
Detailed Experimental and In Silico Investigation of Indomethacin Binding with Human Serum Albumin Considering Primary and Secondary Binding Sites

 ,
, 
 , and
, and
Abstract
1. Introduction
2. Results and Discussions
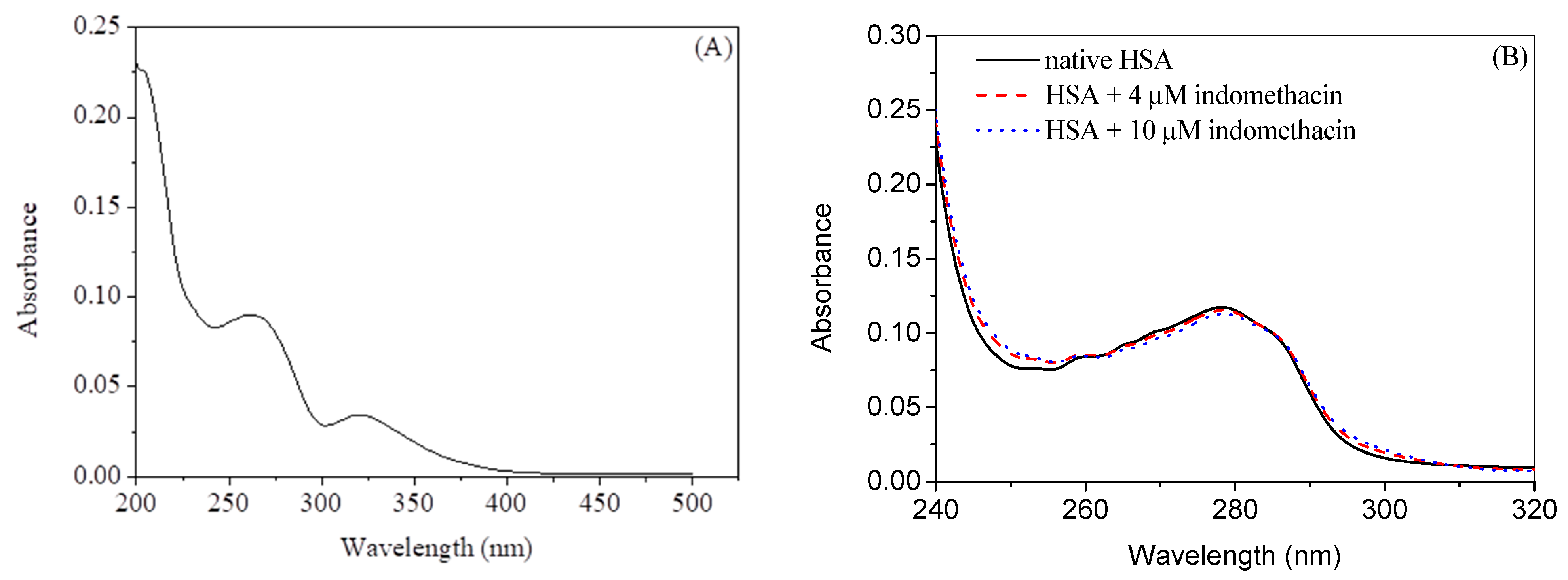
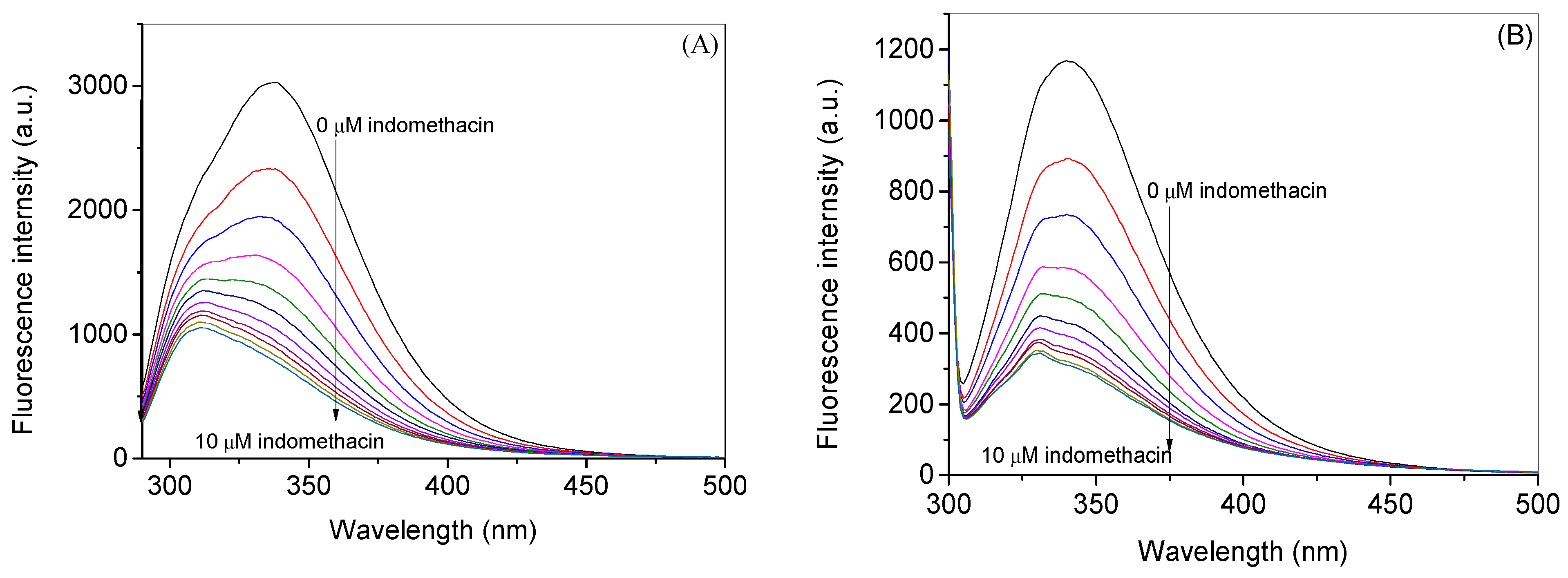
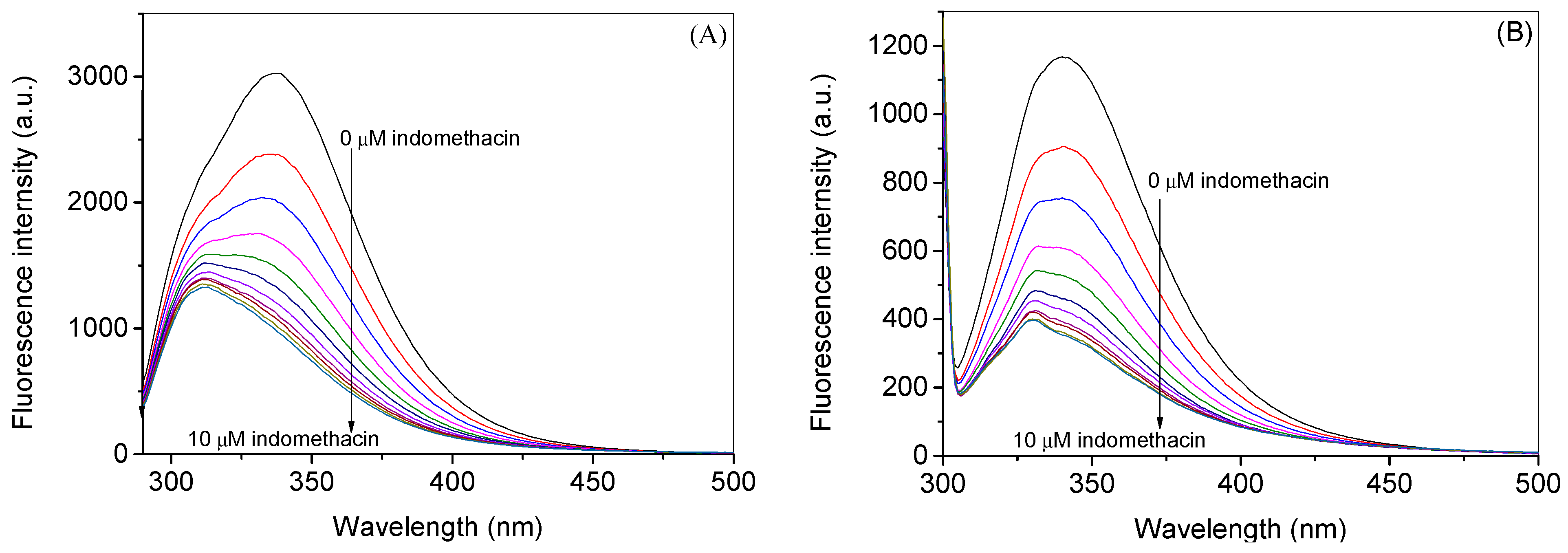
2.1. Experimental Investigation of Indomethacin Binding with HSA
2.2. In Silico Investigation of Indomethacin Binding with HSA
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Soh, J.-W.; Weinstein, I.B. Role of COX-independent targets of NSAIDs and related compounds in cancer prevention and treatment. Prog. Exp. Tumor Res. 2003, 37, 261–283. [Google Scholar]
- Nalamachu, S.; Wortmann, R. Role of Indomethacin in Acute Pain and Inflammation Management: A Review of the Literature. Postgrad. Med. 2014, 126, 92–97. [Google Scholar] [CrossRef]
- Munjal, A.; Allam, A.E. Indomethacin. In StatPearls [Internet]; StatPearls Publishing: Tampa, FL, USA, 2020. [Google Scholar]
- Deb, P.K.; Al-Attraqchi, O.; Prasad, M.R.; Tekade, R.K. Chapter 11—Protein and Tissue Binding: Implication on Pharmacokinetic Parameters. In Dosage Form Design Considerations; Tekade, R.K., Ed.; Academic Press: Cambridge, MA, USA, 2018; pp. 371–399. [Google Scholar]
- Merlot, A.M.; Kalinowski, D.S.; Richardson, D.R. Unraveling the mysteries of serum albumin—More than just a serum protein. Front. Physiol. 2014, 5, 299. [Google Scholar] [CrossRef] [PubMed]
- Fanali, G.; di Masi, A.; Trezza, V.; Marino, M.; Fasano, M.; Ascenzi, P. Human serum albumin: From bench to bedside. Mol. Asp. Med. 2012, 33, 209–290. [Google Scholar] [CrossRef]
- Ali, M.S.; Al-Lohedan, H.A. Spectroscopic and computational evaluation on the binding of safranal with human serum albumin: Role of inner filter effect in fluorescence spectral correction. Spectrochim. Acta A 2018, 203, 434–442. [Google Scholar] [CrossRef]
- Ali, M.S.; Amina, M.; Al-Lohedan, H.A.; Al Musayeib, N.M. Elucidation of the interaction of human serum albumin with anti-cancer sipholane triterpenoid from the Red Sea sponge. Luminescence 2017, 32, 223–230. [Google Scholar] [CrossRef]
- Ali, M.S.; Altaf, M.; Al-Lohedan, H.A. Green synthesis of biogenic silver nanoparticles using Solanum tuberosum extract and their interaction with human serum albumin: Evidence of “corona” formation through a multi-spectroscopic and molecular docking analysis. J. Photochem. Photobiol. B 2017, 173, 108–119. [Google Scholar] [CrossRef]
