
Efficient Synthesis of 1H-Benzo[4,5]imidazo[1,2-c][1,3]oxazin-1-one Derivatives Using Ag2CO3/TFA-Catalyzed 6-endo-dig Cyclization: Reaction Scope and Mechanistic Study

 , ,
, ,  ,
,
Abstract
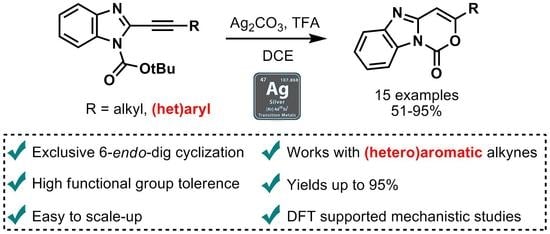
1. Introduction
2. Results and Discussion
Theoretical Calculation (Computational Studies)
3. Materials and Methods
3.1. General Information
3.2. General Procedure for the Synthesis of Tert-butyl 2-alkynyl-1H-benzimidazole-1-carboxylate (4a–o)
3.3. General Procedure for the Synthesis of Benzo[1′,2′:4,5]imidazo[1,2-c][1,3]oxazin-1-one (5a–o)
3.4. Details of DFT Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Chen, K.X.; Venkatraman, S.; Anilkumar, G.N.; Zeng, Q.; Lesburg, C.A.; Vibulbhan, B.; Velazquez, F.; Chan, T.-Y.; Bennett, F.; Jiang, Y.; et al. Discovery of SCH 900188: A potent hepatitis C virus NS5B polymerase inhibitor prodrug as a development candidate. ACS Med. Chem. Lett. 2014, 5, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Venkatraman, S.; Velazquez, F.; Gavalas, S.; Wu, W.; Chen, K.X.; Nair, A.G.; Bennett, F.; Huang, Y.; Pinto, P.; Jiang, Y.; et al. Optimization of potency and pharmacokinetics of tricyclic indole derived inhibitors of HCV NS5B polymerase. Identification of ester prodrugs with improved oral pharmacokinetics. Bioorg. Med. Chem. 2014, 22, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.X.; Lesburg, C.A.; Vibulbhan, B.; Yang, W.; Chan, T.-Y.; Venkatraman, S.; Velazquez, F.; Zeng, Q.; Bennett, F.; Anilkumar, G.N.; et al. A Novel Class of Highly Potent Irreversible Hepatitis C Virus NS5B Polymerase Inhibitors. J. Med. Chem. 2012, 55, 2089–2101. [Google Scholar] [CrossRef]
- Neagoie, C.; Vedrenne, V.; Buron, F.; Mérour, J.-Y.; Rosca, S.; Bourg, S.; Lozach, O.; Meijer, l.; Baldeyrou, B.; Lansiaux, A.; et al. Synthesis of chromeno[3,4-b]indoles as Lamellarin D analogues: A novel DYRK1A inhibitor class. Eur. J. Med. Chem. 2012, 49, 379–396. [Google Scholar] [CrossRef]
- Praveen, C.; Ayyanar, A.; Perumal, P.T. Gold(III) chloride catalyzed regioselective synthesis of pyrano[3,4-b]indol-1(9H)-ones and evaluation of anticancer potential towards human cervix adenocarcinoma. Bioorg. Med. Chem. Lett. 2011, 21, 4170–4173. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Tang, Z.; Ma, C.; Jiao, Y. Synthesis and fungicidal activity of novel 6H-benzimidazo[1,2-c][1,3]benzoxazin-6-ones. Chem. Heterocycl. Compd. 2021, 57, 581–587. [Google Scholar] [CrossRef]
- Wu, C.; Xu, H.; Li, Y.; Xie, R.; Li, P.; Pang, X.; Zhou, Z.; Gu, B.; Li, H.; Zhang, Y. An ESIPT-based fluorescent probe for the detection of phosgene in the solution and gas phases. Talanta 2019, 200, 78–83. [Google Scholar] [CrossRef]
- Shi, H.; Wang, X.; Li, X.; Zhang, B.; Li, X.; Zhang, J.; Yang, J.; Du, Y. Trifluoromethylthiolation/Selenolation and Lactonization of 2-Alkynylbenzoate: The Application of Benzyl Trifluoromethyl Sulfoxide/Selenium Sulfoxides as SCF3/SeCF3 Reagents. Org. Lett. 2022, 24, 2214–2219. [Google Scholar] [CrossRef]
- Zhang, H.; Li, W.; Hu, X.-D.; Liu, W.-B. Enantioselective Synthesis of Fused Isocoumarins via Palladium-Catalyzed Annulation of Alkyne-Tethered Malononitriles. J. Org. Chem. 2021, 86, 10799–10811. [Google Scholar] [CrossRef] [PubMed]
