
Copper-Catalyzed Asymmetric Dearomative [3+2] Cycloaddition of Nitroheteroarenes with Azomethines
 ,
,
Abstract
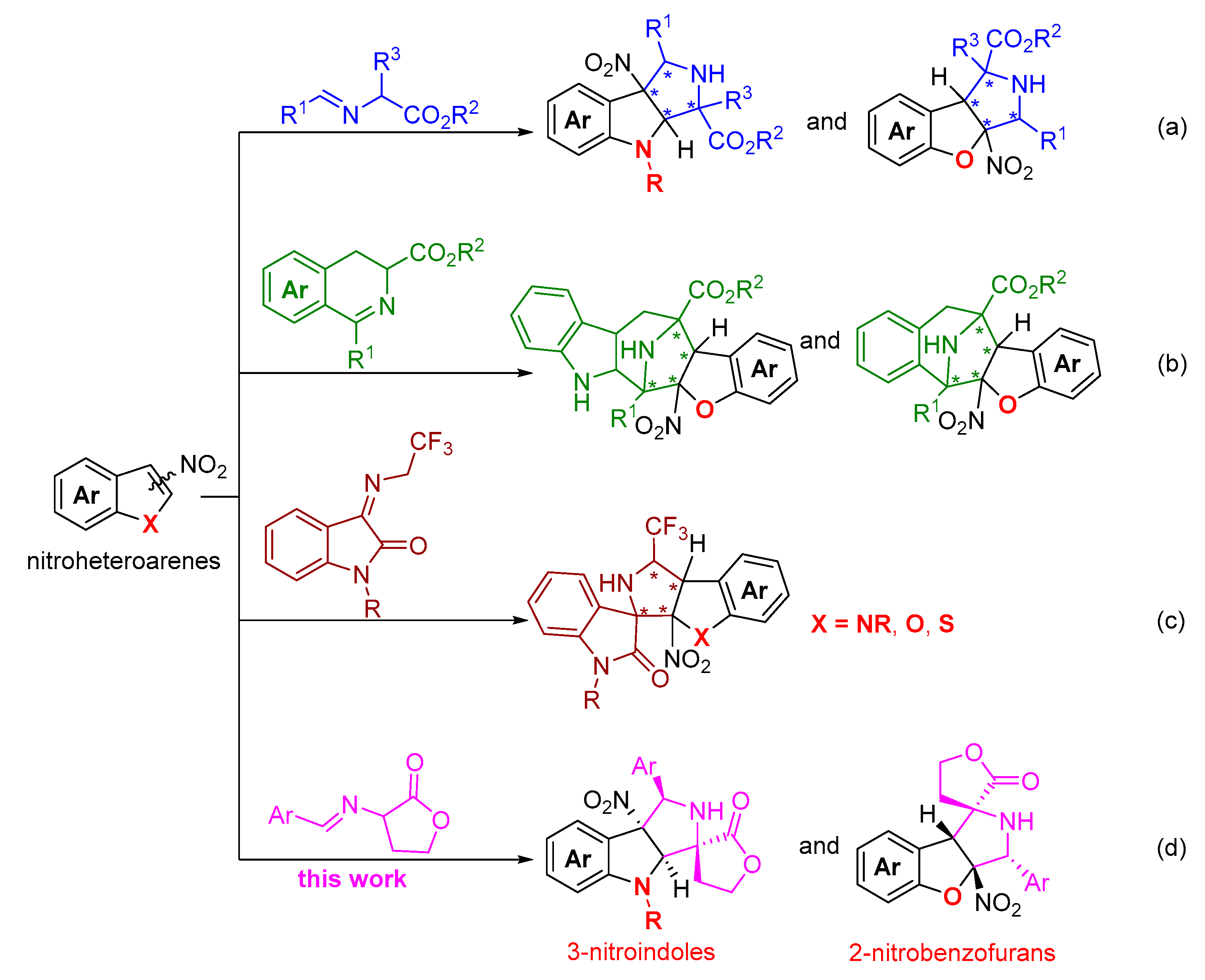
1. Introduction
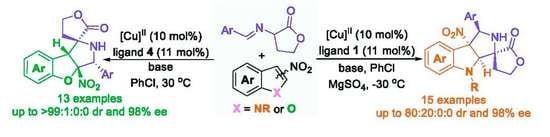
2. Results and Discussion
2.1. Optimization Studies
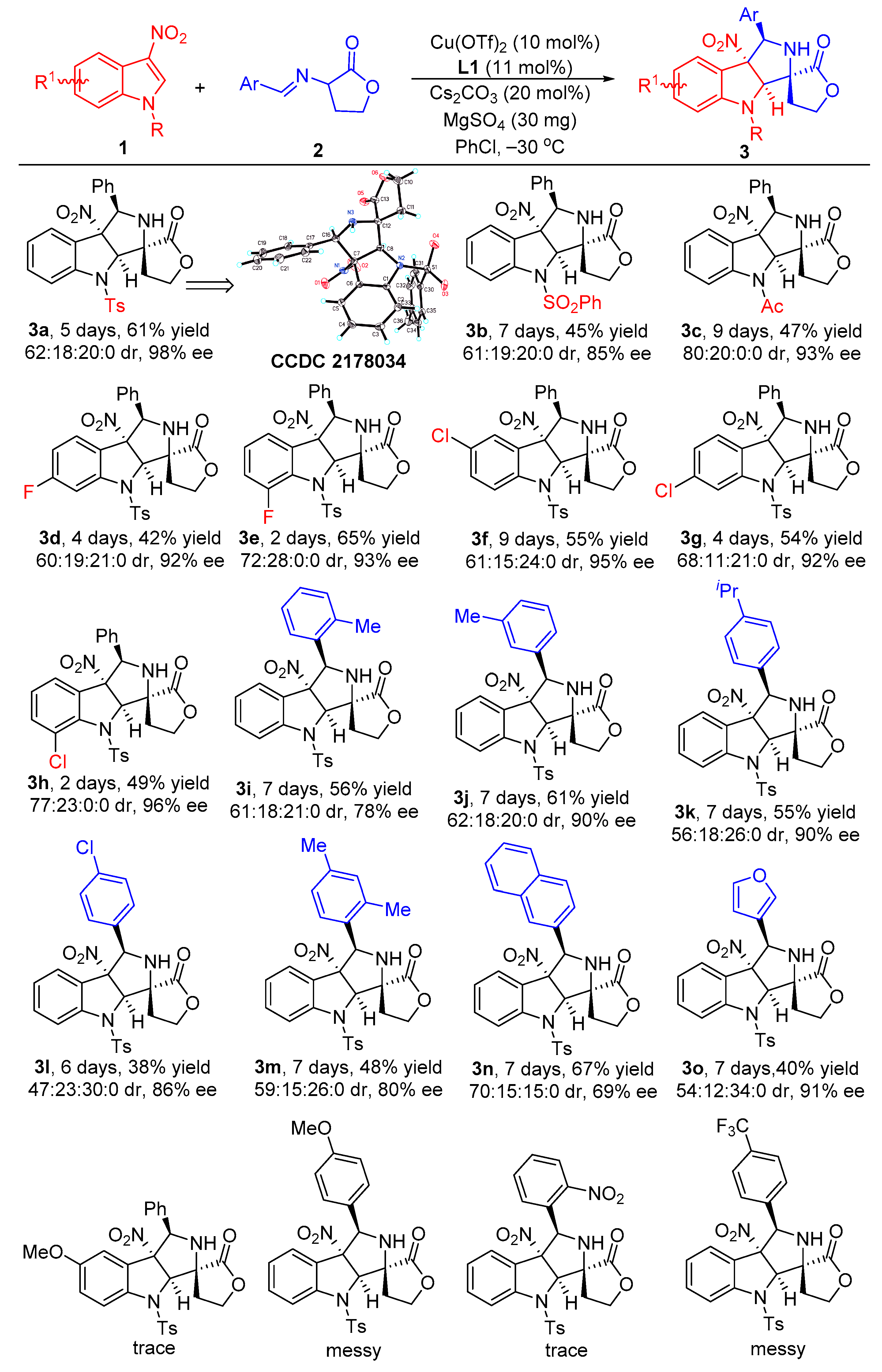
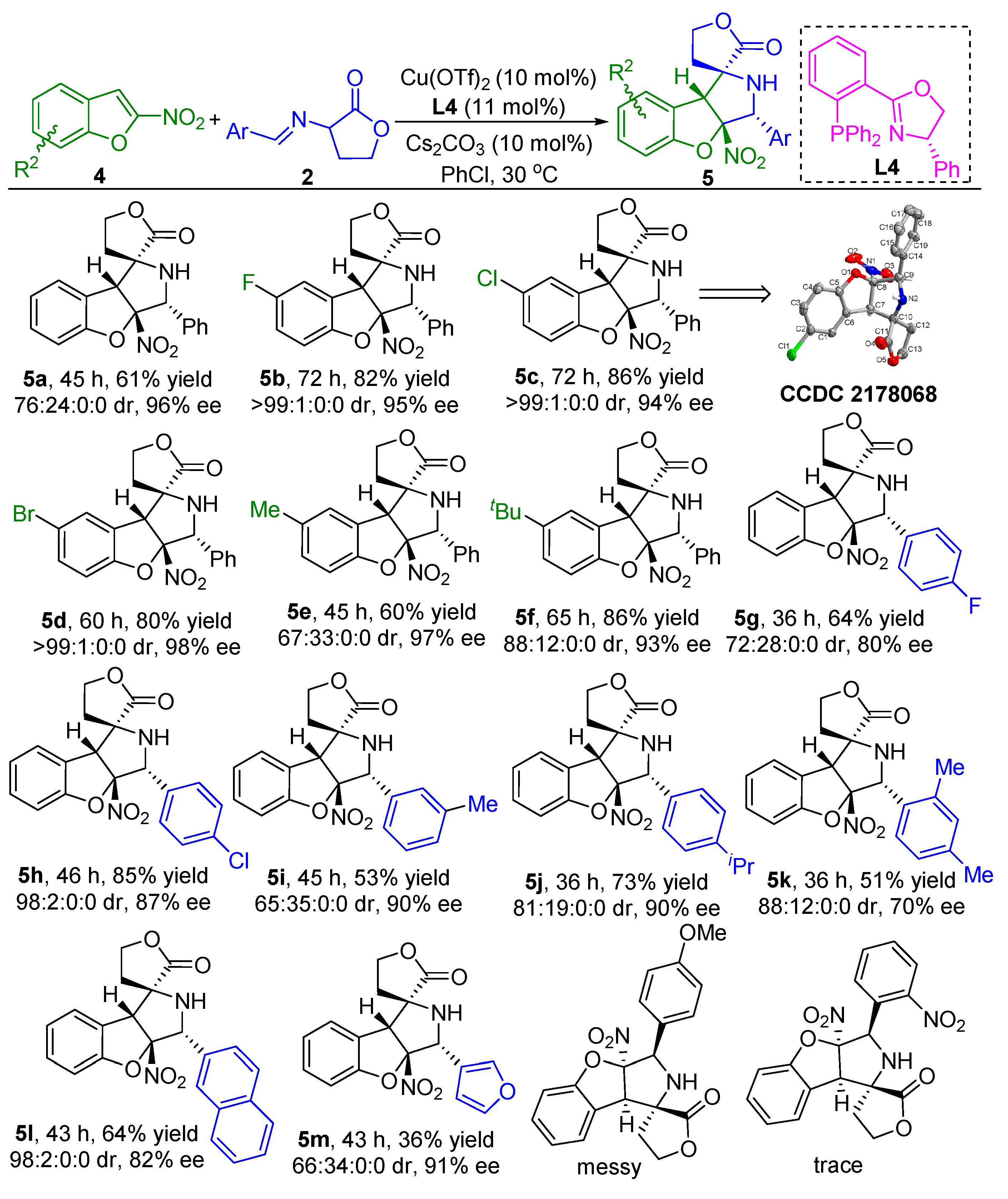
2.2. Substrate Scope Studies
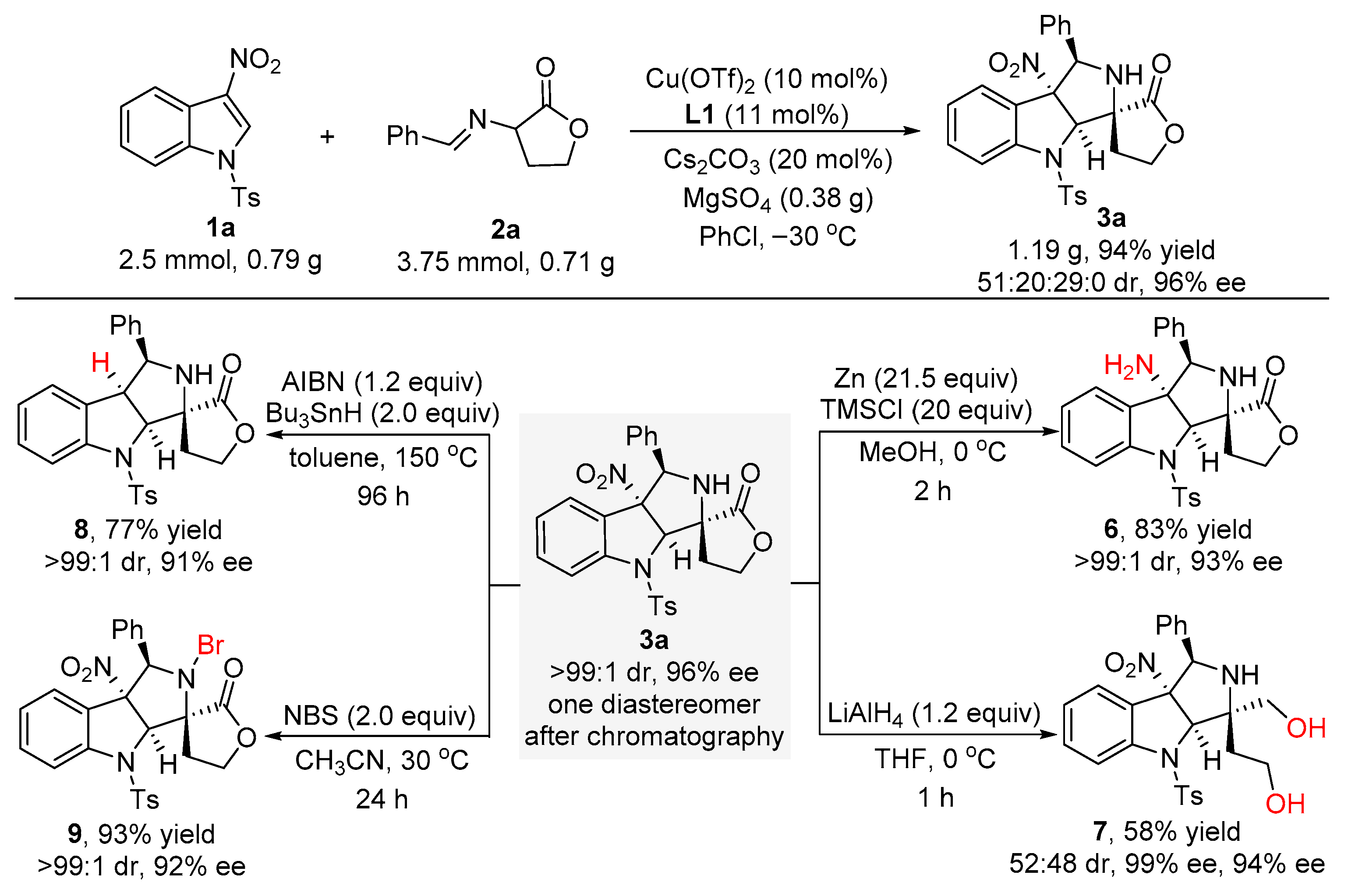
2.3. Scale-Up Experiment and the Versatile Transformations of Product 3a
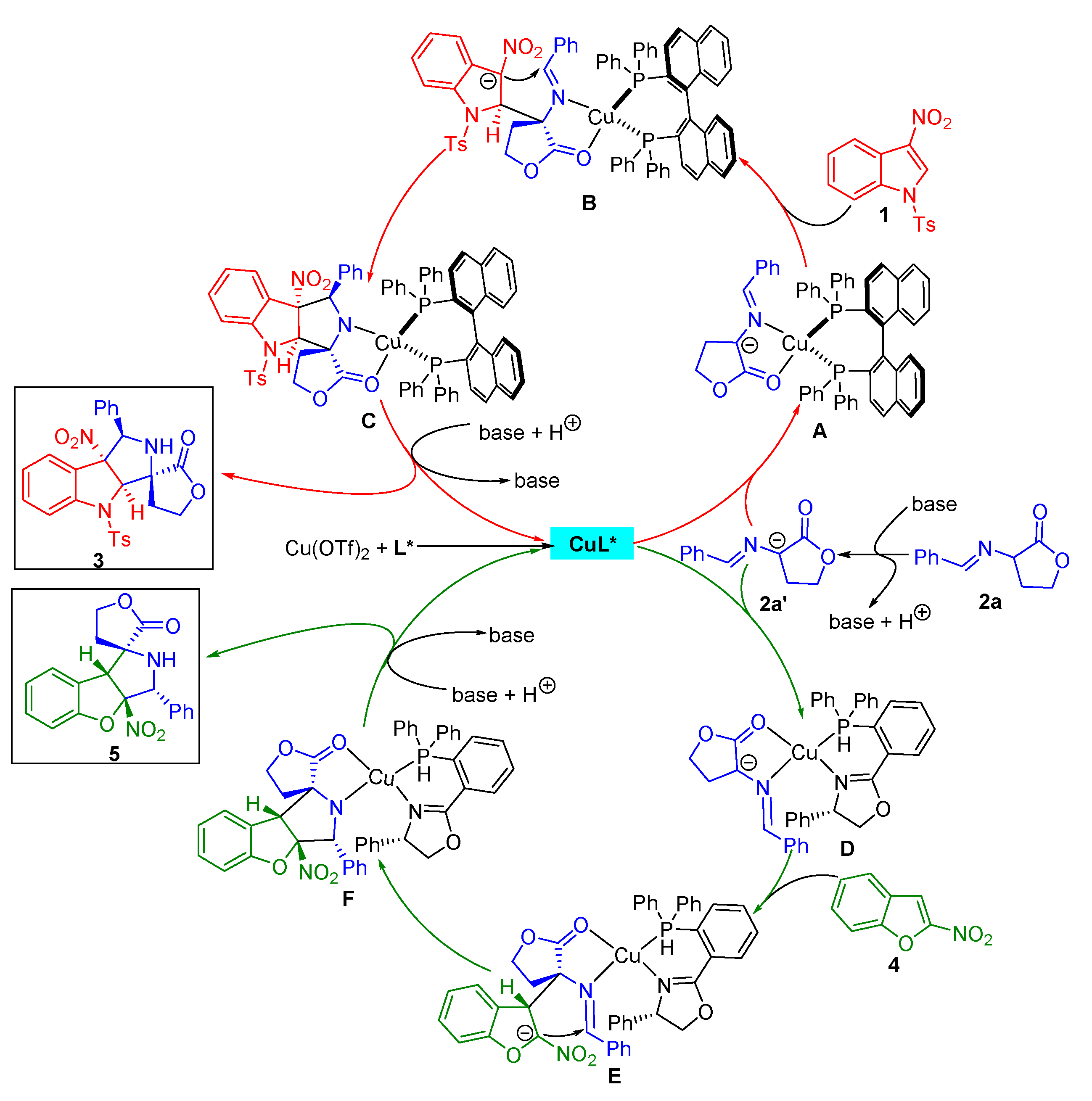
2.4. Proposed Mechanism for the Catalytic Asymmetric Dearomative [3+2] Cycloaddition
3. Materials and Methods
3.1. General Information
3.2. General Experimental Procedure for the Catalytic Asymmetric Dearomative [3+2] Cycloaddtion Reaction of α-imino γ-lactones with 3-nitroindoles for the Synthesis of Products 3 (Scheme 2)
3.3. General Experimental Procedure for the Catalytic Asymmetric Dearomative [3+2] Cycloaddtion Reaction of α-imino γ-lactones with 2-nitrobenzofurans for the Synthesis of Products 5 (Scheme 3)
3.4. Procedure for the Catalytic Asymmetric Synthesis of Product 3a on a 2.5 mmol Scale
3.5. Procedure for the Synthesis of Compound 6
3.6. Procedure for the Synthesis of Compound 7
3.7. Procedure for the Synthesis of Compound 8
3.8. Procedure for the Synthesis of Compound 9
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References and Notes
- Lee, K.K.; Zhou, B.N.; Kingston, D.G.I.; Vaisberg, A.J.; Hammond, G.B. Bioactive Indole Alkaloids from the Bark of Uncaria guianensis. Planta Med. 1999, 65, 759–760. [Google Scholar] [CrossRef]
- Kang, T.-H.; Matsumoto, K.; Murakami, Y.; Takayama, H.; Kitajima, M.; Aimi, N.; Watanabe, H. Pteropodine and isopteropodine positively modulate the function of rat muscarinic M1 and 5-HT2 receptors expressed in Xenopus oocyte. Eur. J. Pharmacol. 2002, 444, 39–45. [Google Scholar] [CrossRef]
