
Heterologous Biosynthesis of Myxobacterial Antibiotic Miuraenamide A


Abstract
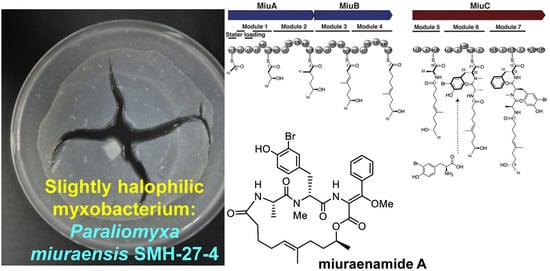
1. Introduction
2. Results
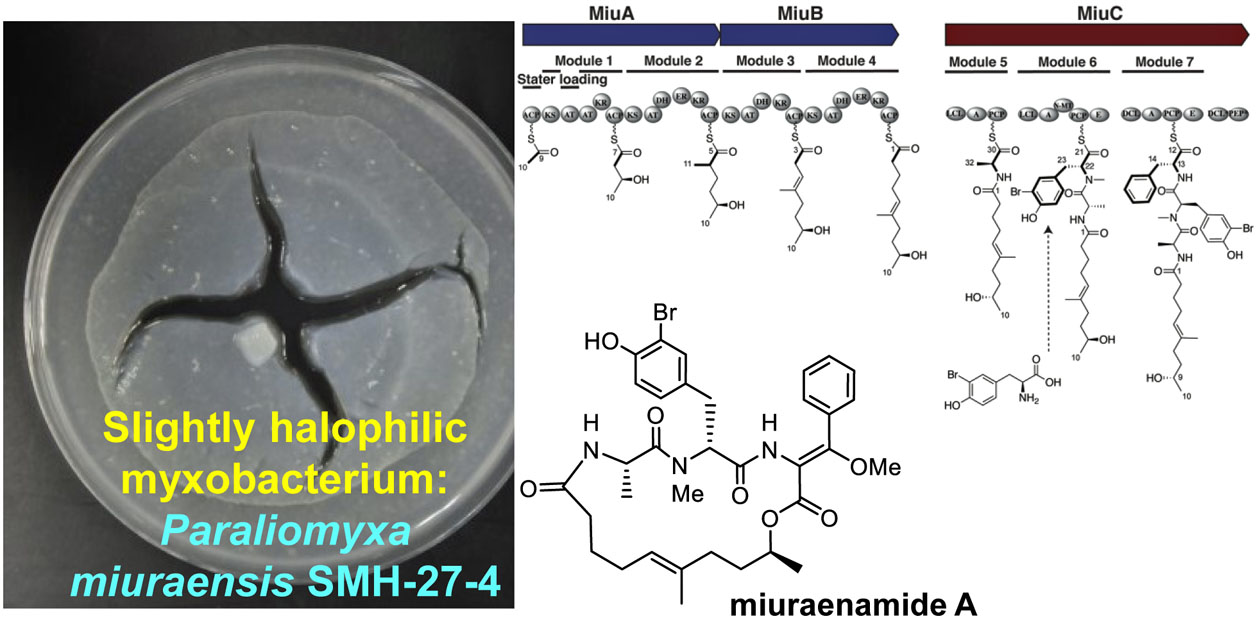
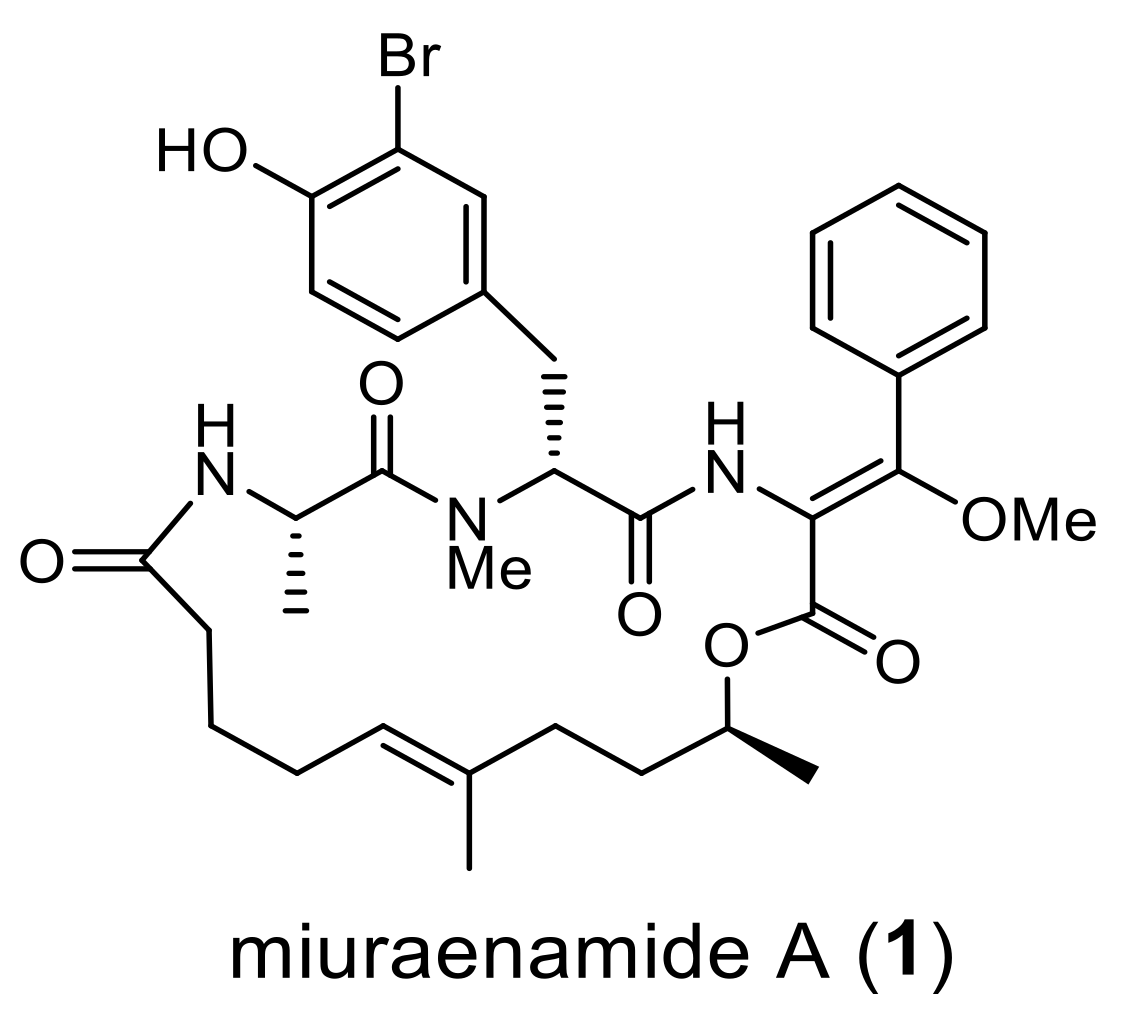
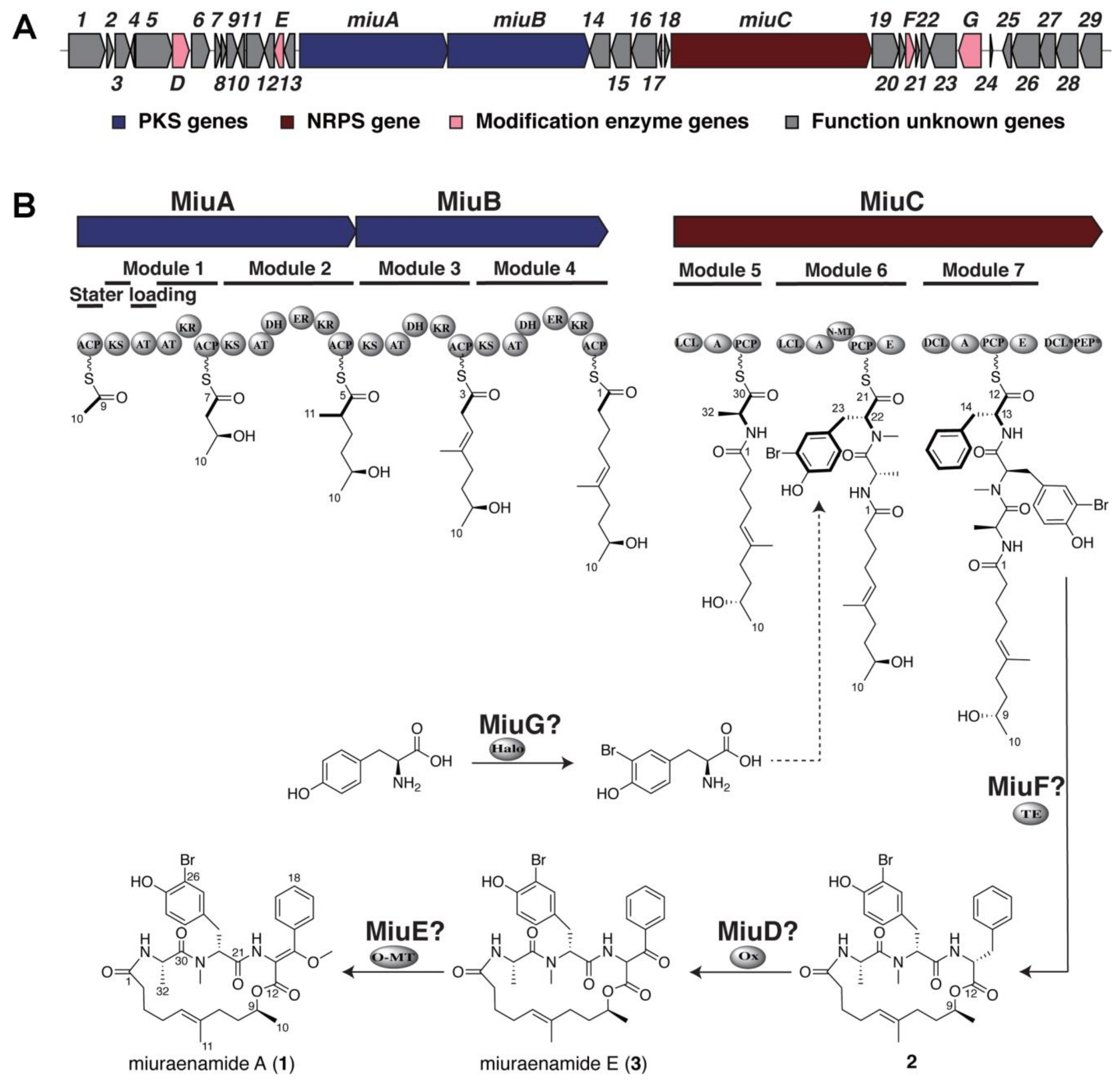
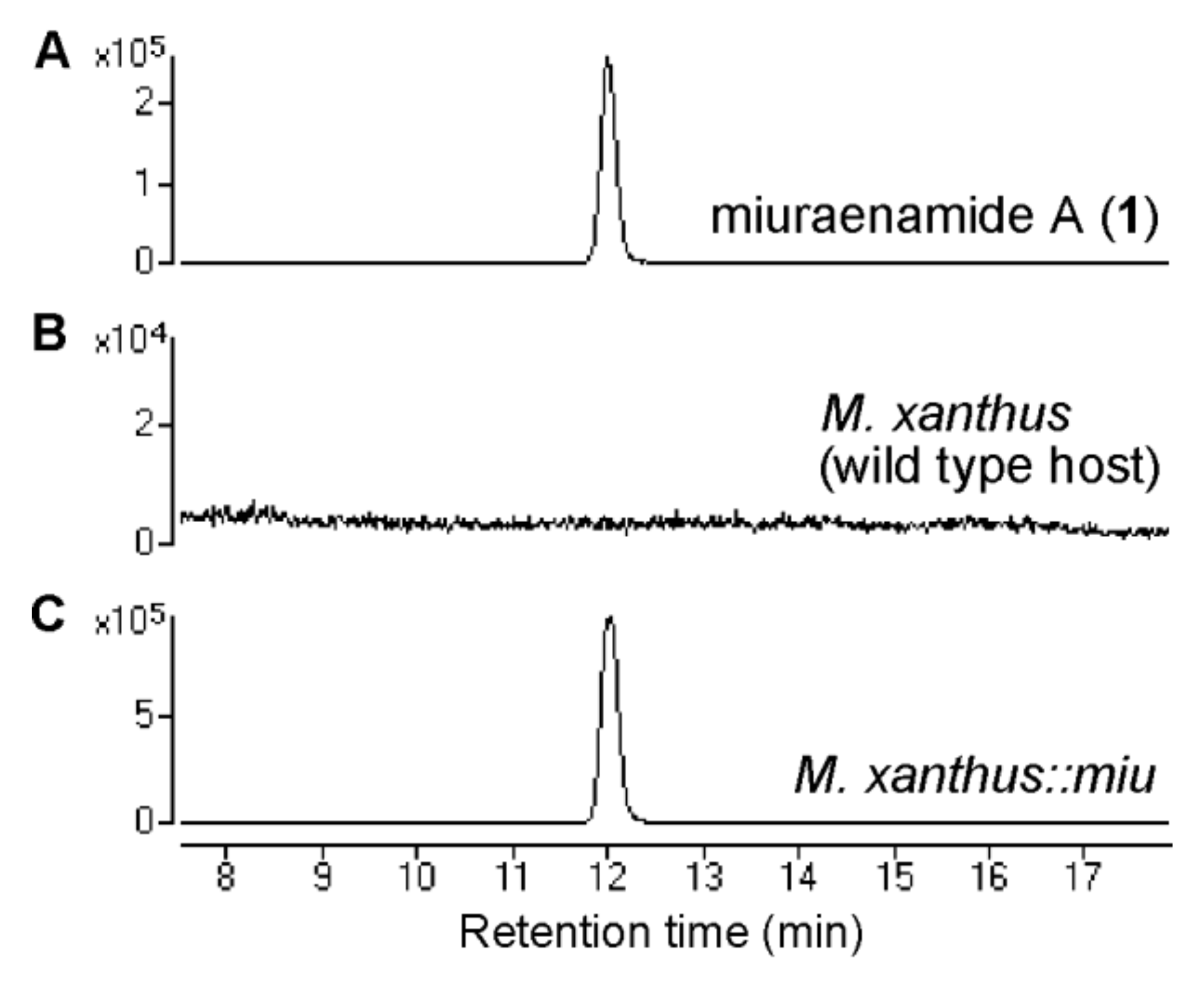
2.1. Identification and Heterologous Expression of BGC for Miuraenamide A
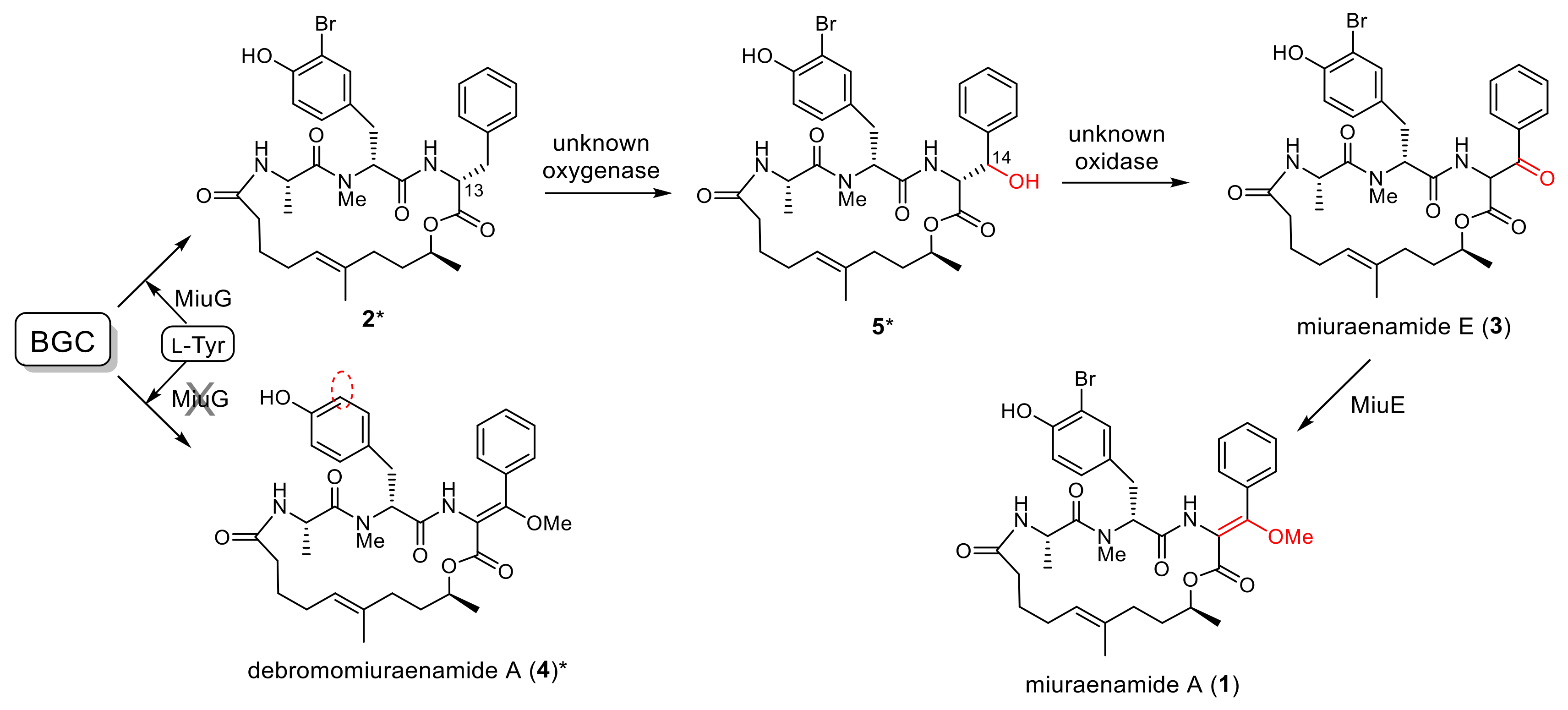
2.2. Proposed Mechanism for the Biosynthesis of Miuraenamide A
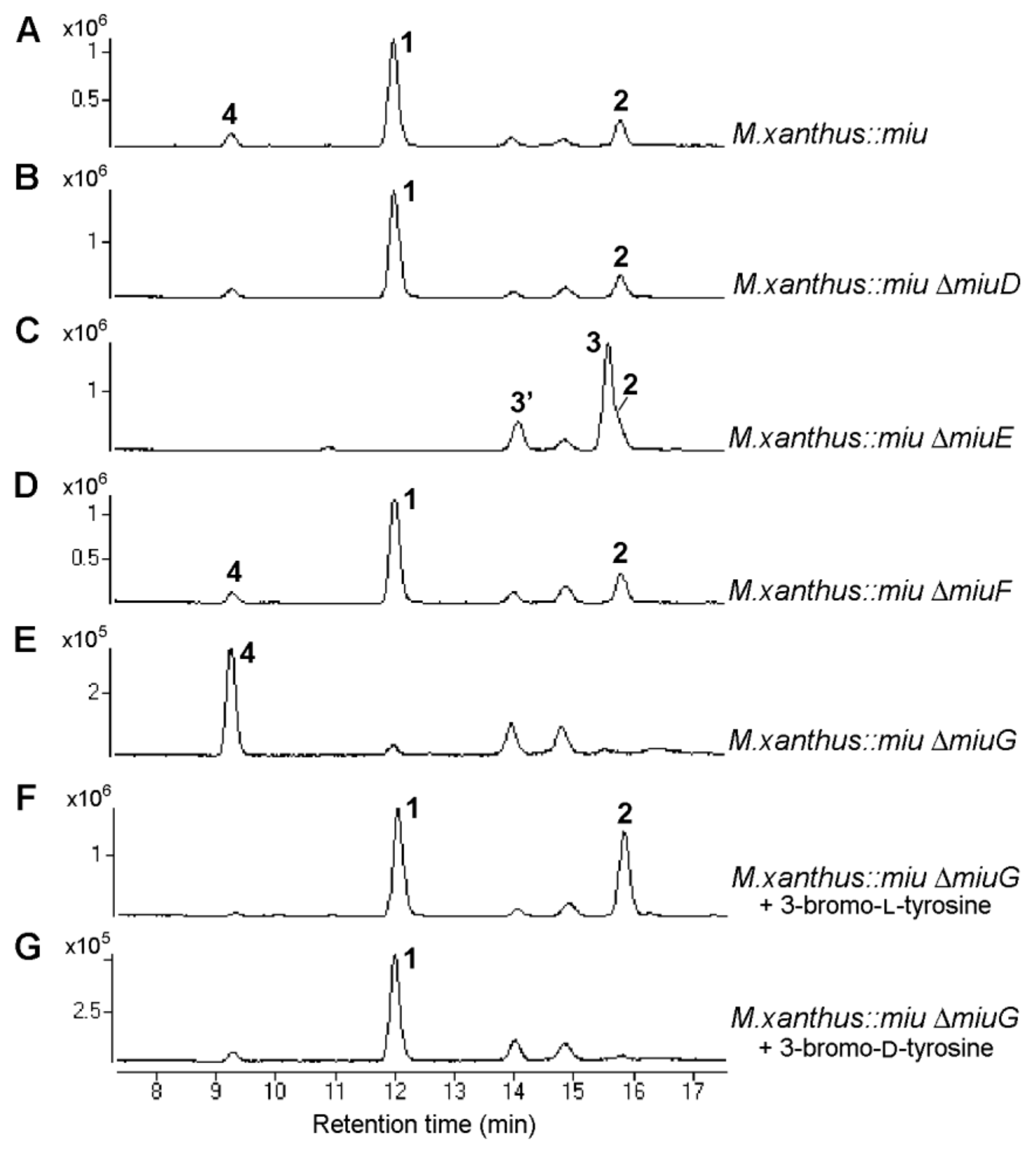
2.3. Verification of Modification Enzyme Genes
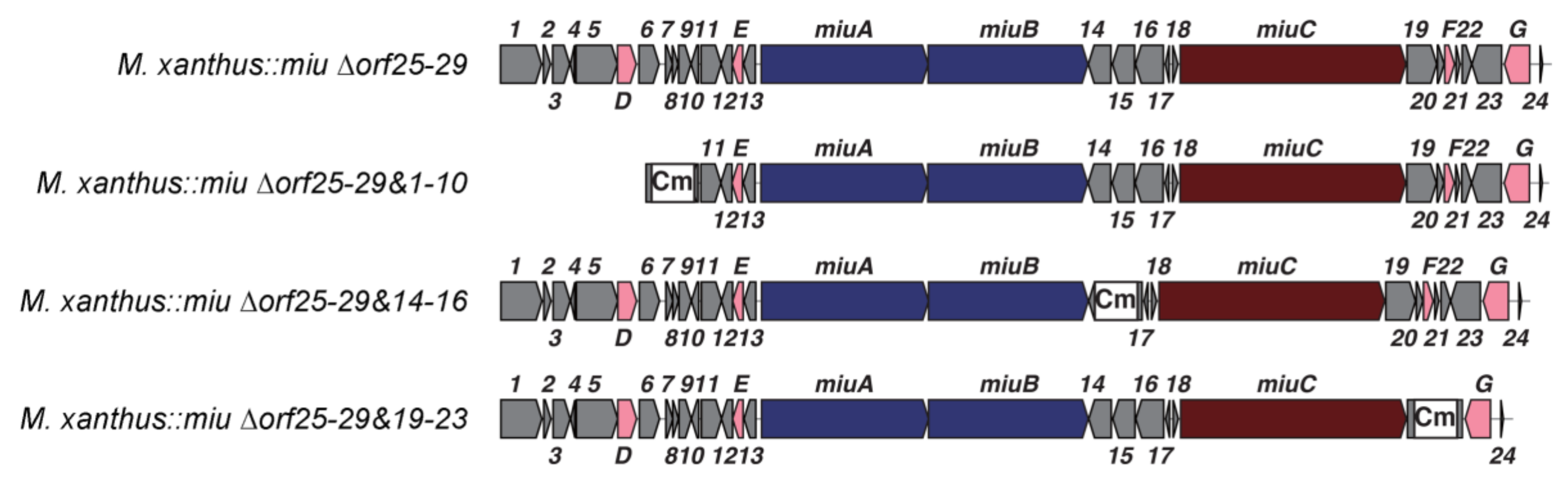
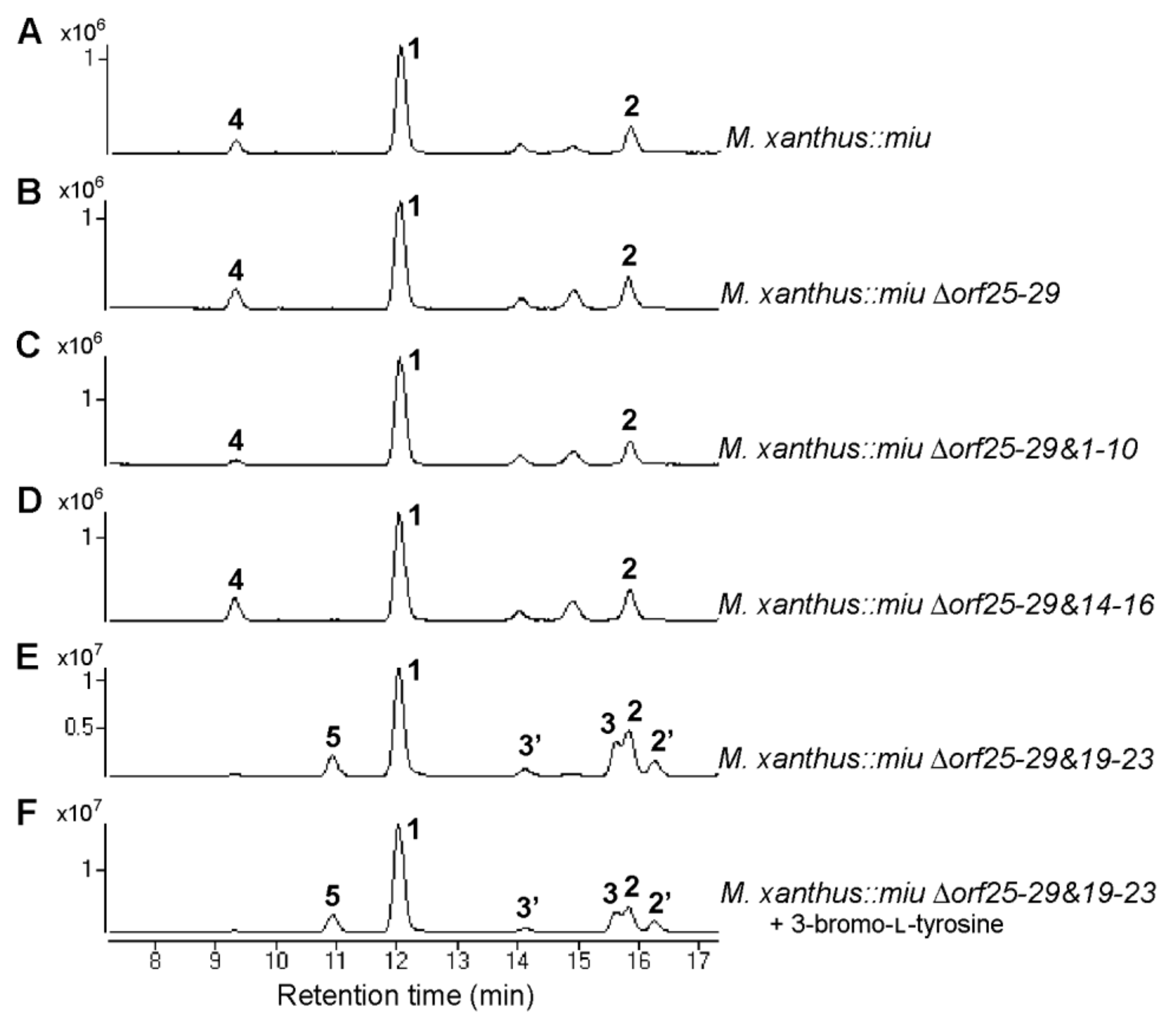
2.4. Verification of Unknown Genes Using Multigene Disruption
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains and Culture Conditions
4.2. Chemicals
4.3. PCR and Products Purification
4.4. Construction and Screening of a Genomic BAC Library
4.5. Construction of Red/ET Recombination Modification Cassette 5TA-KanR
4.6. Modification of BAC Vector to miu BAC via Red/ET Rcombination
4.7. Gene Disruption
4.8. Construction of M. xanthus Transformants
4.9. Production of Miuraenamide A (1) Using M. xanthus Heterologous Mutants
4.10. LC-MS Analysis of Miuraenamide A (1) and Congeners (2–5)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Velicer, G.J.; Vos, M. Sociobiology of the myxobacteria. Annu. Rev. Microbiol. 2009, 63, 599–623. [Google Scholar] [CrossRef]
- Kaiser, D.; Robinson, M.; Kroos, L. Myxobacteria, polarity, and multicellular morphogenesis. Cold Spring Harb. Perspect. Biol. 2010, 2, a000380. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Dorado, J.; Marcos-Torres, F.J.; García-Bravo, E.; Moraleda-muñoz, A.; Pérez, J. Myxobacteria: Moving, killing, feeding, and surviving together. Front. Microbiol. 2016, 7, 781. [Google Scholar] [CrossRef]
