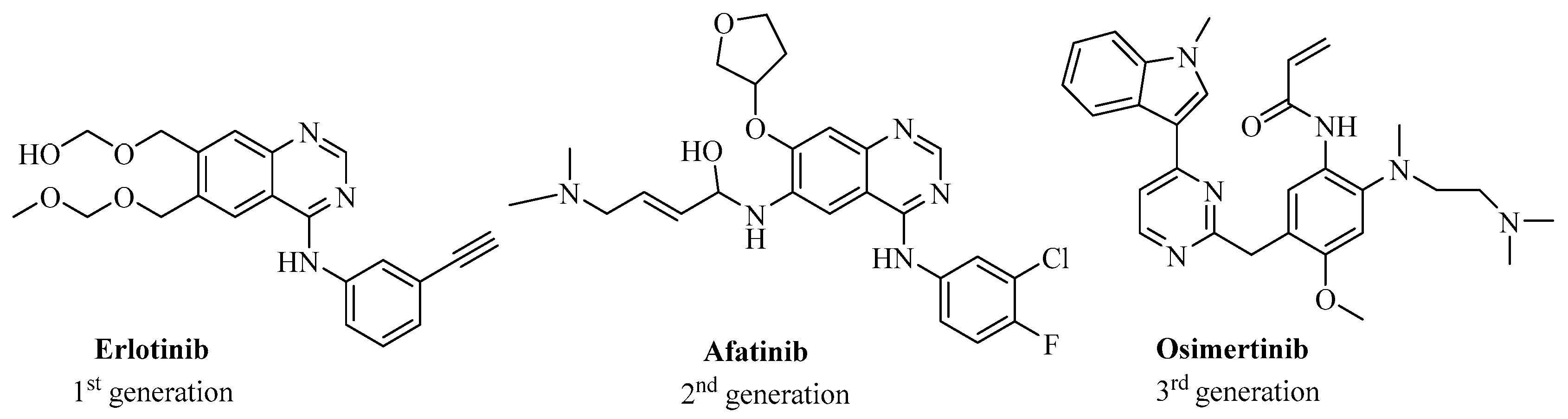
1. Introduction
2. Results
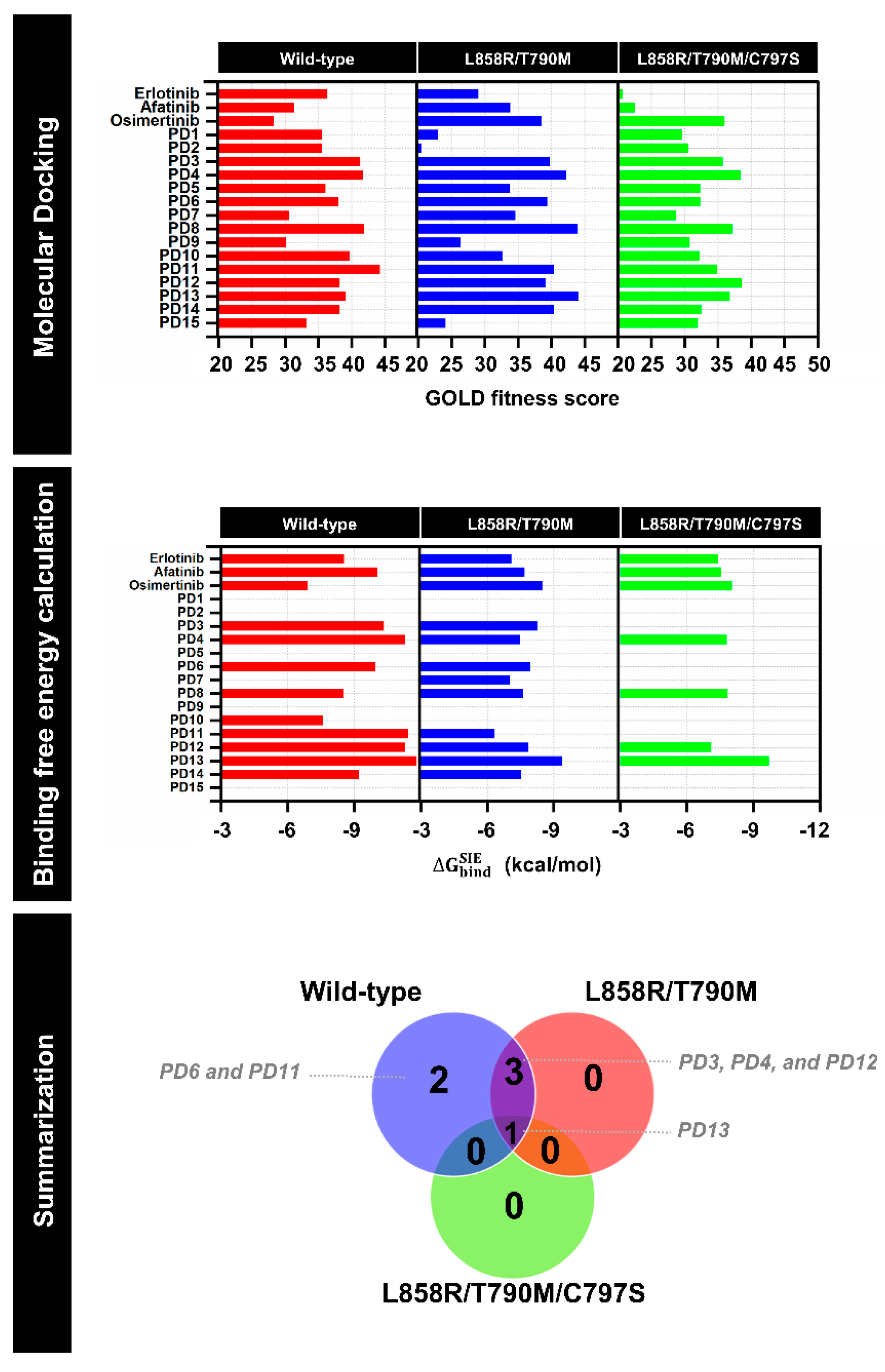
2.1. Virtual Screening
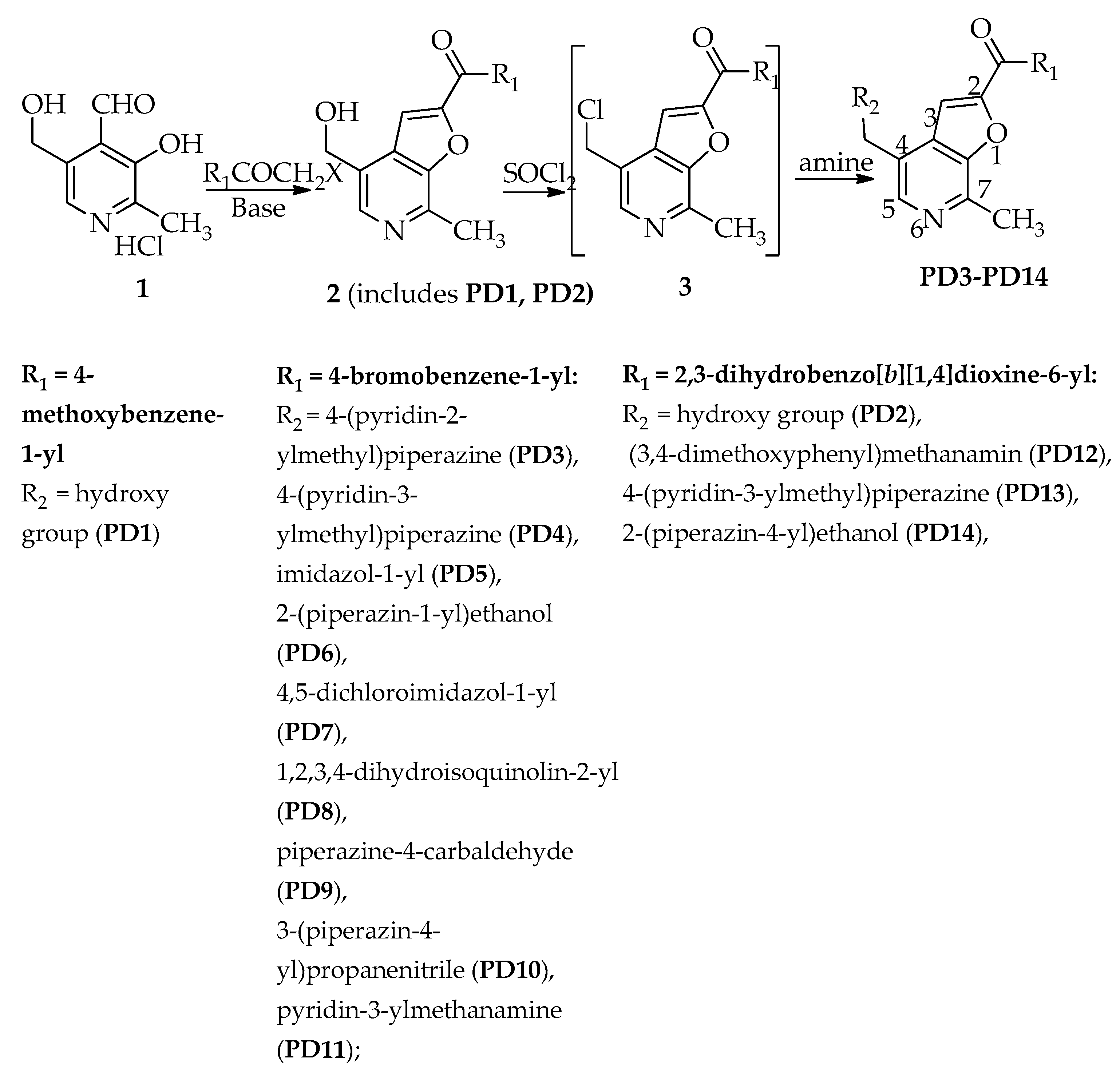
2.2. Chemistry
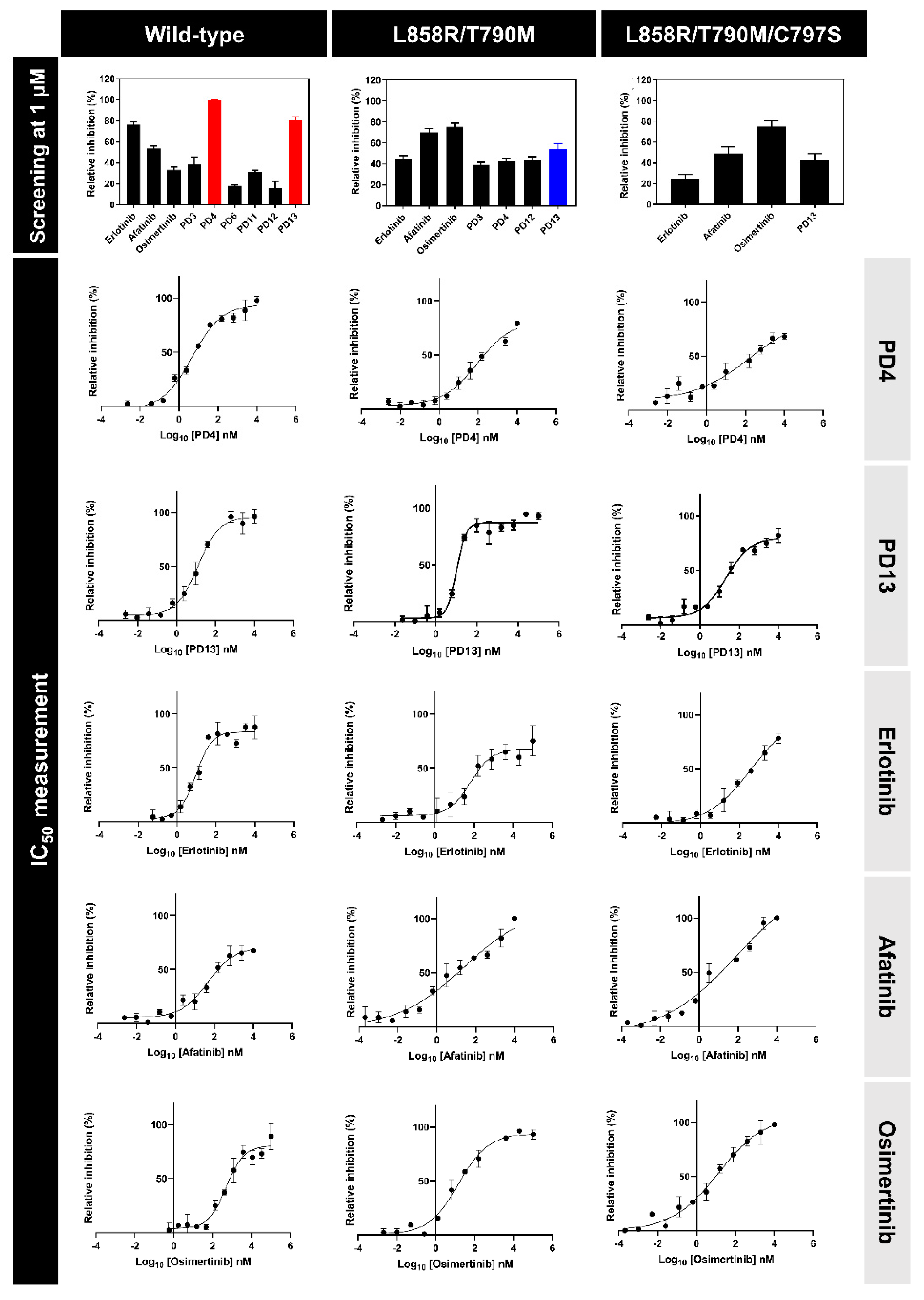
2.3. Kinase Inhibitory Activities of EGFR
2.4. Antiproliferative Activities on NSCLC and Normal Cell Lines
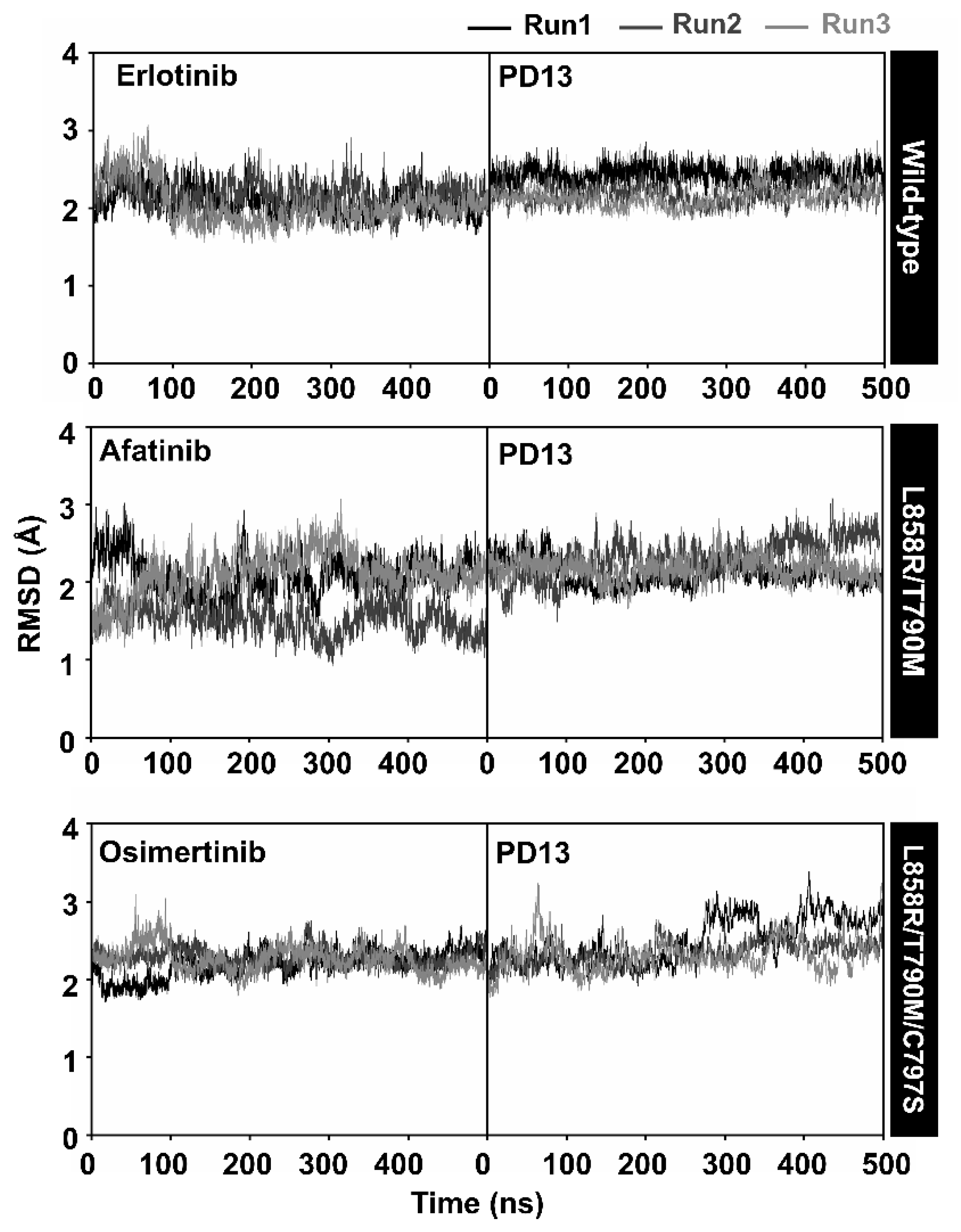
2.5. System Stability
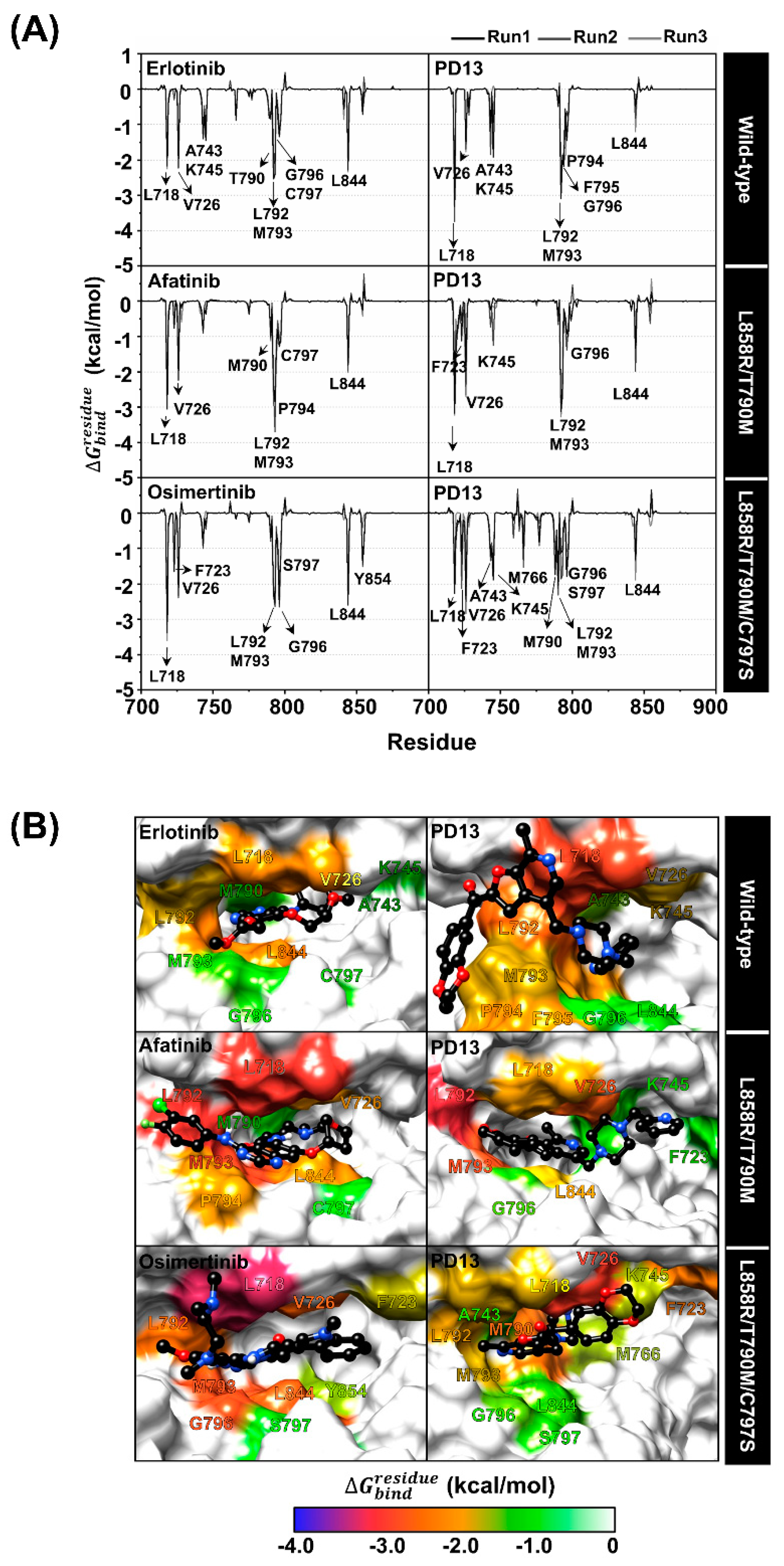
2.6. Key Binding Residues
2.7. Protein-Ligand Hydrogen Bonding
2.8. Binding Affinity
3. Materials and Methods
3.1. General Information
3.2. Chemistry
General Procedure for the Synthesis of 7-Methylfuro[2,3-c]pyridine Derivatives PD3–PD14
3.3. Computational Studies
3.3.1. System Preparation
3.3.2. Molecular Dynamics Simulation and Free Energy Calculation Based on Solvated Interaction Energy (SIE) Method
3.4. Biological Studies
3.4.1. EGFR Inhibitory ACTIVITY
3.4.2. Cell Viability Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Engel, J.; Richters, A.; Getlik, M.; Tomassi, S.; Keul, M.; Termathe, M.; Lategahn, J.; Becker, C.; Mayer-Wrangowski, S.; Grütter, C. Targeting drug resistance in EGFR with covalent inhibitors: A structure-based design approach. J. Med. Chem. 2015, 58, 6844–6863. [Google Scholar] [CrossRef]
- Peters, S.; Zimmermann, S.; Adjei, A.A. Oral epidermal growth factor receptor tyrosine kinase inhibitors for the treatment of non-small cell lung cancer: Comparative pharmacokinetics and drug–drug interactions. Cancer Treat. Rev. 2014, 40, 917–926. [Google Scholar] [CrossRef] [PubMed]
- Oda, K.; Matsuoka, Y.; Funahashi, A.; Kitano, H. A comprehensive pathway map of epidermal growth factor receptor signaling. Mol. Syst. Bbiol. 2005, 1, 2005.0010. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef]
- Chen, P.; Xie, H.; Sekar, M.C.; Gupta, K.; Wells, A. Epidermal growth factor receptor-mediated cell motility: Phospholipase C activity is required, but mitogen-activated protein kinase activity is not sufficient for induced cell movement. J. Cell Biol. 1994, 127, 847–857. [Google Scholar] [CrossRef]
- Kharitonenkov, A.; Chen, Z.; Sures, I.; Wang, H.; Schilling, J.; Ullrich, A. A family of proteins that inhibit signalling through tyrosine kinase receptors. Nature 1997, 386, 181–186. [Google Scholar] [CrossRef]