- Raj, A.A.; Raghunathan, R.; SrideviKumari, M.R.; Raman, N. Synthesis, Antimicrobial and Antifungal Activity of a New Class of Spiro pyrrolidines. Bioorg. Med. Chem. 2003, 11, 407–419. [Google Scholar] [CrossRef]
- Dzedulionytė, K.; Veikšaitė, M.; Morávek, V.; Malinauskienė, V.; Račkauskienė, G.; Šačkus, A.; Žukauskaitė, A.; Arbačiauskienė, E. Convenient Synthesis of N-Heterocycle-Fused Tetrahydro-1,4-diazepinones. Molecules 2022, 27, 8666. [Google Scholar] [CrossRef] [PubMed]
- Hong, K.; Huang, J.; Yao, M.; Xu, X. Recent Advances in Nitrene/Alkyne Metathesis Cascade Reaction. Chin. J. Org. Chem. 2022, 42, 344–352. [Google Scholar] [CrossRef]
- Philippov, I.; Gatilov, Y.; Sonina, A.; Vorob’ev, A. Oxidative [3+2] Cycloaddition of Alkynylphosphonates with Heterocyclic N-Imines: Synthesis of Pyrazolo [1,5-a]Pyridine-3-phosphonates. Molecules 2022, 27, 7913. [Google Scholar] [CrossRef]
- Dong, S.; Fu, X.; Xu, X. [3+2]-Cycloaddition of Catalytically Generated Pyridinium Ylide: A General Access to Indolizine Derivatives. Asian J. Org. Chem. 2020, 9, 1133–1143. [Google Scholar] [CrossRef]
- Hazra, A.; Bharitkar, Y.P.; Chakraborty, D.; Mondal, S.K.; Singal, N.; Mondal, S.; Maity, A.; Paira, R.; Banerjee, S.; Mondal, N.B. Regio- and Stereoselective Synthesis of a Library of Bioactive Dispiro-Oxindolo/Acenaphthoquino Andrographolides via 1,3-Dipolar Cycloaddition Reaction Under Microwave Irradiation. ACS Comb. Sci. 2013, 15, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Becker, M.H.; Chua, P.; Downham, R.; Douglas, C.J.; Garg, N.K.; Hiebert, S.; Jaroch, S.; Matsuoka, R.T.; Middleton, J.A.; Ng, F.W.; et al. Total Synthesis of (-)-Sarain A. J. Am. Chem. Soc. 2007, 129, 11987–12002. [Google Scholar] [CrossRef]
- Sun, M.-R.; Lu, H.-T.; Wang, Y.-Z.; Yang, H.; Liu, H.-M. Highly Efficient Formal Synthesis of Cephalotaxine, Using the Stevens Rearrangement-Acid Lactonization Sequence as A Key Transformation. J. Org. Chem. 2009, 74, 2213–2216. [Google Scholar] [CrossRef]