- Wenzel, S.C.; Müller, R. Myxobacteria—‘Microbial factories’ for the production of bioactive secondary metabolites. Mol. BioSyst. 2009, 5, 567–574. [Google Scholar] [CrossRef]
- Diez, J.; Martinez, J.P.; Mestres, J.; Sasse, F.; Frank, R.; Meyerhans, A. Myxobacteria: Natural pharmaceutical factories. Microb. Cell Fact. 2012, 11, 52. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hug, J.J.; Müller, R. Host development for heterologous expression and biosynthetic studies of myxobacterial natural products. In Comprehensive Natural Products III, 3rd ed.; Liu, H., Begley, T.P., Eds.; Elsevier: San Diego, CA, USA, 2020; Volume 6, pp. 149–216. [Google Scholar]
- Dávila-Céspedes, A.; Hufendiek, P.; Crüsemann, M.; Schäberle, T.F.; König, G.M. Marine-derived myxobacteria of the suborder Nannocystineae: An underexplored source of structurally intriguing and biologically active metabolites. Beilstein J. Org. Chem. 2016, 12, 969–984. [Google Scholar] [CrossRef]
- Albataineh, H.; Stevens, D.C. Marine myxobacteria: A few good halophiles. Mar. Drugs 2018, 16, 209. [Google Scholar] [CrossRef]
- Gemperlein, K.; Zaburannyi, N.; Garcia, R.; La Clair, J.J.; Müller, R. Metabolic and biosynthetic diversity in marine myxobacteria. Mar. Drugs 2018, 16, 314. [Google Scholar] [CrossRef] [PubMed]
- Moghaddam, J.A.; Crüsemann, M.; Alanjary, M.; Harms, H.; Dávila-Céspedes, A.; Blom, J.; Poehlein, A.; Ziemert, N.; König, G.M.; Schäberle, T.F. Analysis of the genome and metabolome of marine myxobacteria reveals high potential for biosynthesis of novel specialized metabolites. Sci. Rep. 2018, 8, 16600. [Google Scholar] [CrossRef]