- Sternberg, M.J.; Gullick, W.J. A sequence motif in the transmembrane region of growth factor receptors with tyrosine kinase activity mediates dimerization. PEDS 1990, 3, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H. Osimertinib making a breakthrough in lung cancer targeted therapy. Onco Targets Ther. 2016, 9, 5489. [Google Scholar] [CrossRef] [PubMed]
- Bethune, G.; Bethune, D.; Ridgway, N.; Xu, Z. Epidermal growth factor receptor (EGFR) in lung cancer: An overview and update. J. Thorac. Dis. 2010, 2, 48–51. [Google Scholar] [PubMed]
- Li, X.; Fan, X.-X.; Jiang, Z.-B.; Loo, W.T.; Yao, X.-J.; Leung, E.L.-H.; Chow, L.W.; Liu, L. Shikonin inhibits gefitinib-resistant non-small cell lung cancer by inhibiting TrxR and activating the EGFR proteasomal degradation pathway. Pharmacol. Res. 2017, 115, 45–55. [Google Scholar] [CrossRef]
- Normanno, N.; De Luca, A.; Bianco, C.; Strizzi, L.; Mancino, M.; Maiello, M.R.; Carotenuto, A.; De Feo, G.; Caponigro, F.; Salomon, D.S. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 2006, 366, 2–16. [Google Scholar] [CrossRef]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Perez, E.A. The role of adjuvant monoclonal antibody therapy for breast cancer: Rationale and new studies. Curr. Oncol. Rep. 2001, 3, 516–522. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, S.; MacLennan, G.T.; Eble, J.N.; Lopez-Beltran, A.; Yang, X.J.; Pan, C.-X.; Zhou, H.; Montironi, R.; Cheng, L. Epidermal growth factor receptor protein expression and gene amplification in small cell carcinoma of the urinary bladder. Clin. Cancer Res. 2007, 13, 953–957. [Google Scholar] [CrossRef]
- Shia, J.; Klimstra, D.S.; Li, A.R.; Qin, J.; Saltz, L.; Teruya-Feldstein, J.; Akram, M.; Chung, K.Y.; Yao, D.; Paty, P.B. Epidermal growth factor receptor expression and gene amplification in colorectal carcinoma: An immunohistochemical and chromogenic in situ hybridization study. Mod. Pathol. 2005, 18, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, F.A.; Rodrigues Pereira, J.; Ciuleanu, T.; Tan, E.H.; Hirsh, V.; Thongprasert, S.; Campos, D.; Maoleekoonpiroj, S.; Smylie, M.; Martins, R. Erlotinib in previously treated non–small-cell lung cancer. NEJM 2005, 353, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2015. CA Cancer J. Clin. 2015, 65, 5–29. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Soler, R.; Chachoua, A.; Hammond, L.A.; Rowinsky, E.K.; Huberman, M.; Karp, D.; Rigas, J.; Clark, G.M.; Santabárbara, P.; Bonomi, P. Determinants of tumor response and survival with erlotinib in patients with non-small-cell lung cancer. J. Clin. Oncol. 2004, 22, 3238–3247. [Google Scholar] [CrossRef]
