Abstract
Two aplysiatoxin derivatives, neo-debromoaplysiatoxin I (1) and neo-debromoaplysiatoxin J (2), were isolated from marine cyanobacterium Lyngbya sp. collected from the South China Sea. Their structures including absolute configurations were assigned by spectroscopic analysis, in combination with GIAO NMR shift calculation and DP4+ analysis. Structures of neo-debromoaplysiatoxin I and neo-debromoaplysiatoxin J contained a decahydro-5H-pyrano [2,3,4-de] chromen-5-one 6/6/6 ring skeleton and an intriguing peroxide bridge group, respectively, which are unprecedented structure scaffold and motif in aplysiatoxins. Two compounds displayed comparable inhibitory activities against Kv1.5 K+ channel with IC50 values of 2.59 ± 0.37 μM (1) and 1.64 ± 0.15 μM (2); however, they presented differential cytotoxic effects. It is worth noting that neo-debromoaplysiatoxin J, containing a peroxide bridge, showed remarkable cytotoxicity against four cancer cell lines including SW480, SGC7901, LoVo and PC-9 compared to the human normal cell line.
Keywords:
cyanobacteria; Lyngbya sp.; aplysiatoxins; peroxide bridge; DP4+; Kv1.5 inhibition; cytotoxicity 1. Introduction
Cyanobacteria (blue-green algae), known as one of the oldest bacteria in evolutionary history, are pioneers to conquer the ancient environments [1,2]. They are widely distributed in oceans, tropical reefs, freshwater ponds, rivers, polar ice and terrestrial substrates, among which the ocean is preferred due to its alkaline environment [3]. They have the ability to produce significant quantities of toxins—cyanotoxins, such as saxitoxins (consisting of a tetrahydropurine group and two guanidinium moieties), ciguatoxins (polyether) [4], microcystins (cyclic peptide) [5], lipopolysaccharide endotoxins [6] and so on. Aplysiatoxins (ATXs) are polyketides existing in cyanobacteria with anti-proliferative, antiviral, pro-inflammatory and Kv1.5 K+ channel inhibition properties [7,8,9,10]. Up to now, there have been about 55 aplysiatoxins isolated from cyanobacteria [11,12,13,14]. Based on structural skeletons, we classified them into four categories: 6/12/6 fused ring system featuring a macrolactone ring; spirobicyclic system; linear structure; and some neo-ATXs displaying uncommon carbon skeletons. Several ATXs were found to be activators of protein kinase C (PKC) [15,16], a vital therapeutic target for cancers [17], such as debromoaplysiatoxin (DAT) and 3-deoxy-DAT, probably owing to the dihydroxyvaleryl moiety in their structures, namely a recognition domain interacting with the PKCδ C1B domain in the form of hydrogen bonds [18]. In our previous studies, the inhibitory activity on the shaker-related subfamily of voltage-gated channels (Kv1.1–Kv1.5) was investigated, and potential drug targets for the treatment of diverse disease processes, ranging from cancer to autoimmune diseases to metabolic, neurological and cardiovascular disorders [19], of ATXs were tested, and it turned out several ATXs had selective and strong blocking inhibitory effects on the Kv1.5 K+ channel [10,11,12,20,21,22]. Among them, neo-debromoaplysiatoxin B (Neo-B) and oscillatoxin E displayed the strongest Kv1.5 inhibition activities with IC50 values of 0.3 μM and 0.79 μM respectively.
Recently, in our ongoing search for new ATXs from cyanobacteria, two undescribed ATX analogues, neo-debromoaplysiatoxin I (1) and neo-debromoaplysiatoxin J (2), were isolated (Figure 1). Compound 1 possessed decahydro-5H-pyrano [2,3,4-de] chromen-5-one structural skeleton, and compound 2 had a 6/14/6 fused-ring system. Although the ABC tricyclic ring system with a macrolide is quite common in ATXs, this is the first example of the introduction of a peroxide bridge in 2. Their inhibitory activities against the Kv1.5 K+ channel and cytotoxic effects against human normal and cancer cell lines were evaluated. Herein, we report the isolation, structure elucidation and bioactivities of these two compounds.
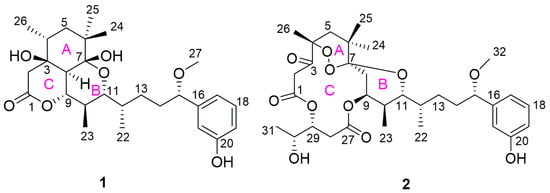
Figure 1.
Structures of neo-debromoaplysiatoxin I (1) and neo-debromoaplysiatoxin J (2).
2. Results and Discussion
2.1. Structure Elucidation of Compounds
Neo-debromoaplysiatoxin I (1) was obtained as a white solid, with a molecular formula of C27H40O7 based on its HRESIMS data (m/z 499.2668 [M + Na]+, calcd. for C27H40O7Na, 499.2672), requiring eight degrees of unsaturation. Compound 1 showed IR absorption bands for hydroxy group (3418 (br), 3260 (br) cm−1), ester linkage (1712 cm−1) and aromatic system (1663, 1646, 1455 cm−1). The 13C and DEPT NMR spectra revealed 27 carbon signals, attributed to 6 quaternary carbons, 11 methines, 4 methylenes and 6 methyls including 1 methoxy (Table 1). Detailed 1H, 13C and HSQC NMR spectra analysis suggested the presence of a 1,3-disubstituted benzene ring, implying it might belong to ATXs.