- Goulart, H.A.; Seto, J.S.S.; Barcellos, A.M.; Silva, K.B.; Moraes, M.C.; Jacob, R.G.; Lenardão, E.J.; Barcellos, T.; Perin, G. Synthesis of 4-Selanyl- and 4-Tellanyl-1H-isochromen-1-ones Promoted by Diorganyl Dichalcogenides and Oxone. J. Org. Chem. 2021, 86, 14016–14027. [Google Scholar] [CrossRef]
- Gandhi, S.; Baire, B. Fe(III)-Catalyzed, Cyclizative Coupling between 2-Alkynylbenzoates and Carbinols: Rapid Generation of Polycyclic Isocoumarins and Phthalides and Mechanistic Study. Adv. Synth. Catal. 2020, 362, 2651–2657. [Google Scholar] [CrossRef]
- Lin, X.; Fang, Z.; Zeng, C.; Zhu, C.; Pang, X.; Liu, C.; He, W.; Duan, J.; Qin, N.; Guo, K. Continuous Electrochemical Synthesis of Iso-Coumarin Derivatives from o-(1-Alkynyl) Benzoates under Metal- and Oxidant-Free. Chem. Eur. J. 2020, 26, 13738–13742. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.; Zhang, Y.; Li, B.; Du, Y. Synthesis of 4-Chloroisocoumarins via Intramolecular Halolactonization of o-Alkynylbenzoates: PhICl2-Mediated C–O/C–Cl Bond Formation. Org. Lett. 2019, 21, 1989–1993. [Google Scholar] [CrossRef] [PubMed]
- Yata, T.; Kita, Y.; Nishimoto, Y.; Yasuda, M. Regioselective Synthesis of 5-Metalated 2-Pyrones by Intramolecular Oxymetalation of Carbonyl-ene-yne Compounds Using Indium Trihalide. J. Org. Chem. 2019, 84, 14330–14341. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wan, X.; Cong, Y.; Zhen, X.; Li, Q.; Zhang-Negrerie, D.; Du, Y.; Zhao, K. Lactonization of 2-Alkynylbenzoates for the Assembly of Isochromenones Mediated by BF3·Et2O. J. Org. Chem. 2019, 84, 10402–10411. [Google Scholar] [CrossRef]
- Norseeda, K.; Chaisan, N.; Thongsornkleeb, C.; Tummatorn, J.; Ruchirawat, S. Metal-Free Synthesis of 4-Chloroisocoumarins by TMSCl-Catalyzed NCS-Induced Chlorinative Annulation of 2-Alkynylaryloate Esters. J. Org. Chem. 2019, 84, 16222–16236. [Google Scholar] [CrossRef]
- Gianni, J.; Pirovano, V.; Abbiati, G. Silver triflate/p-TSA co-catalysed synthesis of 3-substituted isocoumarins from 2-alkynylbenzoates. Org. Biomol. Chem. 2018, 16, 3213–3219. [Google Scholar] [CrossRef]
- Saikia, P.; Gogoi, S. Isocoumarins: General Aspects and Recent Advances in their Synthesis. Adv. Synth. Catal. 2018, 360, 2063–2075. [Google Scholar] [CrossRef]
- Chen, Z.; Zeng, X.; Yan, B.; Zhao, Y.; Fu, Y. Au-catalyzed intramolecular annulations toward fused tricyclic [1,3]oxazino[3,4-a]indol-1-ones under extremely mild conditions. RSC Adv. 2015, 5, 100251–100255. [Google Scholar] [CrossRef]
- Sperança, A.; Godoi, B.; Pinton, S.; Back, D.F.; Menezes, P.H.; Zeni, G. Regioselective Synthesis of Isochromenones by Iron(III)/PhSeSePh-Mediated Cyclization of 2-Alkynylaryl Esters. J. Org. Chem. 2011, 76, 6789–6797. [Google Scholar] [CrossRef]
- Marcellino, R.; Koichi, K.; Toshio, H. Synthetic Studies on Natural Isocoumarins and Isocarbostyril Derivatives Having an Alkyl Substituent at the 3-Position: Total Synthesis of Scoparines A and B, and Ruprechstyril. Heterocycles 2009, 79, 753–764. [Google Scholar]
- Uchiyama, M.; Ozawa, H.; Takuma, K.; Matsumoto, Y.; Yonehara, M.; Hiroya, K.; Sakamoto, T. Regiocontrolled Intramolecular Cyclizations of Carboxylic Acids to Carbon−Carbon Triple Bonds Promoted by Acid or Base Catalyst. Org. Lett. 2006, 8, 5517–5520. [Google Scholar] [CrossRef] [PubMed]
- Mallampudi, N.A.; Choudhury, U.M.; Mohapatra, D.K. Total Synthesis of (−)-Citreoisocoumarin, (−)-Citreoisocoumarinol, (−)-12-epi-Citreoisocoumarinol, and (−)-Mucorisocoumarins A and B Using a Gold(I)-Catalyzed Cyclization Strategy. J. Org. Chem. 2020, 85, 4122–4129. [Google Scholar] [CrossRef]