- Iizuka, T.; Fudou, R.; Jojima, Y.; Ogawa, S.; Yamanaka, S.; Inukai, Y.; Ojika, M. Miuraenamides A and B, novel antimicrobial cyclic depsipeptides from a new slightly halophilic myxobacterium: Taxonomy, production, and biological properties. J. Antibiot. 2006, 59, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Sumiya, E.; Shimogawa, H.; Sasaki, H.; Tsutsumi, M.; Yoshita, K.; Ojika, M.; Suenaga, K.; Uesugi, M. Cell-morphology profiling of a natural product library identifies bisebromoamide and miuraenamide A as actin filament stabilizers. ACS Chem. Biol. 2011, 6, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Moser, C.; Rüdiger, D.; Förster, F.; von Blume, J.; Yu, P.; Kuster, B.; Kazmaier, U.; Vollmar, A.M.; Zahler, S. Persistent inhibition of pore-based cell migration by sub-toxic doses of miuraenamide, an actin filament stabilizer. Sci. Rep. 2017, 7, 16407. [Google Scholar] [CrossRef] [PubMed]
- Gegenfurtner, F.A.; Zisis, T.; Al Danaf, N.; Schrimpf, W.; Kliesmete, Z.; Ziegenhain, C.; Enard, W.; Kazmaier, U.; Lamb, D.C.; Vollmar, A.M.; et al. Transcriptional effects of actin-binding compounds: The cytoplasm sets the tone. Cell. Mol. Life Sci. 2018, 75, 4539–4555. [Google Scholar] [CrossRef]
- Wang, S.; Crevenna, A.H.; Ugur, I.; Marion, A.; Antes, I.; Kazmaier, U.; Hoyer, M.; Lamb, D.C.; Gegenfurtner, F.; Kliesmete, Z.; et al. Actin stabilizing compounds show specific biological effects due to their binding mode. Sci. Rep. 2019, 9, 9731. [Google Scholar] [CrossRef] [PubMed]
- Baltes, C.; Thalla, D.G.; Kazmaier, U.; Lautenschläger, F. Actin stabilization in cell migration. Front. Cell Dev. Biol. 2022, 10, 931880. [Google Scholar] [CrossRef]
- Karmann, L.; Schultz, K.; Herrmann, J.; Müller, R.; Kazmaier, U. Total syntheses and biological evaluation of miuraenamides. Angew. Chem. Int. Ed. 2015, 54, 4502–4507. [Google Scholar] [CrossRef] [PubMed]
- Ojima, D.; Yasui, A.; Tohyama, K.; Tokuzumi, K.; Toriihara, E.; Ito, K.; Iwasaki, A.; Tomura, T.; Ojika, M.; Suenaga, K. Total synthesis of miuraenamides A and D. J. Org. Chem. 2016, 81, 9886–9894. [Google Scholar] [CrossRef]
