

Density Functional Theory Study of CO2 Hydrogenation on Transition-Metal-Doped Cu(211) Surfaces

 , , and
, , and 
Abstract
1. Introduction
2. Results and Discussion
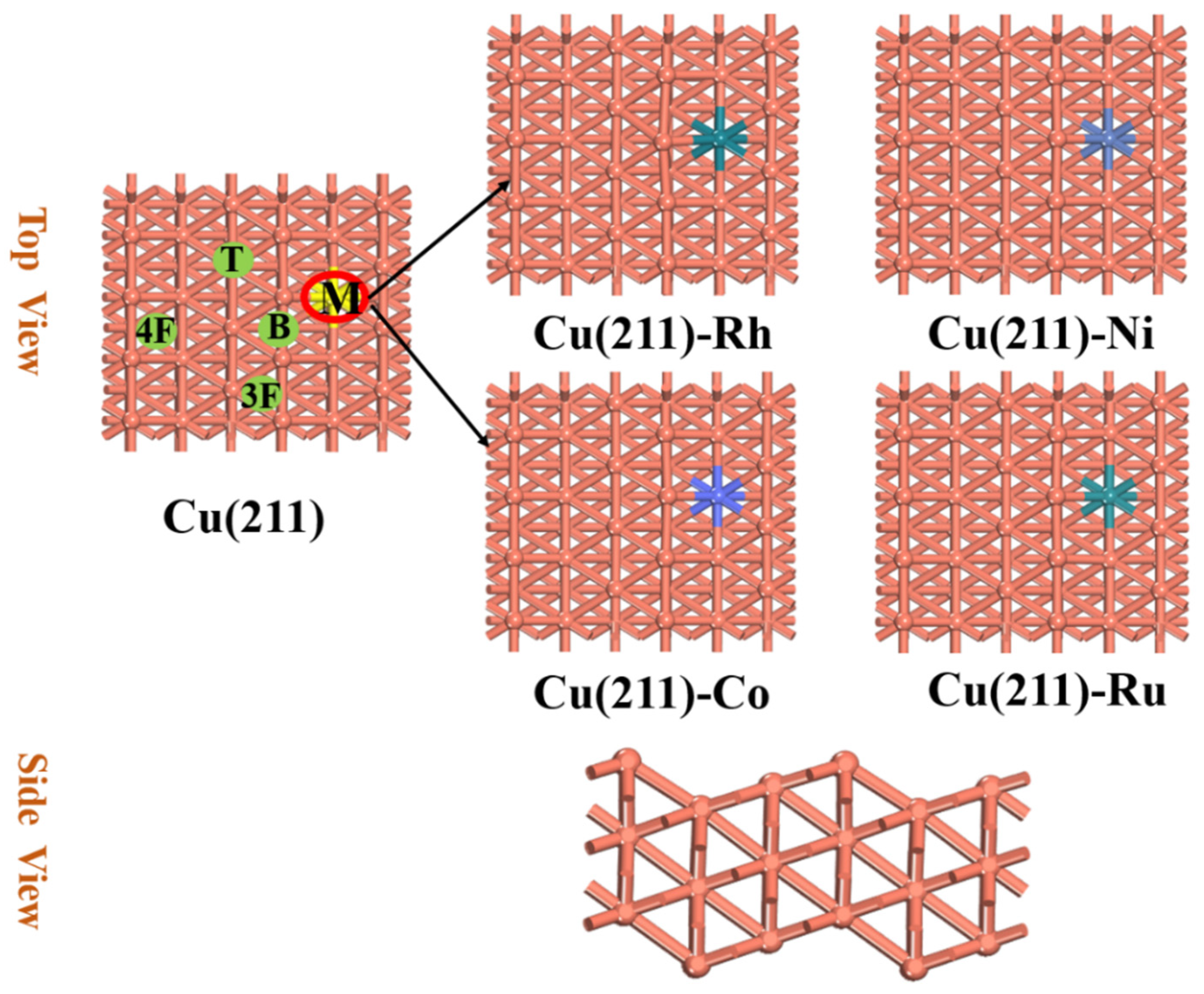
2.1. Formation Energies of Cu(211)-M Surfaces
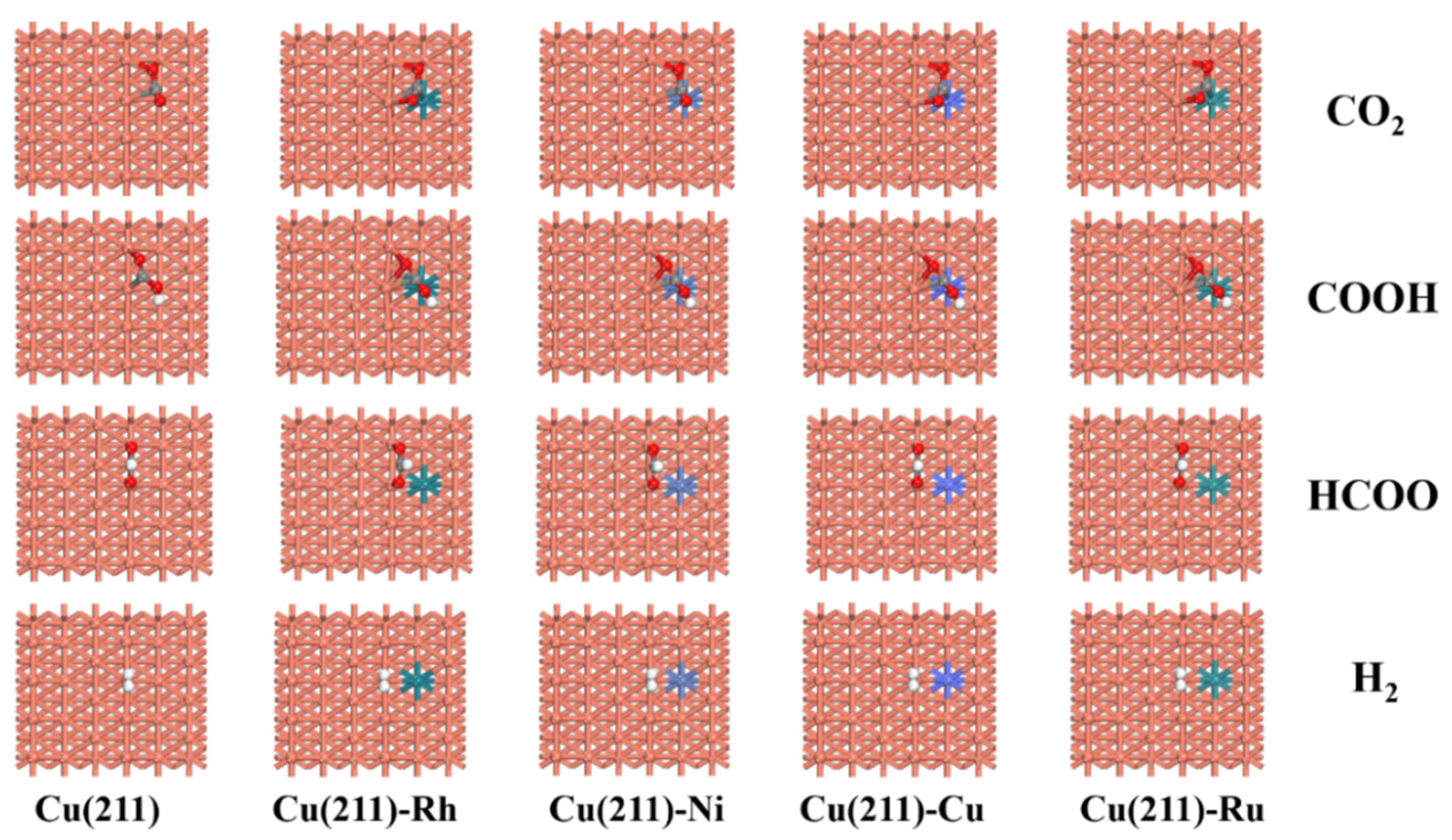
2.2. Adsorption of Intermediates on Cu(211)-M Surfaces
2.3. H2 Dissociation
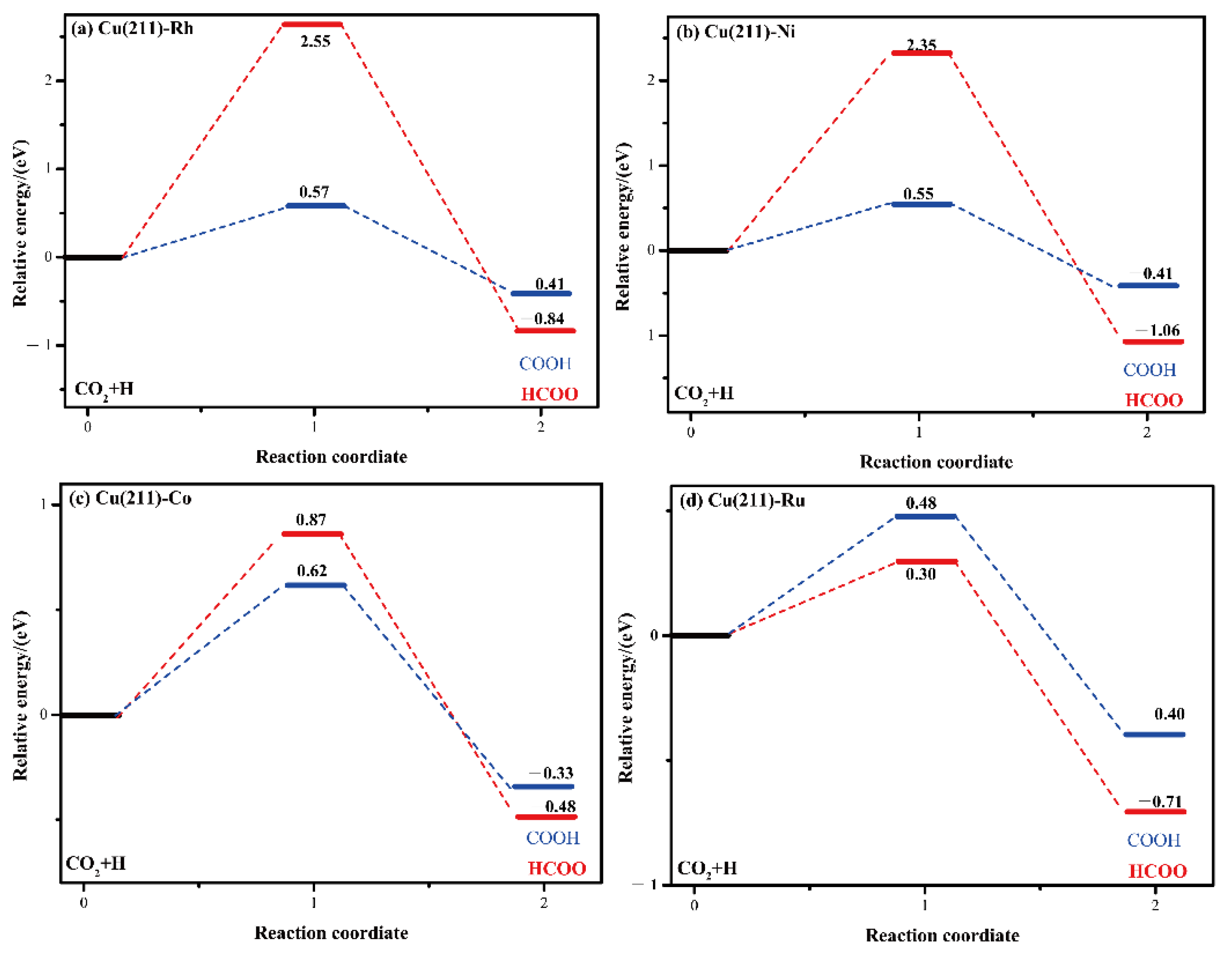
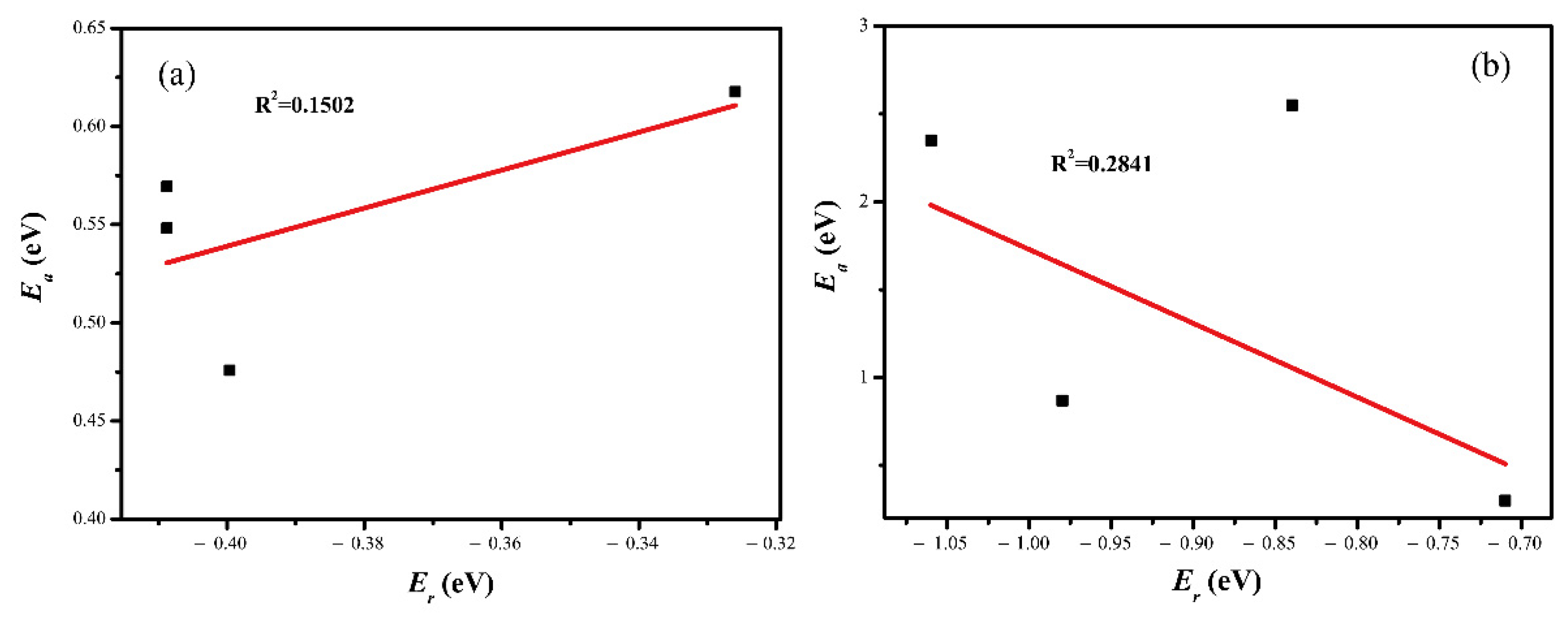
2.4. CO2 Activation
2.5. Electronic Structure Analysis
3. Materials and Methods
3.1. Model
3.2. Calculation Method
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Dimitriou, I.; García-Gutiérrez, P.; Elder, R.H.; Cuéllar-Franca, R.M.; Azapagic, A.; Allen, R.W.K. Carbon dioxide utilisation for production of transport fuels: Process and economic analysis. Energy Environ. Sci. 2015, 8, 1775–1789. [Google Scholar] [CrossRef]
- Sun, R.; Liao, Y.; Bai, S.-T.; Zheng, M.; Zhou, C.; Zhang, T.; Sels, B.F. Heterogeneous catalysts for CO2 hydrogenation to formic acid/formate: From nanoscale to single atom. Energy Environ. Sci. 2021, 14, 1247–1285. [Google Scholar] [CrossRef]
- Bai, X.; Zhao, X.; Zhang, Y.; Ling, C.; Zhou, Y.; Wang, J.; Liu, Y. Dynamic stability of copper single-atom catalysts under working conditions. J. Am. Chem. Soc. 2022, 144, 17140–17148. [Google Scholar] [CrossRef]
- Zhou, W.; Cheng, K.; Kang, J.; Zhou, C.; Subramanian, V.; Zhang, Q.; Wang, Y. New horizon in C1 chemistry: Breaking the selectivity limitation in transformation of syngas and hydrogenation of CO2 into hydrocarbon chemicals and fuels. Chem. Soc. Rev. 2019, 48, 3193–3228. [Google Scholar] [CrossRef]
- Yang, J.; Wang, Z.; Jiang, J.; Chen, W.; Liao, F.; Ge, X.; Zhou, X.; Chen, M.; Li, R.; Xue, Z.; et al. In-situ polymerization induced atomically dispersed manganese sites as cocatalyst for CO2 photoreduction into synthesis gas. Nano Energy 2020, 76, 105059. [Google Scholar] [CrossRef]