Table 1.
1H (600 MHz) and 13C (150 MHz) NMR data for compounds 1 and 2 in CDCl3 (δ in ppm).
With an ester bond and a benzene ring taking five degrees of unsaturation, 1 contained a tricyclic core. The ring A was established by the HMBC correlations from H3-26 to C-3, C-4, C-5 and C-8; from H3-24, H3-25 to C-5, C-6 and C-7; from H2-5 to C-4 and C-6; and from H-8 to C-3, C-7 and C-9. The 1H-1H COSY correlations of H-4/H2-5/H3-26 supported the structure segment. 3-OH (δH 4.35, d, J = 2.0 Hz) exhibited HMBC correlation to C-2, COSY correlation with H-2a and the chemical shifts of C-1 (δC 170.5), C-2 (δC 40.9), C-3 (δC 72.6) and C-9 (δC 77.6), together with HMBC spectrum signals from H2-2 to C-1, C-3 and C-8, indicated that the ring C was composed of C-1, C-2, C-3, C-8 and C-9 through an ester bond to form a six-membered lactone ring with a hydroxy on C-3. Furthermore, IR spectrum showed absorption band at 1712 cm−1, which supported a six-membered lactone ring. As shown in Figure 2, the COSY signals of H-8/H-9/H-10/H3-23/H-11/H-12/H3-22/H2-13/H2-14/H-15 and HMBC correlations from H3-23 to C-9, C-10 and C-11; from H3-22 to C-11, C-12 and C-13; and from H-15 to C-14, C-13, C-17, C-21 and C-27 illustrated the structure segments from C-8 to C-21. The ring B was closed by C7-O-C11, and a hydroxy was attached to C-7 based on the 13C NMR chemical shifts of oxygenated quaternary carbon C-7 (δC 100.1), oxygenated tertiary carbon C-11 (δC 75.5) and unsaturation of 1, as well as structural features of ATXs. Thereby, the planar structure of compound 1 was completed.

Figure 2.
Key 2D correlations of 1 and 2.
Its relative stereochemistry was established by the NOESY experiments and vicinal coupling constants. The key NOE correlations of 3-OH/H-5a, 3-OH/H-9, H-5a/H3-24 and H-5b/H-25/H-26 implicated 3-OH/H-4/H-5a/H3-24/H-9 shared syn relationships, while H-5b/H3-26/H3-25 presented same orientation. The small couplings of H-4 to H-5a and H-5b (J5a,4 = 5.8 Hz, J5b,4 = 2.1 Hz) determined H-4 as the equatorial bond of ring A, which exhibited the opposite configuration with H-4 of Neo-B ((J5a,4 = 6.8 Hz) [23]. The NOE cross-peak of H-8/H-10/H3-22, H-9/H-11/H3-23 defined an anti-relationship between H-8 and H-9, H-9 and H-10 and H-10 and H-11. The large coupling constants of H-9 (J8,9 = J9,10 = 10.7 Hz) and H-11 (J10,11 = 10.4 Hz) showed that H-8, 9, 10 and 11 were axially oriented. A gauche relationship of H-11 and H-12 was deduced from their NOESY correlation and small coupling (J11,12 = 1.9Hz). In addition, the NOE correlations of H-12/H3-23 and H-10/H3-22 shown in Figure 2 also clarified the rationality of this conformation. Agreements of 1H and 13C chemical shifts of C-15 between 1 and other ATXs, such as oscillatoxin D, oscillatoxin F [8] and 3-methoxydebromoaplysiatoxin [7], together with structural features and common biosynthetic origin of ATXs, suggested that the absolute stereochemistry of C-15 was identified as S. In Chembio3D, the three rings exhibited stable chair conformation when C-7 was R, while they showed boat conformation when C-7 was S.
Revolving around C-4 and C-7, δC values of four plausible isomers, 1A, 1B, 1C and 1D (Figure 3 and Figure 4), were calculated at the B3LYP/6-311++G(2d,p) level. The DP4+ probability analysis disclosed that isomer 1B (4R, 7R) was the most likely one with a probability of 100%. Thus, the configuration of compound 1 was inferred as 3S,4R,7R,8S,9S,10R,11R,12S,15S on the basis of the GIAO NMR shift calculation followed by DP4 analysis.
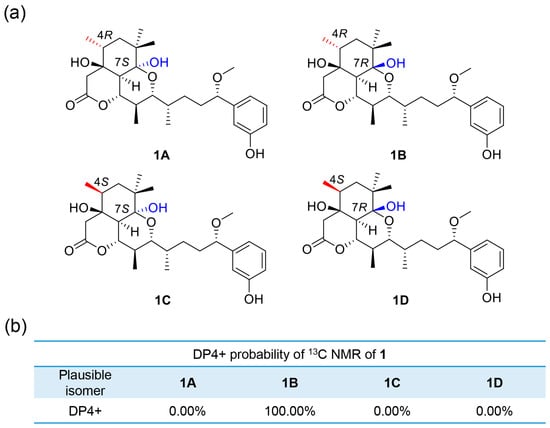
Figure 3.
(a) Possible isomers for 1. (b) DP4+ probability of 13C values of 1.

Figure 4.
13C NMR calculation results of plausible epimers at the B3LYP/6-311++G(2d,p) level. (a) Regression analysis of calculated versus experimental 13C NMR chemical shifts of 1 and 2. (b,c) Relative chemical shift errors between calculated and experimental 13C NMR data for the experimentally produced 1 and 2.