- Kris, M.G.; Natale, R.B.; Herbst, R.S.; Lynch Jr, T.J.; Prager, D.; Belani, C.P.; Schiller, J.H.; Kelly, K.; Spiridonidis, H.; Sandler, A. Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non–small cell lung cancer: A randomized trial. JAMA 2003, 290, 2149–2158. [Google Scholar] [CrossRef]
- Michalczyk, A.; Klüter, S.; Rode, H.B.; Simard, J.R.; Grütter, C.; Rabiller, M.; Rauh, D.; Fritsche, A.; Elfringhoff, A.S.; Fabian, J. Structural insights into how irreversible inhibitors can overcome drug resistance in EGFR. Bioorg. Med. Chem. 2008, 16, 3482–3488. [Google Scholar] [CrossRef] [PubMed]
- Heuckmann, J.M.; Rauh, D.; Thomas, R.K. Epidermal growth factor receptor (EGFR) signaling and covalent EGFR inhibition in lung cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012, 30, 3417–3420. [Google Scholar] [CrossRef] [PubMed]
- Kalgutkar, A.S.; Dalvie, D.K. Drug discovery for a new generation of covalent drugs. Expert Opin. Drug Discov. 2012, 7, 561–581. [Google Scholar] [CrossRef] [PubMed]
- Copeland, R.A.; Pompliano, D.L.; Meek, T.D. Drug–target residence time and its implications for lead optimization. Nat. Rev. Drug Discov. 2006, 5, 730–739. [Google Scholar] [CrossRef] [PubMed]
- Kwak, E.L.; Sordella, R.; Bell, D.W.; Godin-Heymann, N.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Driscoll, D.R.; Fidias, P.; Lynch, T.J. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc. Natl. Acad. Sci. USA 2005, 102, 7665–7670. [Google Scholar] [CrossRef] [PubMed]
- Cross, D.A.; Ashton, S.E.; Ghiorghiu, S.; Eberlein, C.; Nebhan, C.A.; Spitzler, P.J.; Orme, J.P.; Finlay, M.R.; Ward, R.A.; Mellor, M.J.; et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014, 4, 1046–1061. [Google Scholar] [CrossRef]
- Lelais, G.; Epple, R.; Marsilje, T.H.; Long, Y.O.; McNeill, M.; Chen, B.; Lu, W.; Anumolu, J.; Badiger, S.; Bursulaya, B. Discovery of (R, E)-N-(7-Chloro-1-(1-[4-(dimethylamino) but-2-enoyl] azepan-3-yl)-1 H-benzo [d] imidazol-2-yl)-2-methylisonicotinamide (EGF816), a Novel, Potent, and WT Sparing Covalent Inhibitor of Oncogenic (L858R, ex19del) and Resistant (T790M) EGFR Mutants for the Treatment of EGFR Mutant Non-Small-Cell Lung Cancers. J.Med.Chem. 2016, 59, 6671–6689. [Google Scholar]