- Jiang, J.-C.; Chen, J.-C.; Zhao, M.-d.; Yu, Q.; Wang, Y.-G.; Li, J. Rational design of copper-based single-atom alloy catalysts for electrochemical CO2 reduction. Nano Res. 2022, 15, 7116–7123. [Google Scholar] [CrossRef]
- Podrojková, N.; Sans, V.; Oriňak, A.; Oriňaková, R. Recent developments in the modelling of heterogeneous catalysts for CO2 conversion to chemicals. ChemCatChem 2020, 12, 1802–1825. [Google Scholar] [CrossRef]
- Wang, L.X.; Wang, L.; Xiao, F.S. Tuning product selectivity in CO2 hydrogenation over metal-based catalysts. Chem. Sci. 2021, 12, 14660–14673. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, X.; Wang, H.; Cao, L.; Huang, C.; Li, S.; Zhang, X.; Guan, Q.; Shao, X.; Lu, J. Size-dependent strong metal–support interaction in Pd/ZnO catalysts for hydrogenation of CO2 to methanol. Catal. Sci. Technol. 2021, 11, 4398–4405. [Google Scholar] [CrossRef]
- Zhang, Z.; Shen, C.; Sun, K.; Jia, X.; Ye, J.; Liu, C.-j. Advances in studies of the structural effects of supported Ni catalysts for CO2 hydrogenation: From nanoparticle to single atom catalyst. J. Mater. Chem. A 2022, 10, 5792–5812. [Google Scholar] [CrossRef]
- Li, K.; Chen, J.G. CO2 hydrogenation to methanol over ZrO2-containing catalysts: Insights into ZrO2 induced synergy. ACS Catal. 2019, 9, 7840–7861. [Google Scholar] [CrossRef]
- Murthy, P.S.; Liang, W.; Jiang, Y.; Huang, J. Cu-based nanocatalysts for CO2 hydrogenation to methanol. Energy Fuels 2021, 35, 8558–8584. [Google Scholar] [CrossRef]
- Zhao, H.; Yu, R.; Ma, S.; Chen, Y.; Xu, K.; Fang, Y.; Zhu, C.; Liu, X.; Tang, Y.; Wu, L.; et al. The role of Cu1-O3 species in single-atom Cu catalyst for directional methanol synthesis from CO2 hydrogenation. Nat. Catal. 2021, 8, 818–831. [Google Scholar]