- Kappler, S.; Karmann, L.; Prudel, C.; Herrmann, J.; Caddeu, G.; Müller, R.; Vollmar, A.M.; Zahler, S.; Kazmaier, U. Synthesis and biological evaluation of modified miuraenamides. Eur. J. Org. Chem. 2018, 2018, 6952–6965. [Google Scholar] [CrossRef]
- Liu, Y.; Ojika, M. Genomic analysis of the rare slightly halophilic myxobacterium “Paraliomyxa miuraensis” SMH-27-4, the producer of the antibiotic miuraenamide A. Microorganisms 2023, 11, 371. [Google Scholar] [CrossRef]
- Simunovic, V.; Zapp, J.; Rachid, S.; Krug, D.; Meiser, P.; Müller, R. Myxovirescin A biosynthesis is directed by hybrid polyketide synthases/nonribosomal peptide synthetase, 3-hydroxy-3-methylglutaryl-CoA synthases, and trans-acting acyltransferases. Chembiochem 2006, 7, 1206–1220. [Google Scholar] [CrossRef]
- Ojika, M.; Inukai, Y.; Kito, Y.; Hirata, M.; Iizuka, T.; Fudou, R. Miuraenamides: Antimicrobial cyclic depsipeptides isolated from a rare and slightly halophilic myxobacterium. Chem. Asian J. 2008, 3, 126–133. [Google Scholar] [CrossRef]
- Ligon, J.; Hill, S.; Beck, J.; Zirkle, R.; Molnár, I.; Zawodny, J.; Money, S.; Schupp, T. Characterization of the biosynthetic gene cluster for the antifungal polyketide soraphen A from Sorangium cellulosum So Ce26. Gene 2002, 285, 257–267. [Google Scholar] [CrossRef]
- Silakowski, B.; Schairer, H.U.; Ehret, H.; Kunze, B.; Weinig, S.; Nordsiek, G.; Brandt, P.; Blöcker, H.; Höfle, G.; Beyer, S.; et al. New lessons for combinatorial biosynthesis from myxobacteria. the myxothiazol biosynthetic gene cluster of Stigmatella aurantiaca DW4/3-1. J. Biol. Chem. 1999, 274, 37391–37399. [Google Scholar] [CrossRef] [PubMed]