- Zhou, W.; Ercan, D.; Chen, L.; Yun, C.H.; Li, D.; Capelletti, M.; Cortot, A.B.; Chirieac, L.; Iacob, R.E.; Padera, R.; et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 2009, 462, 1070–1074. [Google Scholar] [CrossRef]
- Zhou, W.; Ercan, D.; Janne, P.A.; Gray, N.S. Discovery of selective irreversible inhibitors for EGFR-T790M. Bioorg. Med. Chem. Lett. 2011, 21, 638–643. [Google Scholar] [CrossRef]
- Planken, S.; Behenna, D.C.; Nair, S.K.; Johnson, T.O.; Nagata, A.; Almaden, C.; Bailey, S.; Ballard, T.E.; Bernier, L.; Cheng, H.; et al. Discovery of n-((3 r, 4 r)-4-fluoro-1-(6-((3-methoxy-1-methyl-1 h-pyrazol-4-yl) amino)-9-methyl-9 h-purin-2-yl) pyrrolidine-3-yl) acrylamide (pf-06747775) through structure-based drug design: A high affinity irreversible inhibitor targeting oncogenic egfr mutants with selectivity over wild-type egfr. J. Med. Chem. 2017, 60, 3002–3019. [Google Scholar]
- Eberlein, C.A.; Stetson, D.; Markovets, A.A.; Al-Kadhimi, K.J.; Lai, Z.; Fisher, P.R.; Meador, C.B.; Spitzler, P.; Ichihara, E.; Ross, S.J.; et al. Acquired Resistance to the Mutant-Selective EGFR Inhibitor AZD9291 Is Associated with Increased Dependence on RAS Signaling in Preclinical Models. Cancer Res. 2015, 75, 2489–2500. [Google Scholar] [CrossRef] [PubMed]
- Hung, J.M.; Arabshahi, H.J.; Leung, E.; Reynisson, J.; Barker, D. Synthesis and cytotoxicity of thieno [2, 3-b] pyridine and furo [2, 3-b] pyridine derivatives. Eur. J. Med. Chem. 2014, 86, 420–437. [Google Scholar] [CrossRef] [PubMed]
- Garamvölgyi, R.; Dobos, J.; Sipos, A.; Boros, S.; Illyés, E.; Baska, F.; Kékesi, L.; Szabadkai, I.; Szántai-Kis, C.; Kéri, G. Design and synthesis of new imidazo [1, 2-a] pyridine and imidazo [1, 2-a] pyrazine derivatives with antiproliferative activity against melanoma cells. Eur. J. Med. Chem. 2016, 108, 623–643. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.M.; Al-Refai, M.; Azmi, M.N.; Osman, H.; Bakar, M.H.A.; Geyer, A. Synthesis, characterization and cytotoxicity of new nicotinonitriles and their furo [2, 3-b] pyridine derivatives. JICS 2019, 16, 715–722. [Google Scholar] [CrossRef]