- Tan, Q.; Shi, Z.; Wu, D. CO2 hydrogenation to methanol over a highly active Cu–Ni/CeO2–nanotube catalyst. Ind. Eng. Chem. Res. 2018, 57, 10148–10158. [Google Scholar] [CrossRef]
- Liu, C.; Yang, B.; Tyo, E.; Seifert, S.; DeBartolo, J.; von Issendorff, B.; Zapol, P.; Vajda, S.; Curtiss, L.A. Carbon dioxide conversion to methanol over size-selected Cu4 clusters at low pressures. J. Am. Chem. Soc. 2015, 137, 8676–8679. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Yang, B. Significance of surface formate coverage on the reaction kinetics of methanol synthesis from CO2 hydrogenation over Cu. ACS Catal. 2017, 7, 7187–7195. [Google Scholar] [CrossRef]
- Mandal, S.C.; Rawat, K.S.; Garg, P.; Pathak, B. Hexagonal Cu(111) monolayers for selective CO2 hydrogenation to CH3OH: Insights from density functional theory. ACS Appl. Nano Mater. 2019, 2, 7686–7695. [Google Scholar] [CrossRef]
- Qiu, M.; Li, Y.; Zhang, Y. The mechanism for CO2 reduction over Fe-modified Cu(100) surfaces with thermodynamics and kinetics: A DFT study. RSC Adv. 2020, 10, 32569–32580. [Google Scholar] [CrossRef]
- Yang, Y.; White, M.G.; Liu, P. Theoretical study of methanol synthesis from CO2 hydrogenation on metal-doped Cu(111) surfaces. J. Phys. Chem. C 2011, 116, 248–256. [Google Scholar] [CrossRef]
- Chen, X.; Su, X.; Su, H.-Y.; Liu, X.; Miao, S.; Zhao, Y.; Sun, K.; Huang, Y.; Zhang, T. Theoretical insights and the corresponding construction of supported metal catalysts for highly selective CO2 to CO conversion. ACS Catal. 2017, 7, 4613–4620. [Google Scholar] [CrossRef]
- Shyam Kattel, P.J.R.; Jingguang, G.C.; José, A.; Rodriguez, P.L. Active sites for CO2 hydrogenation to methanol on Cu/ZnO catalysts. Science 2017, 355, 1296–1299. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Ji, W.; Russell, C.K.; Zhang, Y.; Fan, M.; Shen, Z. A new and different insight into the promotion mechanisms of Ga for the hydrogenation of carbon dioxide to methanol over a Ga-doped Ni(211) bimetallic catalyst. Nanoscale 2019, 11, 9969–9979. [Google Scholar] [CrossRef] [PubMed]