Neo-debromoaplysiatoxin J (2) was isolated as a white solid. The molecular formula of C32H46O11 with 10 degrees of unsaturation was inferred from HRESIMS data at m/z 629.2938 [M + Na]+ (calcd. for C32H46O11Na, 629.2938). The NMR data of 2 exhibited a keto carbon (δC 207.2), two carbonyl carbons (δC 169.8 and 165.5), six aromatic carbons (δC 156.4, 143.5, 129.5, 118.4, 114.8 and 114.5), two oxygenated quaternary carbons (δC 105.7 and 87.2) and a methoxy (δC 56.7) (Table 1). The detailed interpretation of NMR spectra indicated that the planar structure of 2 closely resembles those of debromoaplysiatoxins, especially neo-debromoaplysiatoxin A (Neo-A) [10], except for C-2, C-4, C-7 and the atom composition of ring A, C. The HMBC correlations from H2-2 to C-1 and C-3; from H3-26 to C-3, C-4 and C-5; from H2-5 to C-3, C-4, C-6, C-7, C-25 and C-26; and from H3-24 and H3-25 to C-5, C-6 and C-7 decided the structural segment of C-1–C-7 (Figure 2). Considering the requirement of the unsaturation and the molecular formula for 2, the C-4 (δC 87.2) and C-7 (δC 105.7) were linked through a peroxide bridge to form a relatively stable six-membered ring A. Besides, IR absorption bands for 877 cm−1 suggested a peroxide bond [24]. 13C chemical shifts of carbons in a similar chemical environment with C-4 and C-7 of 2, such as C-3 (δC 105.4) of artemisinin [24], C-8 (δC 109.4) of hedychin A [25] and C-7 (δC 89.8) of talaperoxides B [26], verified the rationality of the chemical shifts attribution.
The NOESY spectrum displayed correlations of H-15/H-13b and H-11/H-12/H3-23/H-13b, uncovering these protons shared syn relationships. H-11 was attached axially to the ring B according to the big coupling constants of H-11 (J10, 11= 10.7 Hz). The syn relationship of H-9 and H-10 was deduced from the NOE correlations of H-9/H-10/H3-22. H-9 presented an equatorial orientation with a small coupling constant (J = 2.8 Hz). H-5a exhibited a NOE correlation with H3-24, and H-5b had a NOE correlation with H3-24, H3-25 and H3-26, declaring H3-25 and H3-26 were on the same side of ring A. The common biosynthetic origin of ATXs and the coupling constant of H-29 and H-30 (J29,30 = 4.7 Hz), in accordance with existing ATXs [7,20,27], demonstrated C-29 and C-30 as 29R,30R. Considering J11,12 = 1.9 Hz, the relationship of H-11 and H-12 was determined the same way as 1. The DP4+ analysis following the Gauge-Independent Atomic Orbital (GIAO) NMR shift calculation were performed (Figure 4), reflecting isomer 2B (4R, 7S) (Figure 5) with a probability of 100% was the one fits the bill on the basis of the 13C NMR data. The configuration of compound 2 was settled as 4R,7S,9S,10S,11R,12S,15S,29R,30R.
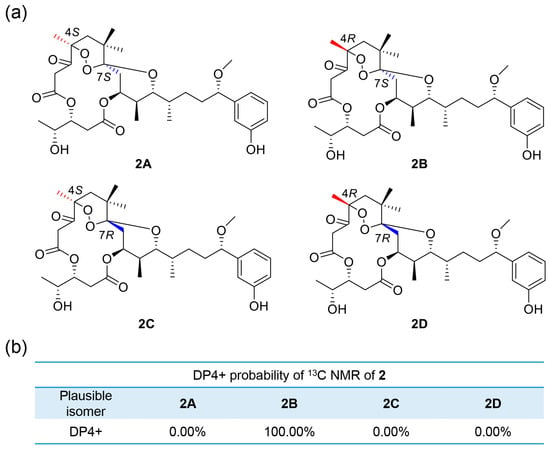
Figure 5.
(a) Possible isomers for 2. (b) DP4+ probability of 13C values of 2.
The two compounds are considered to have common biosynthetic intermediate [13,28], and their plausible biosynthesis pathway is proposed as shown in Scheme 1. Compound 1 is biosynthesized via aldol reaction between C-8 and C-3, followed by nucleophilic addition between 11-OH and C-7, generating 3-OH and 7-OH and ring A, B. Then, 9-OH acting as nucleophilic reagent undergoes transesterification with C-1 to give 1. When it comes to compound 2, nucleophilic addition between 11-OH/C-7 and dehydration of 7-OH/C-8 occurs first, followed by reduction of C-3 and dehydration of 3-OH/C-4. The introduction of peroxide bridge is proposed through endoperoxide-forming oxygenases after careful considerations and in combination with some findings published, such as the cyclooxygenases in the biosynthesis of prostaglandins [29] and fumitremorgin B endoperoxidase (FtmOx1) in the biosynthesis of verruculogen [30]. After oxidation of C-3, the esterification reaction of C-27/9-OH finally establishes compound 2.

Scheme 1.
Postulated biosynthetic pathways of compounds 1 and 2.