- Zhuo, C.-X.; Zhang, W.; You, S.-L. Catalytic Asymmetric Dearomatization Reactions. Angew. Chem. Int. Ed. 2012, 51, 12662–12686. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, C.-X.; Zheng, C.; You, S.-L. Transition-Metal-Catalyzed Asymmetric Allylic Dearomatization Reactions. Acc. Chem. Res. 2014, 47, 2558–2573. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; You, S.-L. Catalytic Asymmetric Dearomatization by Transition-Metal Catalysis: A Method for Transformations of Aromatic Compounds. Chem 2016, 1, 830–857. [Google Scholar] [CrossRef]
- Wu, W.-T.; Zhang, L.; You, S.-L. Recent Progress on Gold-catalyzed Dearomatization Reactions. Acta Chim. Sinica 2017, 75, 419–438. [Google Scholar] [CrossRef]
- Zheng, C.; You, S.-L. Catalytic asymmetric dearomatization (CADA) reaction-enabled total synthesis of indole-based natural products. Nat. Prod. Rep. 2019, 36, 1589–1605. [Google Scholar] [CrossRef]
- Xia, Z.-L.; Xu-Xu, Q.-F.; Zheng, C.; You, S.-L. Chiral phosphoric acid-catalyzed asymmetric dearomatization reactions. Chem. Soc. Rev. 2020, 49, 286–300. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; You, S.-L. Advances in Catalytic Asymmetric Dearomatization. ACS Cent. Sci. 2021, 7, 432–444. [Google Scholar] [CrossRef]
- Wu, W.-T.; Zhang, L.; You, S.-L. Catalytic asymmetric dearomatization (CADA) reactions of phenol and aniline derivatives. Chem. Soc. Rev. 2016, 45, 1570–1580. [Google Scholar] [CrossRef]
- Huang, G.; Yin, B. Recent Developments in Transition Metal-Catalyzed Dearomative Cyclizations of Indoles as Dipolarophiles for the Construction of Indolines. Adv. Synth. Catal. 2019, 361, 405–425. [Google Scholar] [CrossRef]
- Sheng, F.-T.; Wang, J.-Y.; Tan, W.; Zhang, Y.-C.; Shi, F. Progresses in organocatalytic asymmetric dearomatization reactions of indole derivatives. Org. Chem. Front. 2020, 7, 3967–3998. [Google Scholar] [CrossRef]