- Graciani, J.; Mudiyanselage, K.; Xu, F.; Baber, A.E.; Evans, J.; Senanayake, S.D.; Stacchiola, D.J.; Liu, P.; Hrbek, J.; Sanz, J.F.; et al. Highly active copper-ceria and copper-ceria-titania catalysts for methanol synthesis from CO2. Science 2014, 345, 546–549. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Evans, J.; Rodriguez, J.A.; White, M.G.; Liu, P. Fundamental studies of methanol synthesis from CO2 hydrogenation on Cu(111), Cu clusters, and Cu/ZnO(0001). Phys. Chem. Chem. Phys. 2010, 12, 9909–9917. [Google Scholar] [CrossRef] [PubMed]
- Nie, X.; Jiang, X.; Wang, H.; Luo, W.; Janik, M.J.; Chen, Y.; Guo, X.; Song, C. Mechanistic understanding of alloy effect and water promotion for Pd-Cu bimetallic catalysts in CO2 hydrogenation to methanol. ACS Catal. 2018, 8, 4873–4892. [Google Scholar] [CrossRef]
- Ma, L.; Zhao, W.; Wang, B.; Ling, L.; Zhang, R. CO2 activation and conversion on Cu catalysts: Revealing the role of Cu surface defect types in tuning the activity and selectivity. Fuel 2022, 313, 122686. [Google Scholar] [CrossRef]
- Zhao, P.; Cao, Z.; Liu, X.; Ren, P.; Cao, D.B.; Xiang, H.; Jiao, H.; Yang, Y.; Li, Y.-W.; Wen, X.D. Morphology and reactivity evolution of HCP and FCC Ru nanoparticles under CO atmosphere. ACS Catal. 2019, 9, 2768–2776. [Google Scholar] [CrossRef]
- Liao, F.; Huang, Y.; Ge, J.; Zheng, W.; Tedsree, K.; Collier, P.; Hong, X.; Tsang, S.C. Morphology-dependent interactions of ZnO with Cu nanoparticles at the materials’ interface in selective hydrogenation of CO2 to CH3OH. Angew. Chem. Int. Ed. 2011, 50, 2162–2165. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, Y.; Otroshchenko, T.; Han, S.; Lund, H.; Rodemerck, U.; Linke, D.; Jiao, H.; Jiang, G.; Kondratenko, E.V. The effect of phase composition and crystallite size on activity and selectivity of ZrO2 in non-oxidative propane dehydrogenation. J. Catal. 2019, 371, 313–324. [Google Scholar] [CrossRef]