- Rai, S.K.; Singh, P.; Khanam, S.; Tewari, A.K. Polymorphic study and anti-inflammatory activity of a 3-cyano-2-pyridone based flexible model. NJC 2016, 40, 5577–5587. [Google Scholar] [CrossRef]
- Dorsey, B.D.; McDonough, C.; McDaniel, S.L.; Levin, R.B.; Newton, C.L.; Hoffman, J.M.; Darke, P.L.; Zugay-Murphy, J.A.; Emini, E.A.; Schleif, W.A. Identification of MK-944a: A second clinical candidate from the hydroxylaminepentanamide isostere series of HIV protease inhibitors. J. Med. Chem. 2000, 43, 3386–3399. [Google Scholar] [CrossRef]
- Agarwal, R.; Jha, K.K.; Munshi, P.; Adepally, U.; Singh, A.; Charyd, M.T.; Sen, S. Substituted furopyridinediones as novel inhibitors of a-glucosidase. RSC Adv. 2015, 5, 90374. [Google Scholar]
- Salunke, D.B.; Yoo, E.; Shukla, N.M.; Balakrishna, R.; Malladi, S.S.; Serafin, K.J.; Day, V.W.; Wang, X.; David, S.A. Structure–activity relationships in human Toll-like receptor 8-active 2, 3-diamino-furo [2, 3-c] pyridines. J. Med. Chem. 2012, 55, 8137–8151. [Google Scholar] [CrossRef]
- Zubenko, A.; Divaeva, L.; Morkovnik, A.; Fetisov, L.; Sochnev, V.; Kononenko, K.; Bodryakov, A.; Klimenko, A. Structural Modification of Pyridoxal. Synthesis and Evaluation of Anti-Infective Activity of New 4-Chloro-and 4-Alkyl (dialkyl) aminomethyl-2-hetaryl (hetaroyl) furo[2, 3-c] pyridines. Russ. J. Gen. Chem. 2020, 90, 2242–2247. [Google Scholar] [CrossRef]
- Hei, Y.-Y.; Shen, Y.; Wang, J.; Zhang, H.; Zhao, H.-Y.; Xin, M.; Cao, Y.-X.; Li, Y.; Zhang, S.-Q. Synthesis and evaluation of 2, 9-disubstituted 8-phenylthio/phenylsulfinyl-9H-purine as new EGFR inhibitors. Bioorg. Med. Chem. 2018, 26, 2173–2185. [Google Scholar] [CrossRef]
- Karnik, K.S.; Sarkate, A.P.; Tiwari, S.V.; Azad, R.; Burra, P.V.; Wakte, P.S. Computational and Synthetic approach with Biological Evaluation of Substituted Quinoline derivatives as small molecule L858R/T790M/C797S triple mutant EGFR inhibitors targeting resistance in Non-Small Cell Lung Cancer (NSCLC). Bioorg. Chem. 2021, 107, 104612. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Zhang, T.; Zhu, S.-J.; Sun, M.; Tong, L.; Lai, M.; Zhang, R.; Xu, W.; Wu, R.; Ding, J. Structure-based design of 5-methylpyrimidopyridone derivatives as new wild-type sparing inhibitors of the epidermal growth factor receptor triple mutant (EGFRL858R/T790M/C797S). J. Med. Chem. 2019, 62, 7302–7308. [Google Scholar] [CrossRef] [PubMed]