2.2. Bioactivities
2.2.1. Inhibitory Activities against Kv1.5
The Kv1.5 K+ channel is considered as an effective and safe therapeutic target of atrial fibrillation because of its selective existence in atrium [31]. Considering the selective inhibitory effects on Kv1.5 of ATXs, we tested compounds 1 and 2 for their Kv1.5 inhibitory activities. The results showed 1 and 2 exhibited IC50 values of 2.59 ± 0.37 μM and 1.64 ± 0.15 μM, respectively (Figure 6). In all the ATXs tested for Kv1.5 inhibitory activities (Table S3), we preliminarily analyzed the relationship between different ATXs modified by different functional groups and Kv1.5 inhibitory activities (Figure S3). Generally speaking, oxygenated six-membered ring B is necessary for inhibition activities; for example, neo-debromoaplysiatoxin C without six-membered ring B exhibited no significant effect on the Kv1.5 channel. Ring C is not essential for the inhibition activities. Plus, configuration of chiral carbons also plays an important role, such as neo-debromoaplysiatoxin E and neo-debromoapysiatoxin F showing different inhibitory activities against Kv1.5. Different substituent groups on ring A of spiro structures also displayed a difference in activity.

Figure 6.
Kv1.5 currents and concentration–inhibition curve in the absence and presence of 0.3 µM, 1 µM, 3 µM and 10 µM compounds. The abscissa represents the concentration, and the ordinate represents the percentage of Kv1.5 current that is blocked at different concentrations of compounds. Data points represent mean ± SEM of 4 measurements. (a) The inhibitory effect of 1 showed an IC50 value of 2.59 ± 0.37 µM. (b) The inhibitory effect of 2 showed an IC50 value of 1.64 ± 0.15 µM.
2.2.2. Cytotoxic Effects
Since the discovery of artemisinin, natural peroxides have attracted more and more attention. So far, a large number of polyketides and other chemical components with peroxy rings have been discovered from marine organisms [32,33]. Some of their bioactivities are attributed to the presence of endoperoxide. The endoperoxide bridge in artemisinin is essential for its antimalarial activity [34]. A sesquiterpene with an epidioxy bond bridge showed good anti-influenza virus activity, and the peroxy bridge may play a key role in increasing the activity [35]. In the report of Wibowo et al. [36], terpene endoperoxides exhibited cytotoxic activity, while those without an endoperoxide moiety did not show activity. The cytotoxic activity was probably caused by the reactive radical derived from a homolytic cleavage of endoperoxide.
The intriguing structures of 1 and 2, particularly the cyclic peroxide moiety, encouraged us to investigate their cytotoxic activities because many natural peroxides exhibited significant anticancer activities [37]. We assayed the cytotoxicity of two compounds toward the SW480 human colorectal cancer cell line. As expected, compound 2 exhibited obvious cytotoxicity with IC50 values of 4.63 ± 0.20 μM. Compound 1 showed relatively weak activity with IC50 values of 21.14 ± 2.20 μM (Figure 7 and Figure S1). In addition, cytotoxicity against 293A human kidney cells (human normal cell line), SGC7901 human gastric cancer cells, LoVo human colorectal carcinoma cells and PC-9 non-small-cell lung cancer cells were also tested (Figure S2). Similar with effects on SW480 cells, compound 1 mostly exhibited little or weak cytotoxicity against the three cancer cell lines. While compound 2 showed stronger cytotoxicity against the three cancer cells, with cell viability less than 20% at 20 μM. However, both compounds 1 and 2 exhibited no or less cytotoxicity against 293A human kidney cells from 1 μM to 20 μM.

Figure 7.
Cytotoxic effects of two compounds for 72 h on SW480 human colon cancer cells as measured using a cell viability assay. Cisplatin (5 μM) was used as positive control. Asterisks indicate statistical significance at p < 0.05 and ns means no significant differences, compared to the untreated control group. (a) SW480 exposed to different concentrations of 1. (b) SW480 exposed to different concentrations of 2. The data are expressed as the means ± SD of four independent experiments.
Interestingly, among the previously reported cytotoxic tests against cancer cell lines like HeLa cells and L1210 mouse lymphoma cells [28,38,39], most existing ATXs exhibited weak or modest anticancer activities. It seems that debromoaplysiatoxin (IC50 values of 3.03 μM against HeLa cancer cells) [38] and neo-debromoaplysiatoxin J (2) possess relatively strong anticancer activities as natural sources of ATXs. 2 showing cell line-selective antiproliferative activities could be beneficial in cancer treatment and provided an interesting orientation in our following studies.
3. Materials and Methods
3.1. General Experimental Procedures
The UV spectrum was obtained using SPD-M20A diode array detector (Shimadzu, Kyoto, Japan). Optical rotation data were tested on an Autopol IV polarimeter (Rudolph, Hackettstown, NJ, USA). The FT-IR spectra were recorded using a Nicolet iS20 instrument (Thermo Fisher Scientific, Waltham, MA, USA), with a resolution of 4 cm−1 and 32 scans in % transmittance mode. A KBr-disk was used for preparing the samples. 1H and 13C NMR spectra were recorded in CDCl3 containing Me4Si as internal standard on Agilent 600 MHz spectrometer (Agilent Technologies, Santa Clara, CA, USA). HRESIMS was performed with a Bruker micrOTOF-Q II mass spectrometer (Bruker Daltonik GmbH, Bremen, Germany). HPLC was carried out on a Shimadzu LC-16 series instrument (Shimadzu, Kyoto, Japan), equipped with C-18 column (5 µm, 10 mm × 250 mm, SilGreen, Beijing, China) and an SPD-M20A diode array detector (Shimadzu, Kyoto, Japan). As for column chromatography, silica gel 60 (200–300 mesh; Yantai Jiangyou Silica Gel Development Co. LTD, Yantai, China) and octadecylsilyl (ODS) (50 μm, YMC, Kyoto, Japan) were used.