- Tang, Y.; Li, J.; Zhu, Y.; Li, Y.; Yu, B. Mechanistic Insights into the Gold(I)-Catalyzed Activation of Glycosyl ortho-Alkynylbenzoates for Glycosidation. J. Am. Chem. Soc. 2013, 135, 18396–18405. [Google Scholar] [CrossRef]
- Mallampudi, N.A.; Reddy, G.S.; Maity, S.; Mohapatra, D.K. Gold(I)-Catalyzed Cyclization for the Synthesis of 8-Hydroxy-3- substituted Isocoumarins: Total Synthesis of Exserolide F. Org. Lett. 2017, 19, 2074–2077. [Google Scholar] [CrossRef]
- Marchal, E.; Uriac, P.; Legouin, B.; Toupet, L.; Van de Weghe, P. Cycloisomerization of γ- and δ-acetylenic acids catalyzed by gold(I) chloride. Tetrahedron 2007, 63, 9979–9990. [Google Scholar] [CrossRef]
- Pirovano, V.; Marchetti, M.; Carbonaro, J.; Brambilla, E.; Rossi, E.; Ronda, L.; Abbiati, G. Synthesis and photophysical properties of isocoumarin-based D-π-A systems. Dyes Pigm. 2020, 173, 107917. [Google Scholar] [CrossRef]
- Dong, X.; Chen, L.; Zheng, Z.; Ma, X.; Luo, Z.; Zhang, L. Silver-catalyzed stereoselective formation of glycosides using glycosyl ynenoates as donors. Chem. Commun. 2018, 54, 8626–8629. [Google Scholar] [CrossRef] [PubMed]
- Witham, C.A.; Huang, W.; Tsung, C.-K.; Kuhn, J.N.; Somorjai, G.A.; Toste, F.D. Converting homogeneous to heterogeneous in electrophilic catalysis using monodisperse metal nanoparticles. Nat. Chem. 2010, 2, 36–41. [Google Scholar] [CrossRef]
- Lin, H.-P.; Ibrahim, N.; Provot, O.; Alami, M.; Hamze, A. PtO2/PTSA system catalyzed regioselective hydration of internal arylalkynes bearing electron withdrawing groups. RSC Adv. 2018, 8, 11536–11542. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Ma, F.; Shen, Y.; Xie, T.; Gao, S. Convergent Synthesis of Kibdelone, C. Org. Lett. 2018, 20, 2872–2875. [Google Scholar] [CrossRef]
- Li, Y.; Li, G.; Ding, Q. (Trifluoromethyl)thiolation of 2-Alkynylbenzoates: An Efficient Route to 4-[(Trifluoromethyl)thio]-1H-isochromen-1-ones. Eur. J. Org. Chem. 2014, 2014, 5017–5022. [Google Scholar] [CrossRef]
- Hellal, M.; Bourguignon, J.-J.; Bihel, F.J.-J. 6-endo-dig Cyclization of heteroarylesters to alkynes promoted by Lewis acid catalyst in the presence of Brønsted acid. Tetrahedron Lett. 2008, 49, 62–65. [Google Scholar] [CrossRef]
- Chin, L.-Y.; Lee, C.-Y.; Lo, Y.-H.; Wu, M.-J. Halocyclization of Methyl 2-Alkynylbenzoates to Isocoumarins Using Cupric Halides. J. Chin. Chem. Soc. 2008, 55, 643–648. [Google Scholar] [CrossRef]
- Liang, Y.; Xie, Y.-X.; Li, J.-H. Cy2NH·HX-Promoted Cyclizations of o-(Alk-1-ynyl)benzoates and (Z)-Alk-2-en-4-ynoate with Copper Halides to Synthesize Isocoumarins and α-Pyrone. Synthesis 2007, 2007, 400–406. [Google Scholar]
- Rusch, M.; Thevenon, A.; Hoepfner, D.; Aust, T.; Studer, C.; Patoor, M.; Rollin, P.; Livendahl, M.; Ranieri, B.; Schmitt, E.; et al. Design and Synthesis of Metabolically Stable tRNA Synthetase Inhibitors Derived from Cladosporin. ChemBioChem 2019, 20, 644–649. [Google Scholar] [CrossRef]
- Istrate, F.M.; Buzas, A.K.; Jurberg, I.D.; Odabachian, Y.; Gagosz, F. Synthesis of Functionalized Oxazolones by a Sequence of Cu(II)- and Au(I)-Catalyzed Transformations. Org. Lett. 2008, 10, 925–928. [Google Scholar] [CrossRef]