- Rajith, B.; Chakraborty, C.; NagaSundaram, N.; Ali, S.K.; Zhu, H. Structural signature of the G719S-T790M double mutation in the EGFR kinase domain and its response to inhibitors. Sci. Rep. 2014, 4, 5868. [Google Scholar]
- Martínez-Jiménez, F.; Overington, J.P.; Al-Lazikani, B.; Marti-Renom, M.A. Rational design of non-resistant targeted cancer therapies. Sci. Rep. 2017, 7, 46632. [Google Scholar] [CrossRef]
- Stamos, J.; Sliwkowski, M.X.; Eigenbrot, C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem. 2002, 277, 46265–46272. [Google Scholar] [CrossRef]
- Sepay, N.; Mondal, R.; Al-Muhanna, M.K.; Saha, D. Identification of natural flavonoids as novel EGFR inhibitors using DFT, molecular docking, and molecular dynamics. NJC 2022, 46, 9735–9744. [Google Scholar] [CrossRef]
- Zhang, R.; Chen, S.; Zhang, X.; Yu, R.; Wan, S.; Geng, M.; Jiang, T. Synthesis and evaluation of novel non-covalent binding quinazoline glycoside derivatives targeting the L858R and T790M variants of EGFR. RSC Adv. 2016, 6, 36857–36862. [Google Scholar] [CrossRef]
- Lei, H.; Fan, S.; Zhang, H.; Liu, Y.-J.; Hei, Y.-Y.; Zhang, J.-J.; Zheng, A.-Q.; Xin, M.; Zhang, S.-Q. Discovery of novel 9-heterocyclyl substituted 9H-purines as L858R/T790M/C797S mutant EGFR tyrosine kinase inhibitors. Eur. J. Med. Chem. 2020, 186, 111888. [Google Scholar] [CrossRef]
- Bello, M.; Saldaña-Rivero, L.; Correa-Basurto, J.; García, B.; Sánchez-Espinosa, V.A. Structural and energetic basis for the molecular recognition of dual synthetic vs. natural inhibitors of EGFR/HER2. Int. J. Biol. Macromol. 2018, 111, 569–586. [Google Scholar] [CrossRef]
- Kou, S.-B.; Lin, Z.-Y.; Wang, B.-L.; Shi, J.-H.; Liu, Y.-X. Evaluation of the binding behavior of olmutinib (HM61713) with model transport protein: Insights from spectroscopic and molecular docking studies. J. Mol. Struct. 2021, 1224, 129024. [Google Scholar] [CrossRef]
- Zhang, H.-X.; Xiong, H.-X.; Li, L.-W. Investigation on the protein-binding properties of icotinib by spectroscopic and molecular modeling method. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2016, 161, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Kashima, K.; Kawauchi, H.; Tanimura, H.; Tachibana, Y.; Chiba, T.; Torizawa, T.; Sakamoto, H. CH7233163 overcomes osimertinib resistant EGFR-Del19/T790M/C797S mutation. Mol. Cancer Ther. 2020, 19, 2288–2297. [Google Scholar] [CrossRef]