- Ali, M.S.; Al-Lohedan, H.A.; Atta, A.M.; Ezzat, A.O.; Al-Hussain, S.A.A. Interaction of human serum albumin with silver nanoparticles functionalized with polyvinylthiol. J. Mol. Liq. 2015, 204, 248–254. [Google Scholar] [CrossRef]
- Ali, M.S.; Al-Lohedan, H.A. Biophysical characterization of the interaction between human serum albumin and n-dodecyl beta-D-maltoside: A multi-technique approach. Colloids Surf. B. Biointerfaces 2015, 134, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.S.; Al-Lohedan, H.A. Interaction of human serum albumin with sulfadiazine. J. Mol. Liq. 2014, 197, 124–130. [Google Scholar] [CrossRef]
- Ali, M.S.; Amina, M.; Al-Lohedan, H.A.; Al Musayeib, N.M. Human serum albumin binding to the biologically active labdane diterpene “leoheterin”: Spectroscopic and in silico analysis. J. Photochem. Photobiol. B Biol. 2018, 182, 9–17. [Google Scholar] [CrossRef]
- Ali, M.S.; Muthukumaran, J.; Al-Lohedan, H.A. Molecular interactions of ceftazidime with bovine serum albumin: Spectroscopic, molecular docking, and DFT analyses. J. Mol. Liq. 2020, 313, 113490. [Google Scholar] [CrossRef]
- Ali, M.S.; Al-Lohedan, H.A. Experimental and computational investigation on the molecular interactions of safranal with bovine serum albumin: Binding and anti-amyloidogenic efficacy of ligand. J. Mol. Liq. 2019, 278, 385–393. [Google Scholar] [CrossRef]
- Ali, M.S.; Al-Lohedan, H.A. Deciphering the interaction of procaine with bovine serum albumin and elucidation of binding site: A multi spectroscopic and molecular docking study. J. Mol. Liq. 2017, 236, 232–240. [Google Scholar] [CrossRef]
- Amina, M.; Ali, M.S.; Al-Musayeib, N.M.; Al-Lohedan, H.A. Biophysical characterization of the interaction of bovine serum albumin with anticancer sipholane triterpenoid from the Red Sea sponge. J. Mol. Liq. 2016, 220, 931–938. [Google Scholar] [CrossRef]
- Ali, M.S.; Al-Lohedan, H.A. Multi-technique approach on the interaction between sugar-based surfactant n-dodecyl beta-D-maltoside and bovine serum albumin. J. Lumin. 2016, 169, 35–42. [Google Scholar] [CrossRef]
- Ali, M.S.; Al-Lohedan, H.A. Sulfadiazine binds and unfolds bovine serum albumin: An in vitro study. Mol. Biol. Rep. 2013, 40, 6081–6090. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.B.; Khan, J.M.; Ali, M.S.; Khan, R.H.; Kabir-ud-Din. Interaction of amphiphilic drugs with human and bovine serum albumins. Spectrochim. Acta A 2012, 97, 119–124. [Google Scholar] [CrossRef]