3.2. Material
The cyanobacterium sample was collected from Lingshui Port, Sanya, China, in November 2016. According to morphological and molecular identification, the sample belongs to Lyngbya sp. The voucher specimen numbered as BNH-201606 (gene bank accession numbers: MH636576) was deposited by Professor Bingnan Han in Zhejiang Sci-Tech University.
3.3. Extraction and Isolation
The freeze-dried cyanobacterium sample (150 g) was cut into pieces for ultrasonic extraction with CH2Cl2/MeOH (2:1, v/v). Then, the extract was suspended in 1 L of MeOH/H2O (3:2, v/v) and partitioned with CH2Cl2 (1 L) 3 times to yield the CH2Cl2 extract (20 g). Vacuum liquid chromatography (VLC) over silica gel was used for further separation to yield seven subfractions (F. A–G), with gradients of petroleum ether/EtOAc (5:1, 2:1, 1:1, 1:2, 1:5, 0:1, v/v). In which F.B (2000 mg) was subject to purification by reversed-phase octadecylsilyl silica (ODS) (UV detection at 220 nm, flow rate 20 mL/min, 10%–100% MeCN/H2O, 180 min), obtaining 17 subfractions (F.B.1–17). Next, F.B.7 (231mg) was isolated by semi-preparative HPLC (Shimadzu, C-18 (SilGreen), 70% MeCN/H2O, 2.0 mL/min, UV detection at 220 nm), and we acquired our two compounds 1 (6.3 mg, tR = 16.3 min) and 2 (3.2 mg, tR = 18.6 min).
Neo-debromoaplysiatoxin I (1): white solid; +6.7 (c 0.03, MeOH); UV (MeCN) λmax (log ε) 196 (3.27) nm, 276 (2.45) nm; IR (KBr) νmax: 3418 (br), 3260 (br), 2926, 1712, 1663, 1646, 1455, 1253, 1052, 919 cm−1; 1H NMR (600 MHz, CDCl3) and 13C NMR (150 MHz, CDCl3) data see Table 1; (+) HRESIMS m/z 499.2668 [M + Na]+ (calcd. for C27H40O7Na, 499.2672).
Neo-debromoaplysiatoxin J (2): white solid; −35.7 (c 0.07, MeOH); UV (MeCN) λmax (log ε) 196 (3.34) nm, 275 (2.56) nm; IR (KBr) νmax: 3418 (br), 2966, 2929, 1727, 1653, 1599, 1455, 1300, 1223, 1117, 1067, 1047, 877 cm−1; 1H NMR (600 MHz, CDCl3) and 13C NMR (150 MHz, CDCl3) data see Table 1; (+) HRESIMS m/z 629.2938 [M + Na]+ (calcd. for C32H46O11Na, 629.2938).
3.4. Theory and Calculation Details
The calculations, conformational search and optimization were performed as reported [12]. The stable conformations obtained at the B3LYP/6-31G(d) level were further used in magnetic shielding constants at the B3LYP/6-311++G(2d,p) level. The DP4+ calculations were finished with a simplified procedure according to Grimblat [40].
3.5. Cell Culture
The mouse connective tissue fibrocytes (LTK cells) stably expressing human Kv1.5 channels (LTK-Kv1.5) from Professor Weiping Wang at Chinese Academy of Medical Sciences and Peking Union Medical College, China, was cultured for ion channel inhibitory experiment. The 293A human kidney cell line, SW480 human colorectal cancer cell line, SGC7901 human gastric cancer cell line, LoVo human colorectal carcinoma cell line and PC-9 non-small-cell lung cancer cell line, used for cell viability assay, were kindly provided by Professor Yigang Wang and Professor Wenbin Ou at Zhejiang Sci-Tech University, China. Cells were grown in Dulbecco’s Modified Eagle Medium (DMEM) (Gibco, Shanghai, China), except PC-9 in RPMI-1640 medium (Gibco, Shanghai, China), containing 10% fetal bovine serum (FBS) (Wisent bioproducts, Montreal, Canada) and 1% antibiotics (10,000 U/mL penicillin, 10 mg /mL streptomycin) (Solarbio life sciences, Beijing, China) at 37 °C in a humidified atmosphere containing 5% CO2. When the cells grew to 70–85% confluence, they were passaged for subsequent tests. In all the tests, control cells were treated with DMSO (Solarbio Life Sciences, Beijing, China).
3.6. Ion Channel Inhibitory Experiment
The instruments and methods for the experiment mainly refer to previously reported [12]. Whole-oocyte recordings were conducted by electrode patch clamp. The intracellular fluid contained KCl (140 mM), MgCl2 (1 mM), HEPES (10 mM), Mg-ATP (3 mM) and EGTA (10 mM). The pH was adjusted to 7.25 by KOH. The extracellular fluid used to record currents contained NaCl (135 mM), KCl (5.4 mM), MgCl2 (1 mM), CaCl2 (1.8 mM), Glucose (10 mM), HEPES (10 mM) and NaH2PO4 (0.33 mM). pH was adjusted to 7.4 by NaOH.