- Trost, B.M.; Ehmke, V.; O’Keefe, B.M.; Bringley, D.A. Palladium-Catalyzed Dearomative Trimethylenemethane Cycloaddition Reactions. J. Am. Chem. Soc. 2014, 136, 8213–8216. [Google Scholar] [CrossRef]
- Cerveri, A.; Bandini, M. Recent Advances in the Catalytic Functionalization of “Electrophilic” Indoles. Chin. J. Chem. 2020, 38, 287–294. [Google Scholar] [CrossRef]
- Rkein, B.; Bigot, A.; Birbaum, L.; Manneveau, M.; De Paolis, M.; Legros, J.; Chataigner, I. Reactivity of 3-nitroindoles with electron-rich species. Chem. Commun. 2021, 57, 27–44. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.R.; Baire, B. Recent Dearomatization Strategies of Benzofurans and Benzothiophenes. Asian J. Org. Chem. 2021, 10, 932–948. [Google Scholar] [CrossRef]
- Wang, N.; Ren, J.; Li, K. Dearomatization of Nitro(hetero)arenes through Annulation. Eur. J. Org. Chem. 2022, 2022, e202200039. [Google Scholar] [CrossRef]
- Li, Y.-L.; Wang, K.-K.; He, X.-L. Recent Progress of Electron-Withdrawing-Group-Tethered Arenes Involved Asymmetric Nucleophilic Aromatic Functionalizations. Adv. Synth. Catal. 2022, 364, 3630–3650. [Google Scholar] [CrossRef]
- Awata, A.; Arai, T. PyBidine/Copper Catalyst: Asymmetric exo′-Selective [3+2] Cycloaddition using Imino Ester and Electrophilic Indole. Angew. Chem. Int. Ed. 2014, 53, 10462–10465. [Google Scholar] [CrossRef]
- Gerten, A.L.; Stanley, L.M. Enantioselective dearomative [3+2] cycloadditions of indoles with azomethine ylides derived from alanine imino esters. Org. Chem. Front. 2016, 3, 339–343. [Google Scholar] [CrossRef]
- Liang, L.; Niu, H.-Y.; Wang, D.-C.; Yang, X.-H.; Qu, G.-R.; Guo, H.-M. Facile synthesis of chiral [2,3]-fused hydrobenzofuran via asymmetric Cu(I)-catalyzed dearomative 1,3-dipolar cycloaddition. Chem. Commun. 2019, 55, 553–556. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, D.-C.; Xie, M.-S.; Qu, G.-R.; Guo, H.-M. Enantioselective Synthesis of Fused Polycyclic Tropanes via Dearomative [3+2] Cycloaddition Reactions of 2-Nitrobenzofurans. Org. Lett. 2020, 22, 164–167. [Google Scholar] [CrossRef]