- Han, X.; Sun, J.; Niu, T.; Mao, B.; Gao, S.; Zhao, P.; Sun, L. Molecular Insight into the Binding of Astilbin with Human Serum Albumin and Its Effect on Antioxidant Characteristics of Astilbin. Molecules 2022, 27, 4487. [Google Scholar] [CrossRef]
- Aiello, F.; Uccello-Barretta, G.; Picchi, C.; Nazzi, S.; Recchimurzo, A.; Balzano, F. NMR Investigation of the Interaction of Three Non-Steroidal Anti-Inflammatory Drugs with Human Serum Albumin. Molecules 2022, 27, 6647. [Google Scholar] [CrossRef]
- Samperi, M.; Vittorio, S.; De Luca, L.; Romeo, A.; Monsù Scolaro, L. Interaction of Aggregated Cationic Porphyrins with Human Serum Albumin. Int. J. Mol. Sci. 2023, 24, 2099. [Google Scholar] [CrossRef] [PubMed]
- Dombi, G.; Horváth, P.; Fiser, B.; Mirzahosseini, A.; Dobó, M.; Szabó, Z.-I.; Tóth, G. Enantioselective Human Serum Albumin Binding of Apremilast: Liquid Chromatographic, Fluorescence and Molecular Docking Study. Int. J. Mol. Sci. 2023, 24, 2168. [Google Scholar] [CrossRef]
- Erkmen, C.; Bozal-Palabiyik, B.; Tayyab, H.; Kabir, M.Z.; Mohamad, S.B.; Uslu, B. Exploring molecular interaction of cefpirome with human serum albumin: In vitro and in silico approaches. J. Mol. Struct. 2023, 1275, 134723. [Google Scholar] [CrossRef]
- Du, X.; Wang, X.; Yao, J.; Li, H.; Bao, Y.; Lan, J.; Zhao, Z.; Zong, W. Study on the interaction between sulfamerazine and human serum albumin on molecular level using spectral analysis. Colloids Surf. A Physicochem. Eng. Asp. 2023, 661, 130917. [Google Scholar] [CrossRef]
- Perrin, J.H.; Nelson, D.A. Induced optical activity following the binding of warfarin, indomethacin, 4-hydroxycoumarin and salicylic acid to human serum albumin. Life Sci. 1972, 11 Pt 1, 277–283. [Google Scholar] [CrossRef]
- Mason, R.W.; McQueen, E.G. Protein Binding of Indomethacin: Binding of Indomethacin to Human Plasma Albumin and its Displacement from Binding by Ibuprofen, Phenylbutazone and Salicylate, in vitro. Pharmacology 1974, 12, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Zini, R.; d’Athis, P.; Barre, J.; Tillement, J.P. Binding of indomethacin to human serum albumin. Its non displacement by various agents, influence of free fatty acids and the unexpected effect of indomethacin on warfarin binding. Biochem. Pharmacol. 1979, 28, 2661–2665. [Google Scholar] [CrossRef]
- Ekman, B.; Sjödin, T.; Sjöholm, I. Binding of drugs to human serum albumin—XV: Characterization and identification of the binding sites of indomethacin. Biochem. Pharmacol. 1980, 29, 1759–1765. [Google Scholar] [CrossRef]