Kv1.5 currents were evoked by a 300 ms depolarizing pulse from −50 mV to 50 mv in 10 mV increments from a holding potential of −70 mV. The current amplitude at the end of 300 ms pulse at 50 mV was measured. All values were indicated as mean ± SEM, and a value of p < 0.05 was considered to be significant.
3.7. Cell Viability Assay
The cells were seeded on 96-well plates (Nest, Wuxi, China) at a density of 3000 cells in 100 μL medium per well. After 16–20 h, the medium was changed into a fresh medium that had different concentrations of compounds. After a 72 h incubation period, 100 μL fresh medium with 10% CCK-8 reagent (Vazyme, Nanjing, China) was added along the hole-wall into each hole to replace the drug-containing medium. The plates were incubated for 1–2 h before the light absorption values at 450 nm of each group were detected. Cisplatin (5 μM) (Selleckchem, Houston, TX, USA), a kind of broad-spectrum anticancer drug for clinical application, was used as positive control in cell viability assay. Furthermore, the half inhibitory concentration (IC50) was calculated using GraphPad Prism (Version 8.0.2) software.
4. Conclusions
In conclusion, neo-debromoaplysiatoxin I (1), featuring a new skeleton, and neo-debromoaplysiatoxin J (2), with an unprecedented peroxide bridge group, were characterized from cyanobacterium Lyngbya sp. Two compounds all exhibited significant inhibitory activities against the Kv1.5 K+ channel with IC50 values of 2.59 ± 0.37 μM (1) and 1.64 ± 0.15 μM (2), respectively. Surprisingly, in the in vitro cytotoxic assays, 2 exhibited apparent stronger cytotoxic effects against four cancer cell lines, including SW480, SGC7901, LoVo and PC-9, than 1, with cell viability less than 20% at 20 μM, while it exhibited no or less cytotoxicity against 293A human kidney cells (normal cells) from 1 μM to 20 μM.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules28062786/s1, Tables S1 and S2: Detailed NMR data of compounds 1 and 2; Table S3: Kv1.5 inhibitory effects of ATXs; Tables S4 and S5: The calculated 13C NMR data for isomers of compounds 1 and 2; Tables S6–S14: DFT-optimized structures and thermodynamic parameters for low-energy conformers of 1A–1D and 2A–2D; Figure S1: Dose-dependent effects of 1 and 2 on SW480 cell survival rate (72 h); Figure S2: Cytotoxic effects of two compounds for 72 h on different cells as measured using a cell viability assay; Figure S3: Critical structural features of ATXs leading to differential Kv1.5 inhibitory activities; Figures S4–S26: UV, IR, HR-ESI-MS, 1D NMR and 2D NMR spectra of compounds 1 and 2; Figure S27: Morphological and molecular identification of cyanobacterium.
Author Contributions
Conceptualization, Z.C., N.C. and B.H.; methodology, Z.C., P.F. and W.W.; software, Z.C., P.F. and W.W.; data curation, Z.C., N.C., S.B., H.Z. and S.S.; writing—original draft preparation, Z.C.; writing—review and editing, Z.C. and B.H.; funding acquisition, B.H. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (81973233); the Key Research and Development Program of Zhejiang Province, China (2021C02062); the Scientific Research Foundation of Zhejiang Sci-Tech University (17042058-Y) and the Special Program for Research and Development of Zhejiang Sci-Tech University (2019Y009).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article or Supplementary Materials.
Acknowledgments
Thanks to Wenbin Ou and Yigang Wang for supplying cell lines.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hamilton, T.L.; Bryant, D.A.; Macalady, J.L. The role of biology in planetary evolution: Cyanobacterial primary production in low-oxygen Proterozoic oceans. Environ. Microbiol. 2016, 18, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Thuan, N.H.; An, T.T.; Shrestha, A.; Canh, N.X.; Sohng, J.K.; Dhakal, D. Recent advances in exploration and biotechnological production of bioactive compounds in three cyanobacterial genera: Nostoc, Lyngbya, and Microcystis. Front. Chem. 2019, 7, 604. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.Z.; Zhang, T.; Yao, J.X.; Lu, J.; Liu, Z.W.; Ding, L.J. Recent advances in chemistry and bioactivity of marine cyanobacteria Moorea species. Eur. J. Med. Chem. 2020, 201, 112473. [Google Scholar] [CrossRef]
- Sini, P.; Dang, T.B.C.; Fais, M.; Galioto, M.; Padedda, B.M.; Luglie, A.; Iaccarino, C.; Crosio, C. Cyanobacteria, cyanotoxins, and neurodegenerative diseases: Dangerous liaisons. Int. J. Mol. Sci. 2021, 22, 8726. [Google Scholar] [CrossRef]