- Tan, J.-P.; Li, X.; Chen, Y.; Rong, X.; Zhu, L.; Jiang, C.; Xiao, K.; Wang, T. Highly stereoselective construction of polycyclic benzofused tropane scaffolds and their latent bioactivities: Bifunctional phosphonium salt-enabled cyclodearomatization process. Sci. China Chem. 2020, 63, 1091–1099. [Google Scholar] [CrossRef]
- Jiang, Z.; Liu, X.; Zhang, H.; Tan, J.-P.; Ren, X.; Gao, G.; Wang, T. Bifunctinoal Phosphonium Salt-Catalyzed Asymmetric Cyclodearomatization of 2-Nitroindoles and 2-Nitrobenzofurans for Constructing CF3-Containing Spiro-Polycycles. Adv. Synth. Catal. 2021, 363, 3115–3120. [Google Scholar] [CrossRef]
- Saktura, M.; Skrzyńska, A.; Frankowski, S.; Wódka, S.; Albrecht, Ł. Asymmetric Dearomative (3+2)-Cycloaddition Involving Nitro-Substituted Benzoheteroarenes under H-Bonding Catalysis. Molecules 2021, 26, 4992. [Google Scholar] [CrossRef]
- Zhao, J.-Q.; Zhou, S.; Yang, L.; Du, H.-Y.; You, Y.; Wang, Z.-H.; Zhou, M.-Q.; Yuan, W.-C. Catalytic Asymmetric Dearomative 1,3-Dipolar Cycloaddition of 2-Nitrobenzothiophenes and Isatin-Derived Azomethine Ylides. Org. Lett. 2021, 23, 8600–8605. [Google Scholar] [CrossRef]
- Zhou, P.; Yi, Y.; Hua, Y.-Z.; Jia, S.-K.; Wang, M.-C. Dinuclear Zinc Catalyzed Enantioselective Dearomatization [3+2] Annulation of 2-Nitrobenzofurans and 2-Nitrobenzothiophenes. Chem. Eur. J. 2021, 28, e202103688. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.-J.; Zhao, J.-Q.; Lai, Y.-Q.; You, Y.; Wang, Z.-H.; Yuan, W.-C. Organocatalyzed asymmetric dearomative 1,3-dipolar cycloaddition of 2-nitrobenzofurans and N-2,2,2-trifluoroethylisatin ketimines. Chirality 2022, 34, 1019–1034. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.