- Silakowski, B.; Nordsiek, G.; Kunze, B.; Blöcker, H.; Müller, R. Novel features in a combined polyketide synthase/non-ribosomal peptide synthetase: The myxalamid biosynthetic gene cluster of the myxobacterium Stigmatella aurantiaca Sga15. Chem. Biol. 2001, 8, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Rachid, S.; Krug, D.; Kunze, B.; Kochems, I.; Scharfe, M.; Zabriskie, T.M.; Blöcker, H.; Müller, R. Molecular and biochemical studies of chondramide formation—Highly cytotoxic natural products from Chondromyces crocatus Cm c5. Chem. Biol. 2006, 13, 667–681. [Google Scholar] [CrossRef]
- Agarwal, V.; EL Gamal, A.A.; Yamanaka, K.; Poth, D.; Kersten, R.D.; Schorn, M.; Allen, E.E.; Moore, B.S. Biosynthesis of polybrominated aromatic organic compounds by marine bacteria. Nat. Chem. Biol. 2014, 10, 640–647. [Google Scholar] [CrossRef]
- Chang, C.Y.; Lohman, J.R.; Huang, T.; Michalska, K.; Bigelow, L.; Rudolf, J.D.; Jedrzejczak, R.; Yan, X.; Ma, M.; Babnigg, G.; et al. Structural insights into the free-standing condensation enzyme SgcC5 catalyzing ester-bond formation in the biosynthesis of the enediyne antitumor antibiotic C-1027. ACS Biochem. 2018, 57, 3278–3288. [Google Scholar] [CrossRef]
- Hodgkin, J.; Kaiser, D. Cell-to-cell stimulation of movement in nonmotile mutants of Myxococcus. Proc. Natl. Acad. Sci. USA 1977, 74, 2938–2942. [Google Scholar] [CrossRef]
- McCord, T.J.; Smith, D.R.; Winters, D.W.; Grimes, J.F.; Hulme, K.L.; Robinson, L.Q.; Gage, L.D.; Davis, A.L. Synthesis and microbiological activities of some monohalogenated analogs of tyrosine. J. Med. Chem. 1975, 18, 26–29. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Sambrook, J. Molecular Cloning: A Laboratory Manual, 4th ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2012; pp. 21–25. [Google Scholar]
- Sun, Y.; Feng, Z.; Tomura, T.; Suzuki, A.; Miyano, S.; Tsuge, T.; Mori, H.; Suh, J.W.; Iizuka, T.; Fudou, R.; et al. Heterologous production of the marine myxobacterial antibiotic haliangicin and its unnatural analogues generated by engineering of the biochemical pathway. Sci. Rep. 2016, 6, 220911. [Google Scholar] [CrossRef]