- Nie, L.; Li, Z.; Kuang, T.; Lyu, S.; Liu, S.; Zhang, Y.; Peng, B.; Li, J.; Wang, L. Role of well-defined cobalt crystal facets in Fischer-Tropsch synthesis: A combination of experimental and theoretical studies. Chem. Commun. 2019, 55, 10559–10562. [Google Scholar] [CrossRef]
- Zhang, R.; Wang, G.; Wang, B. Insights into the mechanism of ethanol formation from syngas on Cu and an expanded prediction of improved Cu-based catalyst. J. Catal. 2013, 305, 238–255. [Google Scholar] [CrossRef]
- Liu, J.X.; Su, H.Y.; Sun, D.P.; Zhang, B.Y.; Li, W.X. Crystallographic dependence of CO activation on cobalt catalysts: HCP versus FCC. J. Am. Chem. Soc. 2013, 135, 16284–16287. [Google Scholar] [CrossRef] [PubMed]
- Mavrikakis, M.; Bäumer, M.; Freund, H.J.; Nørskov, J.K. Structure sensitivity of CO dissociation on Rh surfaces. Catal. Lett. 2002, 81, 153–156. [Google Scholar] [CrossRef]
- Behrens, M.; Studt, F.; Kasatkin, I.; Kühl, S.; Hävecker, M.; Abild-Pedersen, F.; Zander, S.; Girgsdies, F.; Kurr, P.; Kniep, B.-L.; et al. The active site of methanol synthesis over CuZnO/Al2O3 industrial catalysts. Science 2012, 336, 893–897. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, C.; Tian, P.; Xu, M.; Cao, C.; Yang, Z.; Zhu, M.; Xu, J. Revealing the effect of sodium on iron-based catalysts for CO2 hydrogenation: Insights from calculation and experiment. J. Phys. Chem. C 2021, 125, 7637–7646. [Google Scholar] [CrossRef]
- Zhang, M.; Dou, M.; Yu, Y. Theoretical study of the promotional effect of ZrO2 on In2O3 catalyzed methanol synthesis from CO2 hydrogenation. Appl. Surf. Sci. 2018, 433, 780–789. [Google Scholar] [CrossRef]