-C.; Zhou, X.-J.; Zhao, J.-Q.; Chen, Y.-Z.; You, Y.; Wang, Z.-H. Catalytic Enantioselective Dearomatization/Rearomatization of 2-Nitroindoles to Access 3-Indolyl-3′-Aryl-/Alkyloxindoles: Application in the Formal Synthesis of Cyclotryptamine Alkaloids. Org. Lett. 2020, 22, 7088–7093. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.-Q.; Zhou, S.; Wang, Z.-H.; You, Y.; Chen, S.; Liu, X.-L.; Zhou, M.-Q.; Yuan, W.-C. Catalytic asymmetric dearomative [4 + 2] annulation of 2-nitrobenzofurans and 5H-thiazol-4-ones: Stereoselective construction of dihydrobenzofuran-bridged polycyclic skeletons. Org. Chem. Front. 2021, 8, 6330–6336. [Google Scholar] [CrossRef]
- Zhao, J.-Q.; Zhou, S.; Qian, H.-L.; Wang, Z.-H.; Zhang, Y.-P.; You, Y.; Yuan, W.-C. Higher-order [10 + 2] cycloaddition of 2-alkylidene-1-indanones enables the dearomatization of 3-nitroindoles: Access to polycyclic cyclopenta[b]indoline derivatives. Org. Chem. Front. 2022, 9, 3322–3327. [Google Scholar] [CrossRef]
- Yuan, W.-C.; Chen, X.-M.; Zhao, J.-Q.; Zhang, Y.-P.; Wang, Z.-H.; You, Y. Ag-Catalyzed Asymmetric Interrupted Barton-Zard Reaction Enabling the Enantioselective Dearomatization of 2- and 3-Nitroindoles. Org. Lett. 2022, 24, 826–831. [Google Scholar] [CrossRef]
- Liu, T.-L.; He, Z.-L.; Tao, H.-Y.; Wang, C.-J. Stereoselective Construction of Spiro(butyrolactonepyrrolidines) by Highly Efficient Copper(I)/TF-BiphamPhos-Catalyzed Asymmetric 1,3-Dipolar Cycloaddition. Chem. Eur. J. 2012, 18, 8042–8046. [Google Scholar] [CrossRef] [PubMed]
- Teng, H.-L.; Huang, H.; Wang, C.-J. Catalytic Asymmetric Construction of Spiro(γ-butyrolactam-γ-butyrolactone) Moieties through Sequential Reactions of Cyclic Imino Esters with Morita–Baylis–Hillman Bromides. Chem. Eur. J. 2012, 18, 12614–14628. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Shi, X.-M.; Dong, W.-P.; Zhua, L.-P.; Wang, R. Efficient construction of highly functionalized spiro[γ-butyrolactone-pyrrolidin-3,3′-oxindole] tricyclic skeletons via an organocatalytic 1,3-dipolar cycloaddition. Chem. Commun. 2013, 49, 3458–3460. [Google Scholar] [CrossRef]