- Warming, S.; Costantino, N.; Court, D.L.; Jenkins, N.A.; Copeland, N.G. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res. 2005, 33, e36. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Orfs | Size (aa) | Closest Homolog (BLASTP) | Origin | Accession Number | Identity/Similarity (%) |
|---|---|---|---|---|---|
| orf1 | 1008 | Hypothetical protein | Nannocystis pusilla | WP_224196465.1 | 40/53 |
| orf2 | 173 | Hypothetical protein | Nannocystis exedens | WP_096327445.1 | 29/40 |
| orf3 | 418 | Hypothetical protein | Myxococcales bacterium | MCA9650222.1 | 75/86 |
| orf4 | 104 | Polyhydroxyalkanoic acid system family protein | Deltaproteobacteria bacterium | MCH9682166.1 | 73/86 |
| orf5 | 1020 | Spermidine synthase | Deltaproteobacteria bacterium | MCH9682165.1 | 81/88 |
| miuD | 461 | Cytochrome P450 | Myxococcales bacterium | MCB9753755.1 | 67/77 |
| orf6 | 493 | Peptidase M4 family protein | Deltaproteobacteria bacterium | RME25565.1 | 51/65 |
| orf7 | 171 | Hypothetical protein | Balneola sp. | MBE78502.1 | 27/49 |
| orf8 | 138 | DUF4398 domain-containing protein | bacterium | MCR9159996.1 | 46/67 |
| orf9 | 302 | OmpA family protein | Deltaproteobacteria bacterium | MBC8068113.1 | 47/63 |
| orf10 | 182 | Hypothetical protein | Deltaproteobacteria bacterium | MCH9682164.1 | 69/80 |
| orf11 | 497 | OmpA family protein | Myxococcales bacterium | MCA9650212.1 | 69/84 |
| orf12 | 274 | FHA domain-containing protein | Myxococcales bacterium | MCA9652678.1 | 58/68 |
| miuE | 257 | O-methyltransferase | Stigmatella erecta | SEU19554.1 | 35/54 |
| orf13 | 288 | Lysine-specific demethylase 8 isoform X1 | Micropterus salmoides | XP_038559593.1 | 31/47 |
| miuA | 4105 | Type I polyketide synthase | Pyxidicoccus fallax | WP_169347329.1 | 60/72 |