- Montero, M.T.; Pouplana, R.; GarcÍA, S.; Valls, O. On the binding of cinmetacin and indomethacin to human serum albumin. J. Pharm. Pharm. 1986, 38, 925–927. [Google Scholar] [CrossRef]
- Cots, J.; Pouplana, R.; Estelrich, J. Conformational changes in the human serum albumin-indomethacin binding. Colloid Polym. Sci. 1987, 265, 164–166. [Google Scholar] [CrossRef]
- Tayyab, S.; Haq, S.K.; Sabeeha; Aziz, M.A.; Khan, M.M.; Muzammil, S. Effect of lysine modification on the conformation and indomethacin binding properties of human serum albumin. Int. J. Biol. Macromol. 1999, 26, 173–180. [Google Scholar] [CrossRef]
- Trivedi, V.D.; Vorum, H.; HonorÉ, B.; Qasim, M.A. Molecular Basis of Indomethacin-Human Serum Albumin Interaction. J. Pharm. Pharm. 1999, 51, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.-M.; Du, L.-M. Spectroscopic study on binding of indomethacin to bovine serum albumin. Guang Pu Xue Yu Guang Pu Fen Xi 2007, 27, 973–977. [Google Scholar]
- Bogdan, M.; Pirnau, A.; Floare, C.; Bugeac, C. Binding interaction of indomethacin with human serum albumin. J. Pharm. Biomed. Anal. 2008, 47, 981–984. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Sun, H.-H.; Zhang, Y.-Z.; Yang, L.-Y.; Dai, J.; Liu, Y. Interaction of Human Serum Albumin with Indomethacin: Spectroscopic and Molecular Modeling Studies. J. Solut. Chem. 2012, 41, 422–435. [Google Scholar] [CrossRef]
- Ali, M.S.; Al-Lohedan, H.A. Spectroscopic and Molecular Docking Investigation on the Noncovalent Interaction of Lysozyme with Saffron Constituent “Safranal”. Acs Omega 2020, 5, 9131–9141. [Google Scholar] [CrossRef] [PubMed]
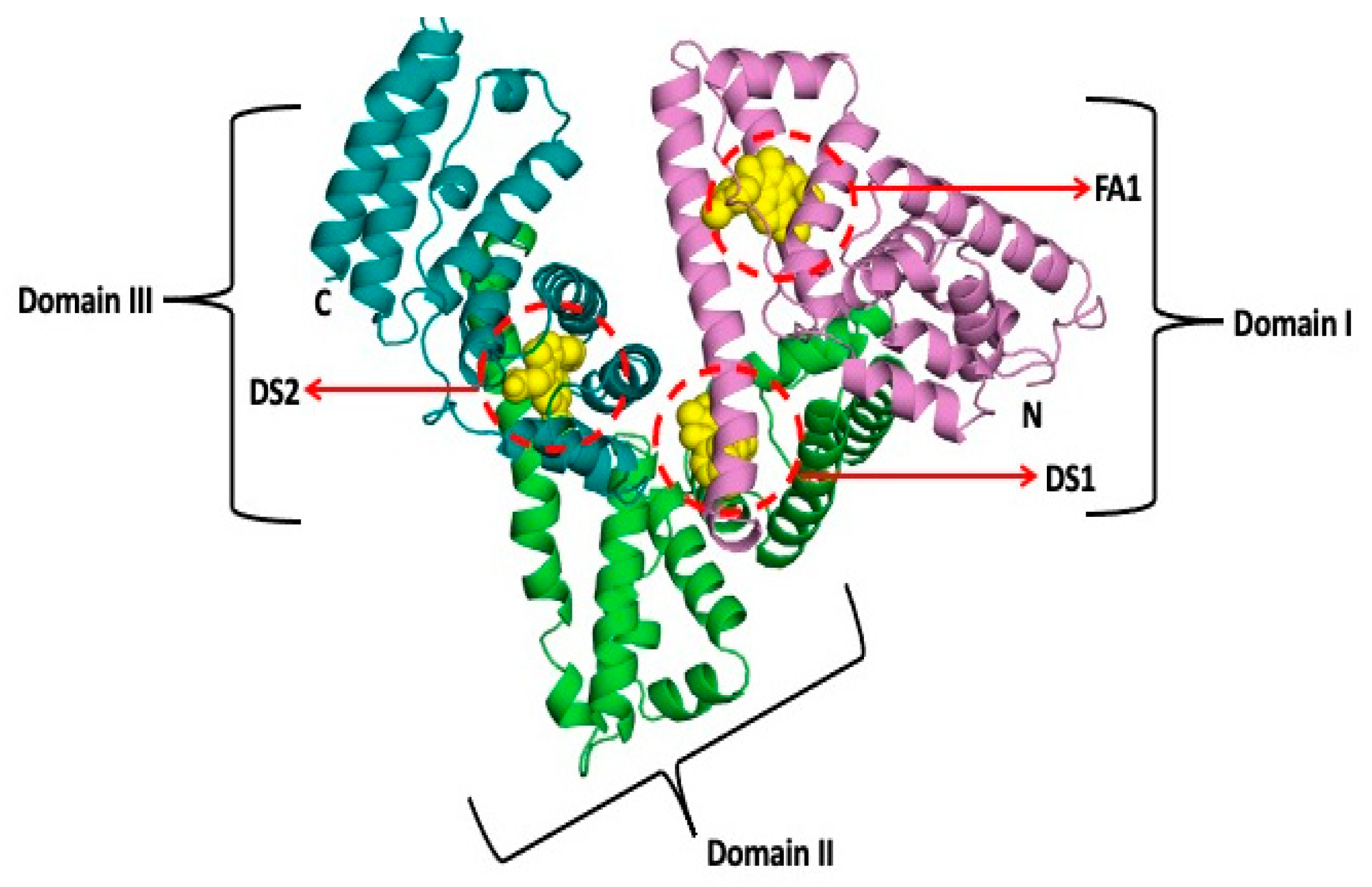
- Ghuman, J.; Zunszain, P.A.; Petitpas, I.; Bhattacharya, A.A.; Otagiri, M.; Curry, S. Structural basis of the drug-binding specificity of human serum albumin. J. Mol. Biol. 2005, 353, 38–52. [Google Scholar] [CrossRef]
- Ali, M.S.; Muthukumaran, J.; Jain, M.; Al-Sanea, A.S.S.; Al-Lohedan, H.A. Experimental and in silico investigation on the interaction of indomethacin with bovine serum albumin: Effect of sodium dodecyl sulfate surfactant monomers on the binding. J. Mol. Liq. 2021, 336, 116858. [Google Scholar]
- Shirley, B.A. Protein Stability and Folding: Theory and Practice; Humana Press: Totowa, NJ, USA, 1995. [Google Scholar]