- Gajiwala, K.S.; Feng, J.; Ferre, R.; Ryan, K.; Brodsky, O.; Weinrich, S.; Kath, J.C.; Stewart, A. Insights into the aberrant activity of mutant EGFR kinase domain and drug recognition. Structure 2013, 21, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Gotz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. Generalized born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Frisch, A. Gaussian 09w Reference; Gaussian, Inc.: Wallingford, CT, USA, 2009; 25p. [Google Scholar]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Miller III, B.R.; McGee Jr, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA. py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Cipher | R1 | R2 | Cipher | R1 | R2 |
| PD1 |  | OH | PD9 |  |  |
| PD2 |  | OH | PD10 |  |  |
| PD3 |  |  | PD11 |  |  |
| PD4 |  |  | PD12 |  |  |
| PD5 |  |  | PD13 |  |  |
| PD6 |  |  | PD14 |  |  |
| PD8 |  |  | |||
| Compounds | IC50 (nM) | ||
|---|---|---|---|
| Wild-Type | L858R/T790M | L858R/T790M/C797S | |
| PD4 | 4.78 ± 0.73 | 91.02 ± 2.01 | 103.70 ± 3.62 |
| PD13 | 11.64 ± 1.30 | 10.51 ± 0.71 | 21.93 ± 1.79 |
| Erlotinib | 14.11 ± 0.19 | 87.96 ± 0.89 | 108.50 ± 1.54 |
| Afatinib | 78.19 ± 1.20 | 20.14 ± 0.58 | 23.86 ± 2.02 |
| Osimertinib | 441.90 ± 3.02 | 17.39 ± 0.25 | 8.98 ± 0.53 |
| Compounds | IC50 (μM) | ||
|---|---|---|---|
| A549 | H1975 | Vero | |
| PD4 | 19.70 ± 0.59 | 27.26 ± 1.02 | >50 |
| PD13 | 18.09 ± 1.57 | 33.87 ± 0.86 | >100 |
| Erlotinib | 19.43 ± 0.79 | 41.84 ± 2.19 | >30 |
| Afatinib | 34.69 ± 0.67 | 27.33 ± 1.87 | >50 |
| Osimertinib | 25.28 ± 1.21 | 18.33 ± 2.00 | >50 |
| ΔEele | ΔEvdW | ΔEMM | TΔS | ΔGsol (PBSA) | ΔGbind (PBSA) | IC50 (nM) | ΔGbind,exp | |
|---|---|---|---|---|---|---|---|---|
| Wild-type | ||||||||
| Erlotinib | −15.43 ± 1.02 | −65.54 ± 1.52 | −80.98 ± 1.87 | −22.69 ± 0.94 | 45.70 ± 0.96 | −12.57 ± 0.69 | 11.44 | 10.83 |
| PD13 | −25.46 ± 2.01 | −98.54 ± 0.89 | −124.01 ± 0.71 | −27.98 ± 0.75 | 77.69 ± 0.05 | −18.33 ± 0.42 | 14.11 | 10.70 |
| L858R/T790M/58 | ||||||||
| Afatinib | −15.56 ± 0.85 | −60.87 ± 0.66 | −79.44 ± 0.09 | −20.47 ± 0.99 | 39.23 ± 0.12 | −19.73 ± 0.95 | 20.14 | 10.49 |
| PD13 | −14.36 ± 0.83 | −91.64 ± 1.99 | −106.00 ± 3.54 | −10.21 ± 0.87 | 73.11 ± 0.93 | −22.68 ± 0.47 | 10.51 | 10.88 |
| L858R/T790M/C797S | ||||||||