- Gong, H.; He, Y.; Yin, J.; Liu, S.; Qing, M.; Peng, Q.; Huo, C.-F.; Wang, H.; Yang, Y.; Wen, X.-D. Electronic effects of transition metal dopants on Fe(100) and Fe5C2(100) surfaces for CO activation. Catal. Sci. Technol. 2020, 10, 2047–2056. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, J.-X.; Zijlstra, B.; Filot, I.A.W.; Zhou, Z.; Sun, S.; Hensen, E.J.M. Optimum Cu nanoparticle catalysts for CO2 hydrogenation towards methanol. Nano Energy 2018, 43, 200–209. [Google Scholar] [CrossRef]
- Bai, S.T.; De Smet, G.; Liao, Y.; Sun, R.; Zhou, C.; Beller, M.; Maes, B.U.W.; Sels, B.F. Homogeneous and heterogeneous catalysts for hydrogenation of CO2 to methanol under mild conditions. Chem. Soc. Rev. 2021, 50, 4259–4298. [Google Scholar] [CrossRef] [PubMed]
- Chi, S.; Huang, H.; Yu, Y.; Zhang, M. Mechanism insight into MnO for CHx(x = 1 to 3) hydrogenation and C1-C1 coupling processes on Co(0001) surface: A DFT and kMC study. Appl. Surf. Sci. 2022, 586, 152840. [Google Scholar] [CrossRef]
- Chen, B.; Wang, D.; Duan, X.; Liu, W.; Li, Y.; Qian, G.; Yuan, W.; Holmen, A.; Zhou, X.; Chen, D. Charge-tuned CO activation over a χ-Fe5C2 Fischer–Tropsch Catalyst. ACS Catal. 2018, 8, 2709–2714. [Google Scholar] [CrossRef]
- Bronsted, J.N. Acid and basic catalysis. Chem. Rev. 1928, 5, 231–338. [Google Scholar] [CrossRef]
- Filot, I.A.W.; Fariduddin, F.; Broos, R.J.P.; Zijlstra, B.; Hensen, E.J.M. A quantum-chemical DFT study of CO dissociation on Fe-promoted stepped Rh surfaces. Catal. Today 2016, 275, 111–118. [Google Scholar] [CrossRef]
- Xie, S.; Zhang, W.; Lan, X.; Lin, H. CO2 Reduction to methanol in the liquid phase: A review. ChemSusChem 2020, 13, 6141–6159. [Google Scholar] [PubMed]