- Ali, M.S.; Waseem, M.; Subbarao, N.; Al-Lohedan, H.A. Noncovalent molecular interactions between antineoplastic drug gemcitabine and a carrier protein identified through spectroscopic and in silico methods. Int. J. Biol. Macromol. 2021, 182, 993–1002. [Google Scholar] [CrossRef]
- Ali, M.S.; Muthukumaran, J.; Jain, M.; Santos-Silva, T.; Al-Lohedan, H.A.; Al-Shuail, N.S. Molecular interactions of cefoperazone with bovine serum albumin: Extensive experimental and computational investigations. J. Mol. Liq. 2021, 337, 116354. [Google Scholar] [CrossRef]
- Ali, M.S.; Waseem, M.; Subbarao, N.; Al-Lohedan, H.A. Dynamic interaction between lysozyme and ceftazidime: Experimental and molecular simulation approaches. J. Mol. Liq. 2021, 328, 115412. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 3rd ed.; Springer: Boston, MA, USA, 2006; p. 954. [Google Scholar]
- Moller, M.; Denicola, A. Study of protein-ligand binding by fluorescence. Biochem. Mol. Biol. Educ. 2002, 30, 309–312. [Google Scholar] [CrossRef]
- Eftink, M.R. Fluorescence Quenching Reactions. In Biophysical and Biochemical Aspects of Fluorescence Spectroscopy; Dewey, T.G., Ed.; Springer: Boston, MA, USA, 1991; pp. 1–41. [Google Scholar]
- Stern, O.; Volmer, M. The extinction period of fluorescence. Phys. Z. 1919, 20, 183–188. [Google Scholar]
- Eftink, M.R.; Ghiron, C.A. Exposure of Tryptophanyl Residues in Proteins—Quantitative-Determination by Fluorescence Quenching Studies. Biochemistry 1976, 15, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Eftink, M.R.; Ghiron, C.A. Fluorescence Quenching Studies with Proteins. Anal. Biochem. 1981, 114, 199–227. [Google Scholar] [CrossRef]
- Maciazek-Jurczyk, M.; Rownicka-Zubik, J.; Dyja, R.; Sulkowska, A. Comparative Analysis of KP-HSA Complex by Spectroscopic Methods. Acta Phys. Pol. A 2013, 123, 673–680. [Google Scholar] [CrossRef]
- Alarcon, E.; Edwards, A.M.; Aspee, A.; Moran, F.E.; Borsarelli, C.D.; Lissi, E.A.; Gonzalez-Nilo, D.; Poblete, H.; Scaiano, J.C. Photophysics and photochemistry of dyes bound to human serum albumin are determined by the dye localization. Photoch. Photobio. Sci. 2010, 9, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Ide, G.; Engelborghs, Y. Fluorescence Quenching and Induced Dissociation of the Tubulin-Colchicine Complex by Iodide. J. Biol. Chem. 1981, 256, 1684–1687. [Google Scholar] [CrossRef]
- Encinas, M.V.; Lissi, E.; Vergara, C. Association of Valdecoxib, a Nonsteroidal Anti-Inflammatory Drug, with Human Serum Albumin. Photochem. Photobiol. 2013, 89, 1399–1405. [Google Scholar] [CrossRef]