- Cayuelas, A.; Ortiz, R.; Nájera, C.; Sansano, J.M.; Larrañaga, O.; de Cózar, A.; Cossío, F.P. Enantioselective Synthesis of Polysubstituted Spiro-nitroprolinates Mediated by a (R,R)-Me-DuPhos·AgF-Catalyzed 1,3-Dipolar Cycloaddition. Org. Lett. 2016, 18, 2926–2929. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.-W.; Li, C.; Zhao, B.-J.; Lan, L.; Zhang, M.; Zhou, Z.-M. Diastereoselective construction of spirocyclic pyrrolidines bearing two quaternary centers via CuII-P, N-Ligand catalyzed 1,3-dipolar cycloaddition. Tetrahedron 2017, 73, 923–930. [Google Scholar] [CrossRef]
- Chen, N.; Zhu, L.; Gan, L.; Liu, Z.; Wang, R.; Cai, X.; Jiang, X. Asymmetric Synthesis of Bispiro[γ-butyrolactone-pyrrolidin-4,4′-pyrazolone] Scaffolds Containing Two Quaternary Spirocenters via an Organocatalytic 1,3-Dipolar Cycloaddition. Eur. J. Org. Chem. 2018, 2018, 2939–2943. [Google Scholar] [CrossRef]
- Guo, D.-G.; Li, Z.; Han, X.-X.; Zhang, L.; Zhang, M.; Liu, X.-L. Decarboxylative, Diastereoselective and exo-Selective 1,3-Dipolar Cycloaddition for Diversity-Oriented Construction of Structural Spiro[Butyrolactone–Pyrrolidine–Chromanone] Hybrids. Synlett 2021, 32, 1447–1452. [Google Scholar]
- According to reviewer’s require, we screened other BINAP derivatives ligands with Cu(CH3CN)4PF6 as the metal source in PhCl at rt for 72 h ((R)-Tol-BINAP: 33% yield, 45:39:16:0 dr, 69% ee; (S)-Di(3,5-xylyl)-BINAP: 21% yield, 70:10:20:0 dr, 45% ee; (R)-Segphos: 15% yield, 83:8:9:0 dr, 73% ee).
- CCDC 2178034 (3a) and CCDC 2178068 (5c) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | [Cu] | Ligand | Solvent | Time (h) | Yield (%) [b] | dr [c] | ee (%) [d] |
|---|---|---|---|---|---|---|---|