| Osimertinib | −20.03 ± 0.79 | −61.65 ± 1.20 | −81.68 ± 0.28 | −19.24 ± 0.88 | 42.57 ± 0.45 | −19.87 ± 0.84 | 8.98 | 10.97 |
| PD13 | −12.02 ± 1.44 | −94.02 ± 0.98 | −106.04 ± 0.95 | −18.21 ± 1.55 | 71.11 ± 0.69 | −16.71 ± 0.69 | 21.93 | 10.44 |
| Furopyridine | Solvent (mL) | Reagent R3NH (mmol) | Reaction Temperature (°C) | Reaction Time (h) | Yield (%) |
|---|---|---|---|---|---|
| PD3 | EtOH 10 | 3 | 25–30 | 6.0 | 62 |
| PD4 | EtOH 10 | 3 | 25–30 | 6.0 | 66 |
| PD5 | EtOH 10 | 12 | boiling | 3.0 | 47 |
| PD6 | EtOH 10 | 10 | 25–30 | 6.0 | 73 |
| PD7 1 | DMF 8 | 3 | 35–40 | 3.0 | 93 |
| PD8 | DMF 10 | 3 | 45–50 | 6.0 | 75 |
| PD9 | EtOH 8, H2O 2 | 3 | boiling | 3.0 | 68 |
| PD10 | EtOH 10 | 3 | 40–45 | 5.0 | 84 |
| PD11 | EtOH 10 | 3 | 35–40 | 5.0 | 44 |
| PD12 | DMF 5 | 3 | 30–40 | 5.0 | 61 |
| PD13 2 | DMF 5 | 3.5 | 30–40 | 5.0 | 58 |
| PD14 2 | DMF 5 | 3.5 | 30–40 | 5.0 | 75 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Todsaporn, D.; Zubenko, A.; Kartsev, V.; Aiebchun, T.; Mahalapbutr, P.; Petrou, A.; Geronikaki, A.; Divaeva, L.; Chekrisheva, V.; Yildiz, I.; et al. Discovery of Novel EGFR Inhibitor Targeting Wild-Type and Mutant Forms of EGFR: In Silico and In Vitro Study. Molecules 2023, 28, 3014. https://doi.org/10.3390/molecules28073014
Todsaporn D, Zubenko A, Kartsev V, Aiebchun T, Mahalapbutr P, Petrou A, Geronikaki A, Divaeva L, Chekrisheva V, Yildiz I, et al. Discovery of Novel EGFR Inhibitor Targeting Wild-Type and Mutant Forms of EGFR: In Silico and In Vitro Study. Molecules. 2023; 28(7):3014. https://doi.org/10.3390/molecules28073014
Chicago/Turabian StyleTodsaporn, Duangjai, Alexander Zubenko, Victor Kartsev, Thitinan Aiebchun, Panupong Mahalapbutr, Anthi Petrou, Athina Geronikaki, Liudmila Divaeva, Victoria Chekrisheva, Ilkay Yildiz, and et al. 2023. "Discovery of Novel EGFR Inhibitor Targeting Wild-Type and Mutant Forms of EGFR: In Silico and In Vitro Study" Molecules 28, no. 7: 3014. https://doi.org/10.3390/molecules28073014
APA StyleTodsaporn, D., Zubenko, A., Kartsev, V., Aiebchun, T., Mahalapbutr, P., Petrou, A., Geronikaki, A., Divaeva, L., Chekrisheva, V., Yildiz, I., Choowongkomon, K., & Rungrotmongkol, T. (2023). Discovery of Novel EGFR Inhibitor Targeting Wild-Type and Mutant Forms of EGFR: In Silico and In Vitro Study. Molecules, 28(7), 3014. https://doi.org/10.3390/molecules28073014