- Silva, D.; Cortez, C.M.; Silva, C.M.C.; Missailidis, S. A fluorescent spectroscopy and modelling analysis of anti-heparanase aptamers-serum protein interactions. J. Photoch. Photobio. B 2013, 127, 68–77. [Google Scholar] [CrossRef]
- Sudlow, G.; Birkett, D.J.; Wade, D.N. The characterization of two specific drug binding sites on human serum albumin. Mol. Pharm. 1975, 11, 824–832. [Google Scholar]
- Hosainzadeh, A.; Gharanfoli, M.; Saberi, M.R.; Chamani, J. Probing the Interaction of Human Serum Albumin with Bilirubin in the Presence of Aspirin by Multi-Spectroscopic, Molecular Modeling and Zeta Potential Techniques: Insight on Binary and Ternary Systems. J. Biomol. Struct. Dyn. 2012, 29, 1013–1050. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Xie, M.X.; Zheng, D.; Liu, Y.; Li, X.Y.; Chen, X. Spectroscopic studies on the interaction of cinnamic acid and its hydroxyl derivatives with human serum albumin. J. Mol. Struct. 2004, 692, 71–80. [Google Scholar]
- Ascenzi, P.; Fasano, M. Allostery in a monomeric protein: The case of human serum albumin. Biophys. Chem. 2010, 148, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.S.; Farah, M.A.; Al-Lohedan, H.A.; Al-Anazi, K.M. Comprehensive exploration of the anticancer activities of procaine and its binding with calf thymus DNA: A multi spectroscopic and molecular modelling study. RSC Adv. 2018, 8, 9083–9093. [Google Scholar] [CrossRef]
- Theodore Peters, J. All About Albumin: Biochemistry, Genetics, and Medical Applications; Academic Press: Cambridge, MA, USA, 1995. [Google Scholar]
- Atkovska, K.; Samsonov, S.A.; Paszkowski-Rogacz, M.; Pisabarro, M.T. Multipose binding in molecular docking. Int. J. Mol. Sci. 2014, 15, 2622–2645. [Google Scholar] [CrossRef]
- Lee, H.S.; Jo, S.; Lim, H.-S.; Im, W. Application of Binding Free Energy Calculations to Prediction of Binding Modes and Affinities of MDM2 and MDMX Inhibitors. J. Chem. Inf. Model. 2012, 52, 1821–1832. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. Software News and Update AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. Ligplot—A Program to Generate Schematic Diagrams of Protein Ligand Interactions. Protein Eng. 1995, 8, 127–134. [Google Scholar] [CrossRef]
- Xue, L.C.; Rodrigues, J.P.; Kastritis, P.L.; Bonvin, A.M.; Vangone, A. PRODIGY: A web server for predicting the binding affinity of protein-protein complexes. Bioinformatics 2016, 32, 3676–3678. [Google Scholar] [CrossRef] [PubMed]