| miuB | 3926 | Amino acid adenylation domain-containing protein | Pyxidicoccus fallax | NPC81269.1 | 51/64 |
| orf14 | 554 | Mechanosensitive ion channel family protein | Vitiosangium sp. GDMCC 1.1324 | WP_108076111.1 | 40/62 |
| orf15 | 545 | Hemopexin repeat-containing protein | Nannocystis sp. fl3 | WP_269038991.1 | 75/87 |
| orf16 | 686 | Heavy metal translocating P-type ATPase | Proteobacteria bacterium | MBU0970734.1 | 41/63 |
| orf17 | 94 | Hypothetical protein | Myxococcales bacterium | MBL8970963.1 | 60/76 |
| orf18 | 125 | Hypothetical protein | Myxococcales bacterium | MBL8970963.1 | 70/83 |
| miuC | 5546 | Non-ribosomal peptide synthetase | Chondromyces crocatus | WP_169796632.1 | 40/55 |
| orf19 | 730 | Bifunctional metallophosphatase/5′-nucleotidase | Chondromyces crocatus | WP_050432501.1 | 59/74 |
| orf20 | 164 | GTPase | Candidatus Methylumidiphilus alinenensis | PZN75038.1 | 49/71 |
| miuF | 241 | Thioesterase | Myxococcales bacterium | MCA9716988.1 | 62/75 |
| orf21 | 95 | Hypothetical protein | Myxococcales bacterium | MBL8970963.1 | 67/81 |
| orf22 | 233 | PEP/pyruvate-binding domain-containing protein | Myxococcales bacterium | MBL8970530.1 | 62/72 |
| orf23 | 728 | Heavy metal translocating P-type ATPase | Nannocystis sp. MB1016 | ALD82534.1 | 61/79 |
| miuG | 621 | FAD-dependent oxidoreductase; | Symploca sp. SIO1A3; | NER47269.1 | 39/54 |
| Bmp5, Flavin-dependent single-component p-hydroxybenzoate brominase/decarboxylase (from MIBiG database) | Pseudoalteromonas phenolica O-BC30 | KF540211.1 | 35/52 | ||
| orf24 | 58 | No significant homology | |||
| orf25 | 235 | OmpA family protein | Myxococcales bacterium | MCA9650211.1 | 61/79 |
| orf26 | 771 | LysM peptidoglycan-binding domain-containing protein | Myxococcales bacterium | MCA9705957.1 | 66/78 |