- Metcalf, J.S.; Tischbein, M.; Cox, P.A.; Stommel, E.W. Cyanotoxins and the nervous system. Toxins 2021, 13, 660. [Google Scholar] [CrossRef] [PubMed]
- Codd, G.A.; Morrison, L.F.; Metcalf, J.S. Cyanobacterial toxins: Risk management for health protection. Toxicol. Appl. Pharmacol. 2005, 203, 264–272. [Google Scholar] [CrossRef]
- Gupta, D.K.; Kaur, P.; Leong, S.T.; Tan, L.T.; Prinsep, M.R.; Chu, J.J. Anti-Chikungunya viral activities of aplysiatoxin-related compounds from the marine cyanobacterium Trichodesmium erythraeum. Mar. Drugs 2014, 12, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Araki, Y.; Hanaki, Y.; Kita, M.; Hayakawa, K.; Irie, K.; Nokura, Y.; Nakazaki, A.; Nishikawa, T. Total synthesis and biological evaluation of oscillatoxins D, E, and F. Biosci. Biotechnol. Biochem. 2021, 85, 1371–1382. [Google Scholar] [CrossRef]
- Kikumori, M.; Yanagita, R.C.; Tokuda, H.; Suenaga, K.; Nagai, H.; Irie, K. Structural optimization of 10-methyl-aplog-1, a simplified analog of debromoaplysiatoxin, as an anticancer lead. Biosci. Biotechnol. Biochem. 2016, 80, 221–231. [Google Scholar] [CrossRef]
- Han, B.N.; Liang, T.T.; Keen, L.J.; Fan, T.T.; Zhang, X.D.; Xu, L.; Zhao, Q.; Wang, S.P.; Lin, H.W. Two marine cyanobacterial aplysiatoxin polyketides, neo-debromoaplysiatoxin A and B, with K(+) channel inhibition activity. Org. Lett. 2018, 20, 578–581. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.H.; Zhang, X.K.; Si, R.R.; Shen, S.C.; Liang, T.T.; Fan, T.T.; Chen, W.; Xu, L.H.; Han, B.N. Chemical and biological study of novel aplysiatoxin derivatives from the marine cyanobacterium Lyngbya sp. Toxins 2020, 12, 733. [Google Scholar] [CrossRef]
- Shen, S.C.; Wang, W.P.; Chen, Z.J.; Zhang, H.H.; Yang, Y.C.; Wang, X.L.; Fu, P.; Han, B.N. Absolute structure determination and Kv1.5 ion channel inhibition activities of new debromoaplysiatoxin analogues. Mar. Drugs 2021, 19, 630. [Google Scholar] [CrossRef] [PubMed]
- Satake, M.; Iguchi, K.; Watanabe, R.; Uchida, H.; Nagai, H. Aplysiadione and aplysiaenal: Truncated biosynthetic intermediates of aplysiatoxins from a cyanobacterium. Results Chem. 2021, 3, 100206. [Google Scholar] [CrossRef]
- Iguchi, K.; Satake, M.; Nishio, Y.; Zhang, B.T.; Kawashima, K.; Uchida, H.; Nagai, H. Debromooscillatoxins G and I from the Cyanobacterium Moorea producens. Int. J. Rev. Commun. Heterocycl. Chem. 2021, 102, 1287–1293. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Yanagita, R.C.; Hamada, N.; Murakami, A.; Takahashi, H.; Saito, N.; Nagai, H.; Irie, K. A simple analogue of tumor-promoting aplysiatoxin is an antineoplastic agent rather than a tumor promoter: Development of a synthetically accessible protein kinase C activator with bryostatin-like activity. J. Am. Chem. Soc. 2009, 131, 7573–7579. [Google Scholar] [CrossRef] [PubMed]
- Kishi, Y.; Rando, R.R. Structural basis of protein kinase C activation by tumor promoters. Acc. Chem. Res. 1998, 31, 163–172. [Google Scholar] [CrossRef]
- Griner, E.M.; Kazanietz, M.G. Protein kinase C and other diacylglycerol effectors in cancer. Nat. Rev. Cancer. 2007, 7, 281–294. [Google Scholar] [CrossRef]
- Ashida, Y.; Yanagita, R.C.; Takahashi, C.; Kawanami, Y.; Irie, K. Binding mode prediction of aplysiatoxin, a potent agonist of protein kinase C, through molecular simulation and structure-activity study on simplified analogs of the receptor-recognition domain. Bioorg. Med. Chem. 2016, 24, 4218–4227. [Google Scholar] [CrossRef] [PubMed]
- Wulff, H.; Castle, N.A.; Pardo, L.A. Voltage-gated potassium channels as therapeutic targets. Nat. Rev. Drug Discov. 2009, 8, 982–1001. [Google Scholar] [CrossRef]
- Tang, Y.H.; Wu, J.; Fan, T.T.; Zhang, H.H.; Gong, X.X.; Cao, Z.Y.; Zhang, J.; Lin, H.W.; Han, B.N. Chemical and biological study of aplysiatoxin derivatives showing inhibition of potassium channel Kv1.5. RSC Adv. 2019, 9, 7594–7600. [Google Scholar] [CrossRef]
- Fan, T.T.; Zhang, H.H.; Tang, Y.H.; Zhang, F.Z.; Han, B.N. Two new neo-debromoaplysiatoxins-a pair of stereoisomers exhibiting potent Kv1.5 ion channel inhibition activities. Mar. Drugs 2019, 17, 652. [Google Scholar] [CrossRef]