- Van der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Grigera, J.R.; Straatsma, T.P. The missing term in effective pair potentials. J. Phys. Chemi. 1987, 91, 6269–6271. [Google Scholar] [CrossRef]
- Schuttelkopf, A.W.; van Aalten, D.M.F. PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. Sect. D-Struct. Biol. 2004, 60, 1355–1363. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
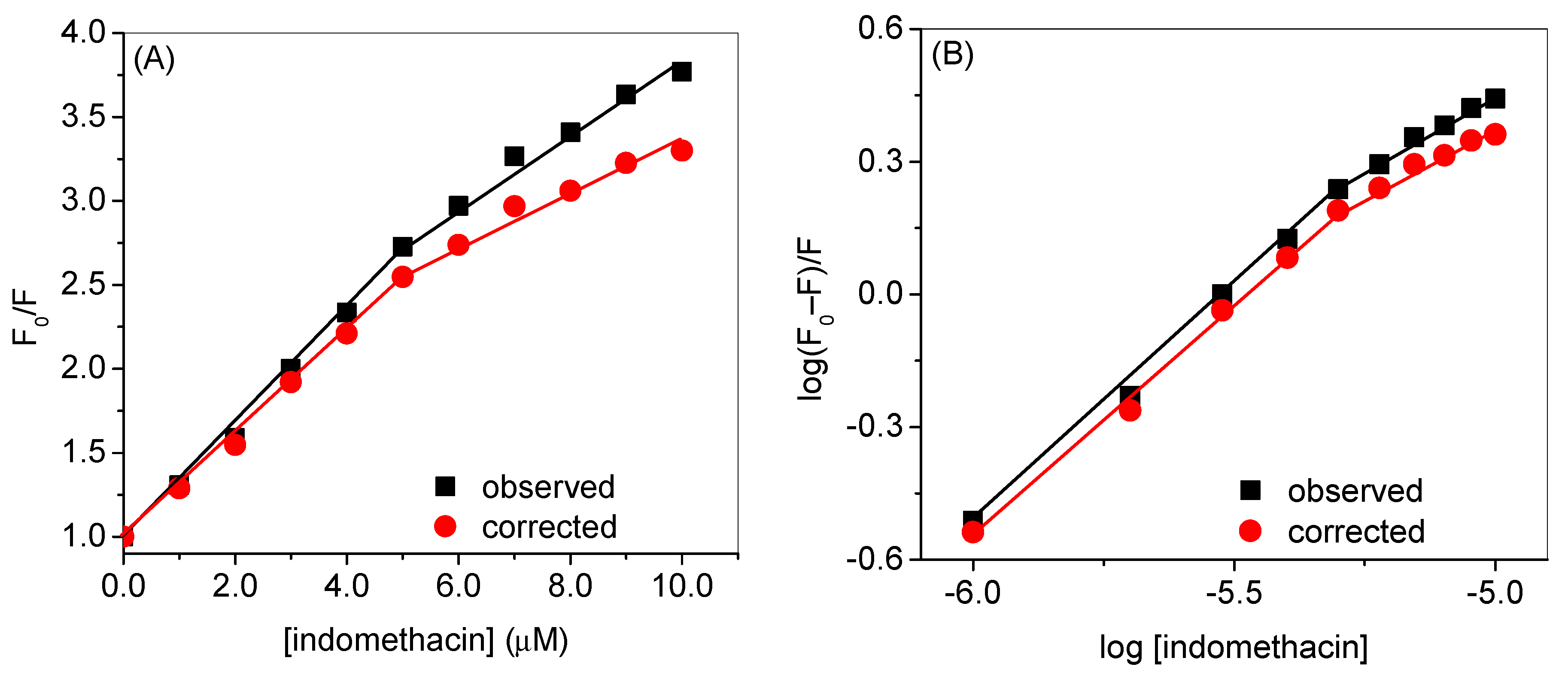
| Observed | Corrected | |
|---|---|---|
| KSV1 (mol−1) | 3.4 × 105 (R2 = 0.9952) | 3.0 × 105 (R2 = 0.9971) |
| KSV2 (mol−1) | 2.1 × 105 (R2 = 0.9861) | 1.5 × 105 (R2 = 0.9739) |
| kb1 (mol−1) | 9.3 × 105 (R2 = 0.9965) | 5.9 × 105 (R2 = 0.9966) |
| kb2 (mol−1) | 5.7 × 103 (R2 = 0.9863) | 1.9 × 103 (R2 = 0.9854) |
| n1 | 1.08 | 1.05 |
| n2 | 0.7 | 0.6 |
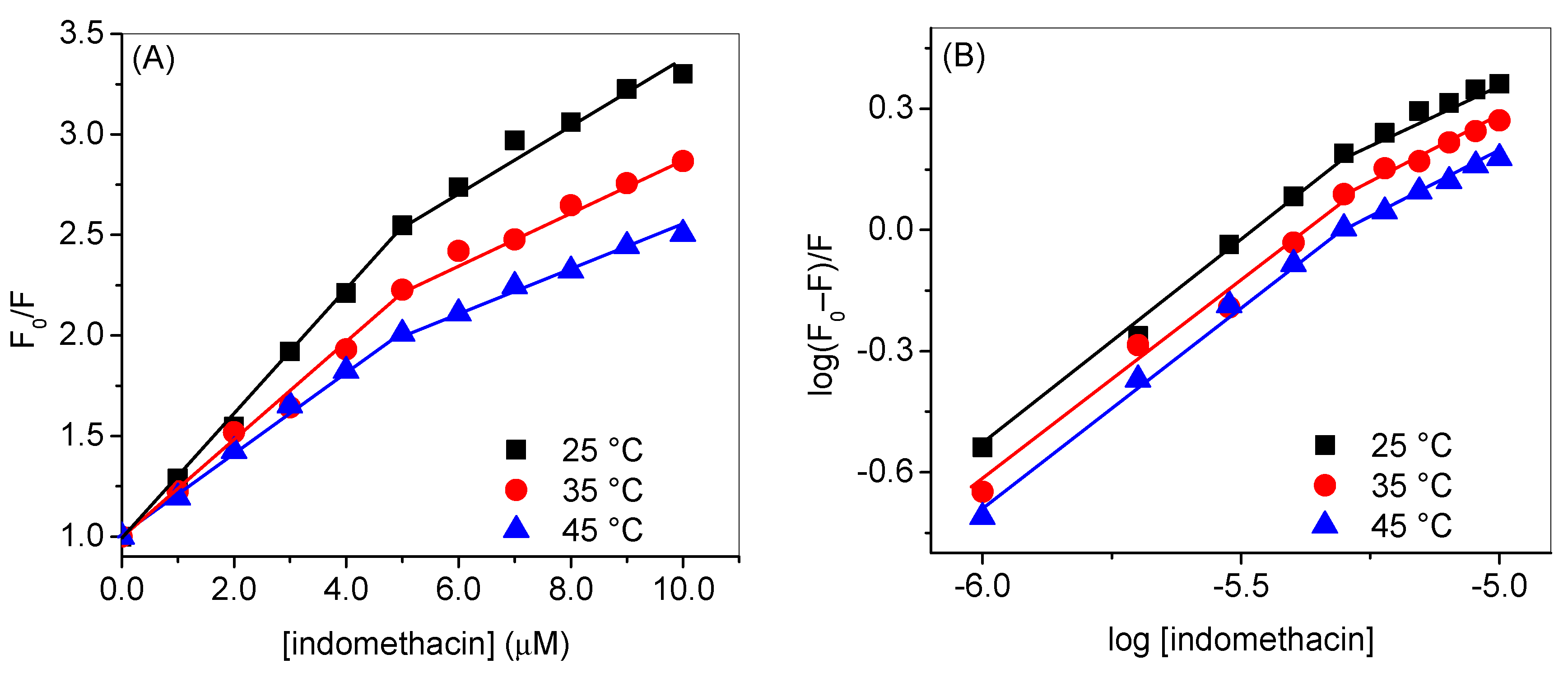
| 35 °C | 45 °C | |
|---|---|---|
| KSV1 (mol−1) | 2.4 × 105 (R2 = 0.9952) | 2.1 × 105 (R2 = 0.9971) |
| KSV2 (mol−1) | 1.3 × 105 (R2 = 0.9861) | 1.0 × 105 (R2 = 0.9739) |
| kb1 (mol−1) | 2.9 × 105 (R2 = 0.9965) | 1.8 × 105 (R2 = 0.9966) |
| kb2 (mol−1) | 1.7 × 103 (R2 = 0.9863) | 1.4 × 103 (R2 = 0.9854) |
| n1 | 1.00 | 0.98 |
| n2 | 0.6 | 0.6 |
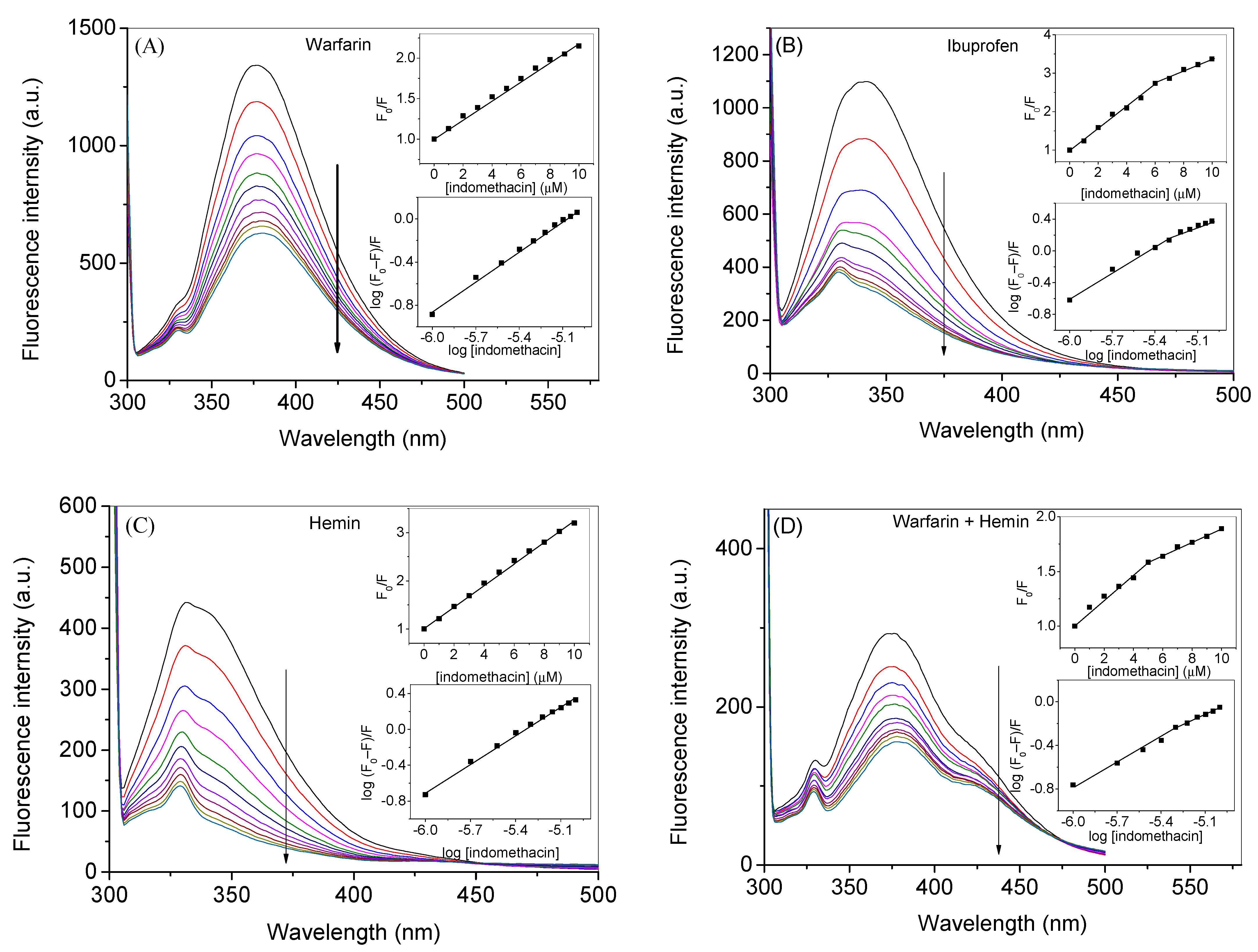
| Warfarin | Ibuprofen | Hemin | Warfarin + Hemin | |
|---|---|---|---|---|
| KSV1 (mol−1) | 1.2 × 105 | 2.8 × 105 | 2.1 × 105 | 1.1 × 105 |
| KSV2 (mol−1) | - | 1.6 × 105 | - | 6.0 × 104 |
| kb1 (mol−1) | 2.8 × 104 | 4.7 × 105 | 1.4 × 105 | 3.4 × 103 |
| kb2 (mol−1) | - | 1.5 × 103 | - | 9.8 × 102 |
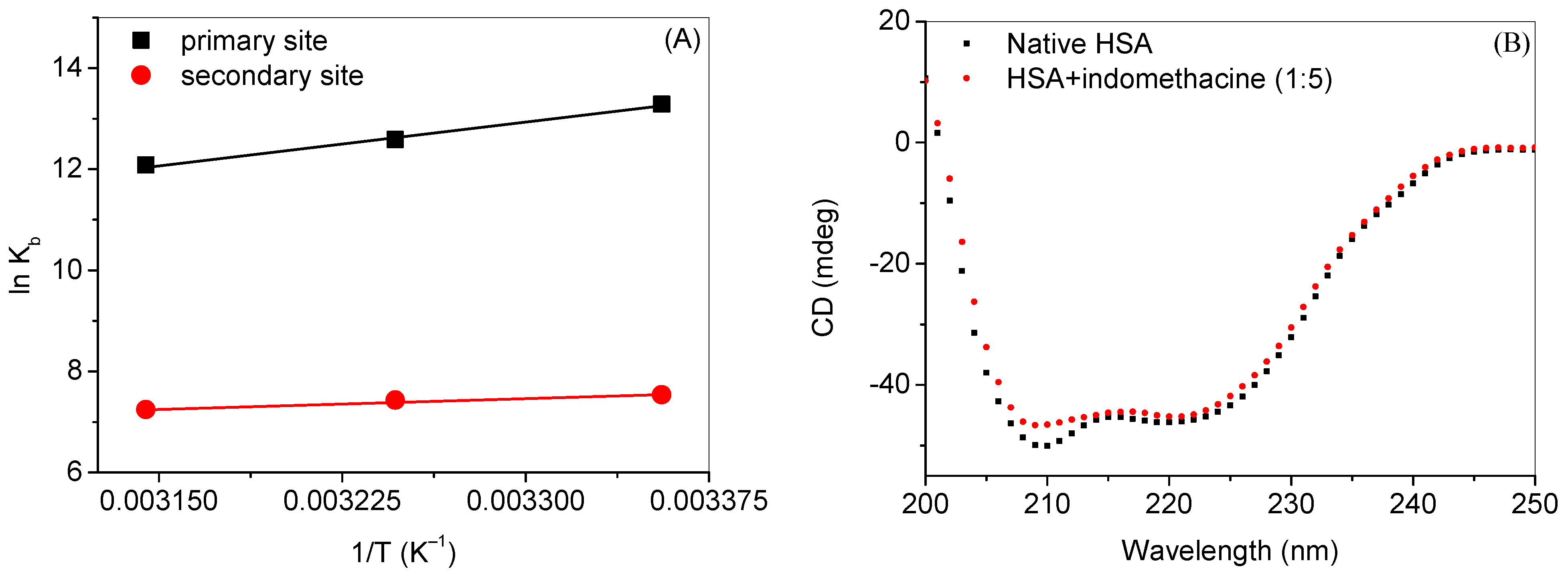
| Primary Site | Secondary Site | |||||
|---|---|---|---|---|---|---|
| Temp (°C) | 25 | 35 | 45 | 25 | 35 | 45 |
| ∆G (KJ mol−1) | –32.8 | –32.3 | –31.9 | –18.7 | –19.0 | –19.2 |
| ∆H (KJ mol−1) | –47.4 | –11.6 | ||||
| ∆S (J mol−1 K−1) | –48.9 | 23.9 | ||||
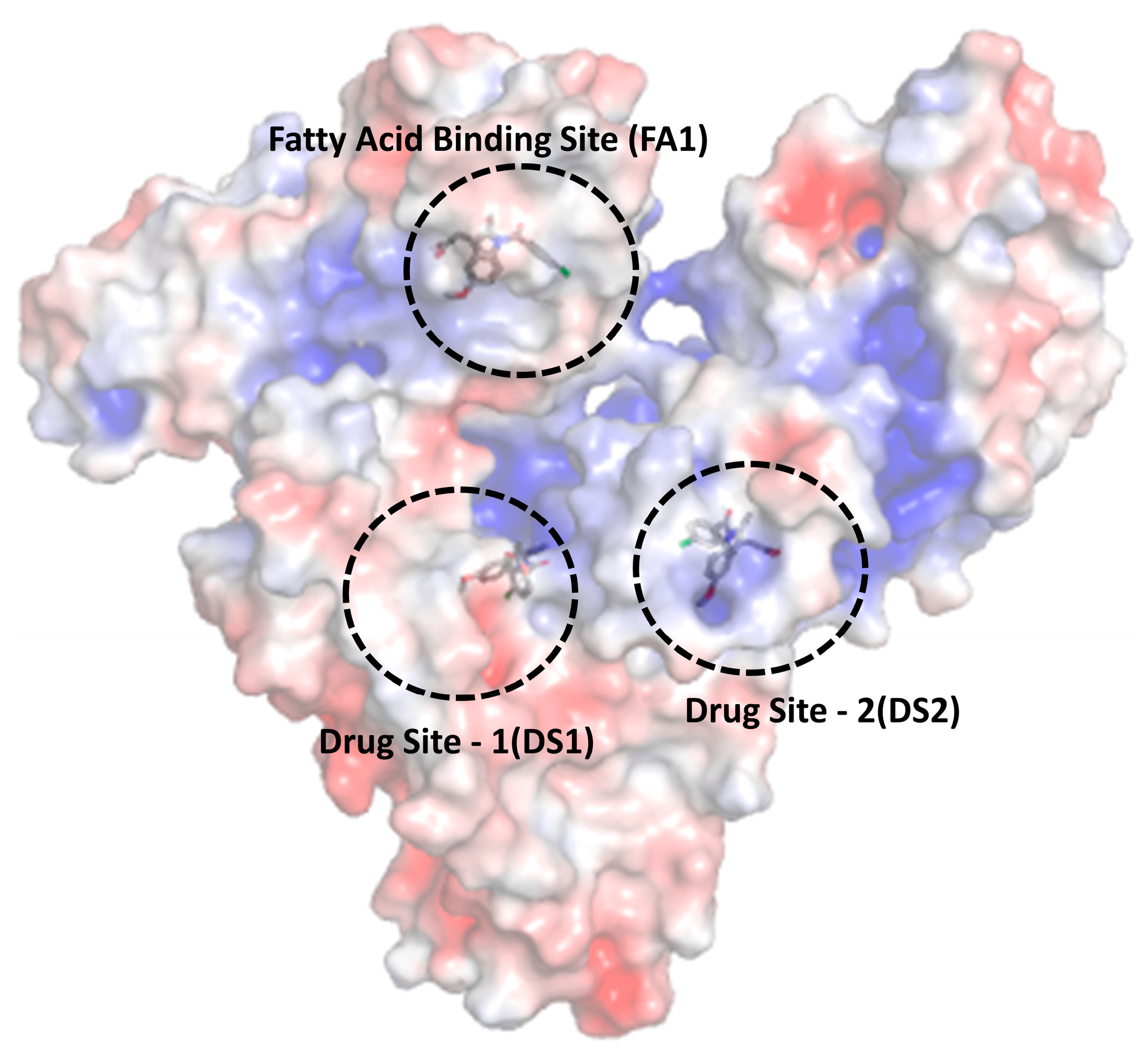
| Name of the Sites | ΔG (kcal/mol) (Program: Auto Dock Vina) | Ki (μM) | Estimated Binding Affinity (kcal/mol) (Program: Prodigy) |
|---|---|---|---|
| Drug site 1 | −10.1 | 0.04 | −6.5 |
| Drug site 2 | −7.96 | 1.46 | −5.4 |
| Fatty acid binding site 1 | −8.4 | 0.69 | −5.8 |
| Name of the Sites | Hydrogen Bonds | Hydrophobic Interactions |
|---|---|---|
| Drug site 1 (DS1) | W214 and R218 | K195, K199, S202, L203, A210, F211, A215, L238, D451, and L481 |
| Drug site 2 (DS2) | N391 | P384, L387, I388, C392, R410, Y411, V433, C437, R445, A449, and R485 |
| Fatty acid binding site 1 (FA1) | None | Leu115, Met123, Phe134, Leu135, Tyr138, Leu139, Ile142, Leu154, Ala158, Tyr161, Phe165, and R186 |