- Wesselbaum, S.; Moha, V.; Meuresch, M.; Brosinski, S.; Thenert, K.M.; Kothe, J.; Stein, T.V.; Englert, U.; Holscher, M.; Klankermayer, J.; et al. Hydrogenation of carbon dioxide to methanol using a homogeneous ruthenium-Triphos catalyst: From mechanistic investigations to multiphase catalysis. Chem. Sci. 2015, 6, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Kresseav, G.; Furthmiiller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Sheppard, D.; Xiao, P.; Chemelewski, W.; Johnson, D.D.; Henkelman, G. A generalized solid-state nudged elastic band method. J. Chem. Phys. 2012, 136, 074103. [Google Scholar] [CrossRef]
- Sheppard, D.; Terrell, R.; Henkelman, G. Optimization methods for finding minimum energy paths. J. Chem. Phys. 2008, 128, 134106. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Surface | Formation Energy | q |
|---|---|---|
| Cu (211)-Rh | −2.75 | −0.32 |
| Cu (211)-Ni | −1.62 | −0.03 |
| Cu (211)-Co | −2.28 | 0.02 |
| Cu (211)-Ru | −4.02 | −0.16 |
| CO2 | COOH | HCOO | H2 | |||||
|---|---|---|---|---|---|---|---|---|
| Surface | Eads | q | Eads | q | Eads | q | Eads | q |
| Cu(211) | −0.26 | 0.80 | −1.76 | 0.39 | −3.32 | 0.65 | −0.30 | 0.02 |
| Cu(211)-Rh | −0.31 | 0.84 | −2.37 | 0.51 | −3.16 | 0.61 | −0.18 | 0.01 |
| Cu(211)-Ni | −0.34 | 0.85 | −2.05 | 0.43 | −3.20 | 0.62 | −0.11 | −0.02 |
| Cu(211)-Co | −0.38 | 0.97 | −2.35 | 0.50 | −3.32 | 0.65 | −0.01 | −0.02 |
| Cu(211)-Ru | −0.35 | 0.91 | −2.61 | 0.55 | −3.18 | 0.61 | −0.01 | −0.02 |
| Surface | Activation Barrier | Reaction Energy | dH-H | v(cm−1) |
|---|---|---|---|---|
| Cu(211) [26] | 0.44 | −0.51 | 1.3349 | 1054i |
| Cu (211)-Rh | 0.41 | −0.84 | 1.321 | 1186i |
| Cu (211)-Ni | 0.42 | −0.57 | 1.307 | 1254i |
| Cu (211)-Co | 0.19 | −0.86 | 1.320 | 1130i |
| Cu (211)-Ru | 0.22 | −1.01 | 0.969 | 465i |
| Surface | Activation Barrier | Reaction Energy | dC-H | v(cm−1) |
|---|---|---|---|---|
| Cu(211) | 2.02 | 0.50 | / | / |
| Cu (211)-Rh | 0.57 | −0.41 | 1.481 | 1189i |