| 1 | Cu(CH3CN)4PF6 | L1 | MTBE | 72 | 94 (59) | 63:15:22:0 | 60 |
| 2 | Cu(CH3CN)4PF6 | L2 | MTBE | 72 | 96 (64) | 67:20:13:0 | 42 |
| 3 | Cu(CH3CN)4PF6 | L3 | MTBE | 66 | 93 (47) | 50:15:35:0 | 35 |
| 4 | CuBr | L1 | MTBE | 70 | 85 (54) | 63:11:26:0 | 21 |
| 5 | Cu(OAc)2 | L1 | MTBE | 70 | 72 (47) | 65:10:24:0 | 31 |
| 6 | Cu(OTf)2 | L1 | MTBE | 70 | 91 (52) | 57:12:31:0 | 81 |
| 7 | Cu(OTf)2 | L1 | THF | 68 | 87 (60) | 69:9:22:0 | 17 |
| 8 | Cu(OTf)2 | L1 | CH2Cl2 | 68 | 86 (52) | 61:11:28:0 | 46 |
| 9 | Cu(OTf)2 | L1 | toluene | 68 | 92 (46) | 50:15:35:0 | 78 |
| 10 | Cu(OTf)2 | L1 | PhCl | 68 | 82 (45) | 55:16:29:0 | 85 |
| 11 [e] | Cu(OTf)2 | L1 | PhCl | 90 | 99 (51) | 52:16:32:0 | 89 |
| 12 [e,f] | Cu(OTf)2 | L1 | PhCl | 120 | 99 (49) | 49:18:33:0 | 91 |
| 13 [e,g] | Cu(OTf)2 | L1 | PhCl | 120 | 99 (61) | 62:18:20:0 | 98 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Zhao, J.-Q.; Zhang, Y.-P.; Zhou, M.-Q.; Zhang, X.-M.; Yuan, W.-C. Copper-Catalyzed Asymmetric Dearomative [3+2] Cycloaddition of Nitroheteroarenes with Azomethines. Molecules 2023, 28, 2765. https://doi.org/10.3390/molecules28062765
Chen Y, Zhao J-Q, Zhang Y-P, Zhou M-Q, Zhang X-M, Yuan W-C. Copper-Catalyzed Asymmetric Dearomative [3+2] Cycloaddition of Nitroheteroarenes with Azomethines. Molecules. 2023; 28(6):2765. https://doi.org/10.3390/molecules28062765
Chicago/Turabian StyleChen, Yan, Jian-Qiang Zhao, Yan-Ping Zhang, Ming-Qiang Zhou, Xiao-Mei Zhang, and Wei-Cheng Yuan. 2023. "Copper-Catalyzed Asymmetric Dearomative [3+2] Cycloaddition of Nitroheteroarenes with Azomethines" Molecules 28, no. 6: 2765. https://doi.org/10.3390/molecules28062765
APA StyleChen, Y., Zhao, J.-Q., Zhang, Y.-P., Zhou, M.-Q., Zhang, X.-M., & Yuan, W.-C. (2023). Copper-Catalyzed Asymmetric Dearomative [3+2] Cycloaddition of Nitroheteroarenes with Azomethines. Molecules, 28(6), 2765. https://doi.org/10.3390/molecules28062765