- Tang, Y.H.; Liang, T.T.; Fan, T.T.; Keen, L.J.; Zhang, X.D.; Xu, L.; Zhao, Q.; Zeng, R.; Han, B.N. Neo-debromoaplysiatoxin C, with new structural rearrangement, derived from debromoaplysiatoxin. Nat. Prod. Res. 2020, 34, 2151–2156. [Google Scholar] [CrossRef] [PubMed]
- Hada, K.; Araki, Y.; Nokura, Y.; Urabe, D.; Nishikawa, T. Collective synthesis of aplysiatoxin/oscillatoxin analogues by a bioinspired strategy. J. Org. Chem. 2022, 87, 15618–15633. [Google Scholar] [CrossRef] [PubMed]
- Acton, N.; Roth, R.J. On the conversion of dihydroartemisinic acid into artemisinin. J. Org. Chem. 1992, 57, 3610–3614. [Google Scholar] [CrossRef]
- Zhao, Q.; Gao, J.J.; Qin, X.J.; Hao, X.J.; He, H.P.; Liu, H.Y. Hedychins A and B, 6,7-dinorlabdane diterpenoids with a peroxide bridge from Hedychium forrestii. Org. Lett. 2018, 20, 704–707. [Google Scholar] [CrossRef] [PubMed]
- Li, H.X.; Huang, H.B.; Shao, C.L.; Huang, H.R.; Jiang, J.Y.; Zhu, X.; Liu, Y.Y.; Liu, L.; Lu, Y.J.; Li, M.F.; et al. Cytotoxic norsesquiterpene peroxides from the endophytic fungus Talaromyces flavus isolated from the mangrove plant Sonneratia apetala. J. Nat. Prod. 2011, 74, 1230–1235. [Google Scholar] [CrossRef]
- Entzeroth, M.; Blackman, A.J.; Mynderse, J.S.; Moore, R.E. Structures and stereochemistries of oscillatoxin B, 31-noroscillatoxin B, oscillatoxin D, and 30-methyloscillatoxin D. J. Org. Chem. 1985, 50, 1255–1259. [Google Scholar] [CrossRef]
- Nagai, H.; Watanabe, M.; Sato, S.; Kawaguchi, M.; Xiao, Y.-Y.; Hayashi, K.; Watanabe, R.; Uchida, H.; Satake, M. New aplysiatoxin derivatives from the Okinawan cyanobacterium Moorea producens. Tetrahedron 2019, 75, 2486–2494. [Google Scholar] [CrossRef]
- Rouzer, C.A.; Marnett, L.J. Cyclooxygenases: Structural and functional insights. J. Lipid Res. 2009, 50, S29–S34. [Google Scholar] [CrossRef]
- Steffan, N.; Grundmann, A.; Afiyatullov, S.; Ruan, H.; Li, S.M. FtmOx1, a non-heme Fe(II) and alpha-ketoglutarate-dependent dioxygenase, catalyses the endoperoxide formation of verruculogen in Aspergillus fumigatus. Org. Biomol. Chem. 2009, 7, 4082–4087. [Google Scholar] [CrossRef]
- Guo, X.K.; Chen, W.L.; Sun, H.P.; You, Q.D. Kv1. 5 inhibitors for treatment of atrial fibrillation: A tradeoff between selectivity and non-selectivity. Curr. Top. Med. Chem. 2016, 16, 1843–1854. [Google Scholar] [CrossRef] [PubMed]
- Norris, M.D.; Perkins, M.V. Structural diversity and chemical synthesis of peroxide and peroxide-derived polyketide metabolites from marine sponges. Nat. Prod. Rep. 2016, 33, 861–880. [Google Scholar] [CrossRef]
- Fattorusso, E.; Taglialatela-Scafati, O. Marine endoperoxides as antimalarial lead compounds. Phytochem. Rev. 2010, 9, 515–524. [Google Scholar] [CrossRef]
- Cheng, F.; Shen, J.H.; Luo, X.M.; Zhu, W.L.; Gu, J.D.; Ji, R.Y.; Jiang, H.L.; Chen, K.X. Molecular docking and 3-D-QSAR studies on the possible antimalarial mechanism of artemisinin analogues. Bioorgan. Med. Chem. 2002, 10, 2883–2891. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.Y.; Ma, X.Y.; Cai, X.Q.; Yan, P.C.; Yue, L.; Lin, C.; Shao, W.W. Sesquiterpenoids from Curcuma wenyujin with anti-influenza viral activities. Phytochemistry 2013, 85, 122–128. [Google Scholar] [CrossRef]
- Wibowo, M.; Prachyawarakorn, V.; Aree, T.; Mahidol, C.; Ruchirawat, S.; Kittakoop, P. Cytotoxic sesquiterpenes from the endophytic fungus Pseudolagarobasidium acaciicola. Phytochemistry 2016, 122, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Dembitsky, V.M.; Gloriozova, T.A.; Poroikov, V.V. Natural peroxy anticancer agents. Mini Rev. Med. Chem. 2007, 7, 571–589. [Google Scholar] [CrossRef]
- Youssef, D.T.; Shaala, L.A.; Mohamed, G.A.; Ibrahim, S.R.; Banjar, Z.M.; Badr, J.M.; McPhail, K.L.; Risinger, A.L.; Mooberry, S.L. 2,3-seco-2,3-dioxo-lyngbyatoxin A from a Red Sea strain of the marine cyanobacterium Moorea producens. Nat. Prod. Res. 2015, 29, 703–709. [Google Scholar] [CrossRef] [PubMed]
- Kikumori, M.; Yanagita, R.C.; Tokuda, H.; Suzuki, N.; Nagai, H.; Suenaga, K.; Irie, K. Structure-activity studies on the spiroketal moiety of a simplified analogue of debromoaplysiatoxin with antiproliferative activity. J. Med. Chem. 2012, 55, 5614–5626. [Google Scholar] [CrossRef]
- Grimblat, N.; Zanardi, M.M.; Sarotti, A.M. Beyond DP4: An improved probability for the stereochemical assignment of isomeric compounds using quantum chemical calculations of NMR shifts. J. Org. Chem. 2015, 80, 12526–12534. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).