| Cu (211)-Ni | 0.55 | −0.41 | 1.491 | 1270i |
| Cu (211)-Co | 0.62 | −0.33 | 1.467 | 1254i |
| Cu (211)-Ru | 0.48 | −0.40 | 2.362 | 1070i |
| Surface | Activation Barrier | Reaction Energy | dC-H | v(cm−1) |
|---|---|---|---|---|
| Cu(211) [26] | 0.74 | 0.46 | / | / |
| Cu(211)-Rh | 2.55 | −0.84 | 4.994 | 1001.5i |
| Cu(211)-Ni | 2.35 | −1.06 | 3.206 | 498.5i |
| Cu(211)-Co | 0.87 | −0.98 | 1.820 | 867.7i |
| Cu(211)-Ru | 0.30 | −0.71 | 2.721 | 1160i |
| Surface Slabs | Cut-Energy | k-Points | Eads (eV) |
|---|---|---|---|
| Cu(211)-Ru | 400 | 3 × 2 × 1 | −2.61 |
| 400 | 3 × 3 × 1 | −2.57 | |
| 400 | 4 × 4 × 1 | −2.59 | |
| Cu(211)-Ru | 400 | 3 × 2 × 1 | −2.61 |
| 500 | 3 × 2 × 1 | −2.57 | |
| 600 | 3 × 2 × 1 | −2.57 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Yu, M.; Zhang, X.; Gao, Y.; Liu, J.; Zhang, X.; Gong, C.; Cao, X.; Ju, Z.; Peng, Y. Density Functional Theory Study of CO2 Hydrogenation on Transition-Metal-Doped Cu(211) Surfaces. Molecules 2023, 28, 2852. https://doi.org/10.3390/molecules28062852
Wang Y, Yu M, Zhang X, Gao Y, Liu J, Zhang X, Gong C, Cao X, Ju Z, Peng Y. Density Functional Theory Study of CO2 Hydrogenation on Transition-Metal-Doped Cu(211) Surfaces. Molecules. 2023; 28(6):2852. https://doi.org/10.3390/molecules28062852
Chicago/Turabian StyleWang, Yushan, Mengting Yu, Xinyi Zhang, Yujie Gao, Jia Liu, Ximing Zhang, Chunxiao Gong, Xiaoyong Cao, Zhaoyang Ju, and Yongwu Peng. 2023. "Density Functional Theory Study of CO2 Hydrogenation on Transition-Metal-Doped Cu(211) Surfaces" Molecules 28, no. 6: 2852. https://doi.org/10.3390/molecules28062852
APA StyleWang, Y., Yu, M., Zhang, X., Gao, Y., Liu, J., Zhang, X., Gong, C., Cao, X., Ju, Z., & Peng, Y. (2023). Density Functional Theory Study of CO2 Hydrogenation on Transition-Metal-Doped Cu(211) Surfaces. Molecules, 28(6), 2852. https://doi.org/10.3390/molecules28062852