| orf27 | 441 | Protein kinase | Deltaproteobacteria bacterium | MCH9682160.1 | 69/80 |
| orf28 | 599 | HAMP domain-containing protein | Myxococcales bacterium | MCA9650207.1 | 76/86 |
| orf29 | 594 | PAS domain S-box protein | Myxococcales bacterium | MCA9650206.1 | 66/76 |
| Heterologous Host | Yield of 1 (mg/L) | Produced Congeners |
|---|---|---|
| M. xanthus::miu | 0.06 | 2, 4 |
| M. xanthus::miu ΔmiuD | 0.13 | 2, 4 |
| M. xanthus::miu ΔmiuE | - | 2, 3, 3′ |
| M. xanthus::miu ΔmiuF | 0.08 | 2, 4 |
| M. xanthus::miu ΔmiuG | - | 4 |
| M. xanthus::miu ΔmiuG + 3-bromo-L-tyrosine | 0.09 | 2 |
| M. xanthus::miu ΔmiuG + 3-bromo-D-tyrosine | 0.02 | 4 |
| Heterologous Host | Yield of 1 (mg/L) | Produced Congeners |
|---|---|---|
| M. xanthus::miu ∆orf25–29 (31 orfs, 77.0 kbp) | 0.07 | 2, 4 |
| M. xanthus::miu ∆orf25–29&1–10 (20 orfs, 62.1 kbp) | 0.10 | 2, 4 |
| M. xanthus::miu ∆orf25–29&14–16 (28 orfs, 72.6 kbp) | 0.07 | 2, 4 |
| M. xanthus::miu ∆orf25–29&19–23 (25 orfs, 70.9 kbp) | 0.70 | 2, 2′, 3, 3′, 5 |
| M. xanthus::miu ∆orf25–29&19–23 + 3-bromo-L-tyrosine | 1.21 | 2, 2′, 3, 3′, 5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Yamazaki, S.; Ojika, M. Heterologous Biosynthesis of Myxobacterial Antibiotic Miuraenamide A. Molecules 2023, 28, 2815. https://doi.org/10.3390/molecules28062815
Liu Y, Yamazaki S, Ojika M. Heterologous Biosynthesis of Myxobacterial Antibiotic Miuraenamide A. Molecules. 2023; 28(6):2815. https://doi.org/10.3390/molecules28062815
Chicago/Turabian StyleLiu, Ying, Satoshi Yamazaki, and Makoto Ojika. 2023. "Heterologous Biosynthesis of Myxobacterial Antibiotic Miuraenamide A" Molecules 28, no. 6: 2815. https://doi.org/10.3390/molecules28062815
APA StyleLiu, Y., Yamazaki, S., & Ojika, M. (2023). Heterologous Biosynthesis of Myxobacterial Antibiotic Miuraenamide A. Molecules, 28(6), 2815. https://doi.org/10.3390/molecules28062815