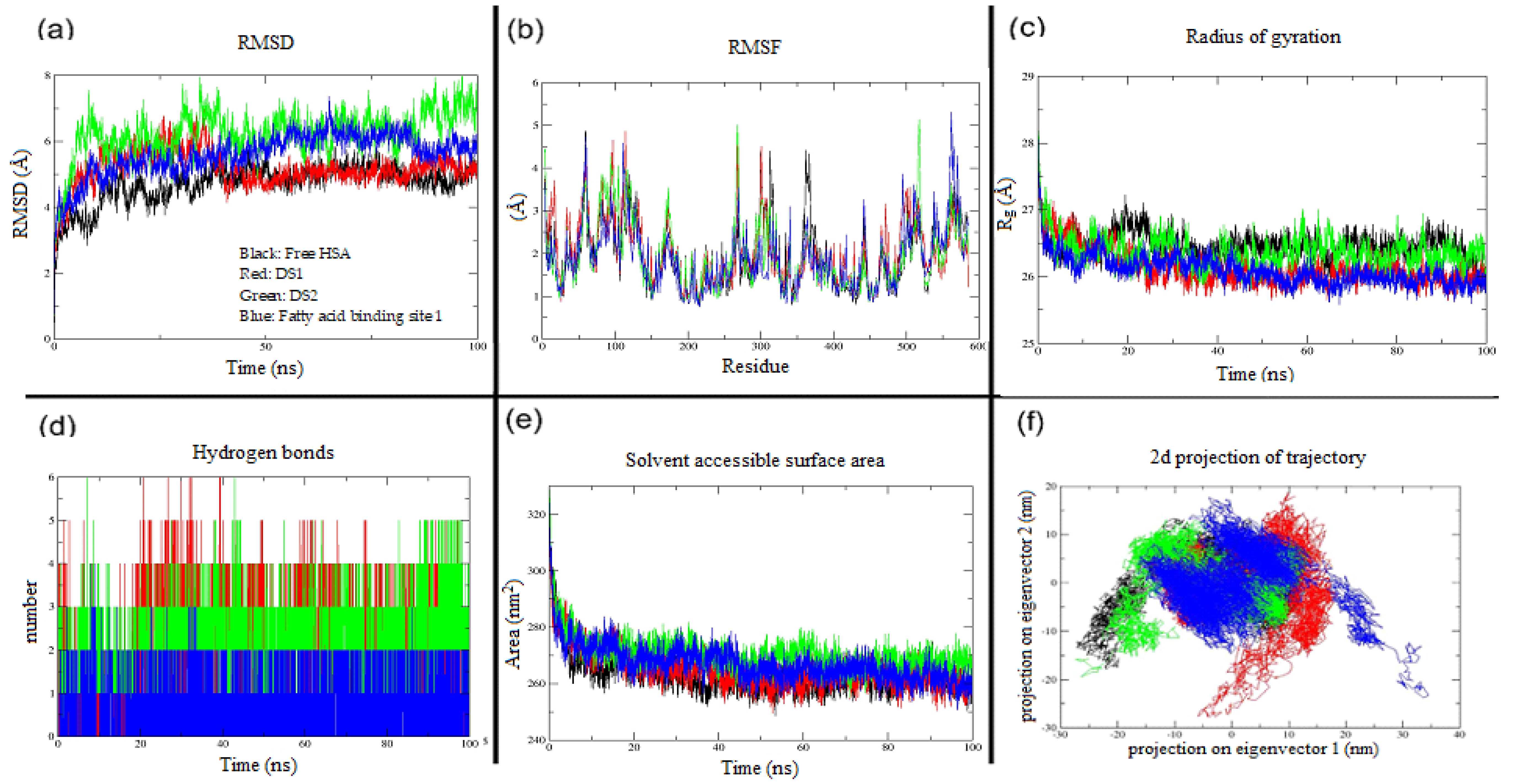
| Protein and Protein–Ligand Complexes | RMSD (Å) | RMSF (Å) | Rg (Å) | HBonds (between HSA & Indomethacin | SASA (nm2) | Trace of Covariance Matrix Values (nm2) |
|---|---|---|---|---|---|---|
| Free HSA | 4.7 | 1.9 | 26.5 | NA | 263.50 | 278.72 |
| Drug site 1 | 5.1 | 1.9 | 26.1 | 3 | 265.12 | 297.98 |
| Drug site 2 | 6.2 | 1.9 | 26.3 | 2 | 270.65 | 287.46 |
| Fatty acid binding site 1 | 5.6 | 1.9 | 26 | 1 | 267.18 | 273.18 |
| Protein and Protein–Ligand Complexes | Coil | β-Bridge | Bend | Turn | α-Helix | π-Helix | 310-Helix |
|---|---|---|---|---|---|---|---|
| Free HSA | 0.13 | 0.00 | 0.00 | 0.07 | 0.71 | 0.01 | 0.01 |
| Drug site 1 | 0.14 | 0.00 | 0.00 | 0.08 | 0.70 | 0.00 | 0.01 |
| Drug site 2 | 0.14 | 0.00 | 0.00 | 0.07 | 0.70 | 0.00 | 0.01 |
| Fatty acid binding site 1 | 0.14 | 0.00 | 0.07 | 0.07 | 0.71 | 0.01 | 0.01 |
| Name of the Sites | Number of Water Molecules Added | Number of Counter Ions Added |
|---|---|---|
| Free HSA | 43,298 | 16 NA |
| Drug site 1 | 43,272 | 17 NA |
| Drug site 2 | 43,293 | 17 NA |
| Fatty acid binding site 1 | 43,299 | 17 NA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, M.S.; Muthukumaran, J.; Jain, M.; Tariq, M.; Al-Lohedan, H.A.; Al-Sanea, A.S.S. Detailed Experimental and In Silico Investigation of Indomethacin Binding with Human Serum Albumin Considering Primary and Secondary Binding Sites. Molecules 2023, 28, 2979. https://doi.org/10.3390/molecules28072979
Ali MS, Muthukumaran J, Jain M, Tariq M, Al-Lohedan HA, Al-Sanea ASS. Detailed Experimental and In Silico Investigation of Indomethacin Binding with Human Serum Albumin Considering Primary and Secondary Binding Sites. Molecules. 2023; 28(7):2979. https://doi.org/10.3390/molecules28072979
Chicago/Turabian StyleAli, Mohd Sajid, Jayaraman Muthukumaran, Monika Jain, Mohammad Tariq, Hamad A. Al-Lohedan, and Abdullah Saad S. Al-Sanea. 2023. "Detailed Experimental and In Silico Investigation of Indomethacin Binding with Human Serum Albumin Considering Primary and Secondary Binding Sites" Molecules 28, no. 7: 2979. https://doi.org/10.3390/molecules28072979
APA StyleAli, M. S., Muthukumaran, J., Jain, M., Tariq, M., Al-Lohedan, H. A., & Al-Sanea, A. S. S. (2023). Detailed Experimental and In Silico Investigation of Indomethacin Binding with Human Serum Albumin Considering Primary and Secondary Binding Sites. Molecules, 28(7), 2979. https://doi.org/10.3390/molecules28072979