- Oppedisano, A.; Prandi, C.; Venturello, P.; Deagostino, A.; Goti, G.; Scarpi, S.; Occhiato, E.G. Synthesis of Vinylogous Amides by Gold(I)-Catalyzed Cyclization of N-Boc-Protected 6-Alkynyl-3,4-dihydro-2H-pyridines. J. Org. Chem. 2013, 78, 11007–11016. [Google Scholar] [CrossRef]
- Lee, E.-S.; Yeom, H.-S.; Hwang, J.-H.; Shin, S. A Practical Gold-Catalyzed Route to 4-Substituted Oxazolidin-2-ones from N-Boc Propargylamines. Eur. J. Org. Chem. 2007, 2007, 3503–3507. [Google Scholar] [CrossRef]
- El Qami, A.; Jismy, B.; Akssira, M.; Jacquemin, J.; Tikad, A.; Abarbri, M. A Ag2CO3/TFA-catalyzed intramolecular annulation approach to imidazo[1,2-c][1,3]oxazin-5-one derivatives. Org. Biomol. Chem. 2022, 20, 1518–1531. [Google Scholar] [CrossRef]
- Veltri, L.; Amuso, R.; Petrilli, M.; Cuocci, C.; Chiacchio, M.A.; Vitale, P.; Gabriele, B. A Zinc-Mediated Deprotective Annulation Approach to New Polycyclic Heterocycles. Molecules 2021, 26, 2318. [Google Scholar] [CrossRef]
- Habert, L.; Sallio, R.; Durandetti, M.; Gosmini, C.; Gillaizeau, I. Zinc Chloride Mediated Synthesis of 3H-Oxazol-2-one and Pyrrolo-oxazin-1-one from Ynamide. Eur. J. Org. Chem. 2019, 2019, 5175–5179. [Google Scholar] [CrossRef]
- Koy, M.; Bellotti, P.; Katzenburg, F.; Daniliuc, C.G.; Glorius, F. Synthesis of All-Carbon Quaternary Centers by Palladium-Catalyzed Olefin Dicarbofunctionalization. Angew. Chem. Int. Ed. 2020, 59, 2375–2379. [Google Scholar] [CrossRef]
- Andrzejewska, M.; Pagano, M.A.; Meggio, F.; Brunatib, A.M.; Kazimierczuk, Z. Polyhalogenobenzimidazoles: Synthesis and Their inhibitory activity against casein kinases. Bioorg. Med. Chem. 2003, 11, 3997–4002. [Google Scholar] [CrossRef] [PubMed]
- Ellingboe, J.W.; Spinelli, W.; Winkley, M.W.; Nguyen, T.T.; Parsons, R.W.; Moubarak, I.F.; Kitzen, J.M.; Von Engen, D.; Bagli, J.F. Class III antiarrhythmic activity of novel substituted 4-[(methylsulfonyl)amino]benzamides and sulfonamides. J. Med. Chem. 1992, 35, 705. [Google Scholar] [CrossRef]
- Bacauanu, V.; Cardinal, S.; Yamauchi, M.; Kondo, M.; Fernández, D.F.; Remy, R.; MacMillan, D.W.C. Metallaphotoredox Difluoromethylation of Aryl Bromides. Angew. Chem. 2018, 130, 12723–12728. [Google Scholar] [CrossRef]
- Nadipuram, A.K.; David, W.M.; Kumar, D.; Kerwin, S.M. Synthesis and Thermolysis of Heterocyclic 3-Aza-3-ene-1,5-diynes1. Org. Lett. 2002, 4, 4543–4546. [Google Scholar] [CrossRef] [PubMed]
- Ahlrichs, R.; Bär, M.; Häser, M.; Horn, H.; Kölmel, C. Electronic Structure Calculations on Workstation Computers: The Program System Turbomole. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar] [CrossRef]
- Jiménez-Hoyos, C.A.; Janesko, B.G.; Scuseria, G.E. Evaluation of range-separated hybrid density functionals for the prediction of vibrational frequencies, infrared intensities, and Raman activities. Phys. Chem. Chem. Phys. 2008, 10, 6621–6629. [Google Scholar] [CrossRef]
- Weigend, F.; Häser, M. RI-MP2: First Derivatives and Global Consistency. Theor. Chem. Acc. 1997, 97, 331–340. [Google Scholar] [CrossRef]
- Weigend, F.; Häser, M.; Patzelt, H.; Ahlrichs, R. RI-MP2: Optimized Auxiliary Basis Sets and Demonstration of Efficiency. Chem. Phys. Lett. 1998, 294, 143–152. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A. 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-Type Density Functional Constructed with a Long-Range Dispersion Correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | R | Solvent | Catalyst (Equiv.) | Additive (Equiv.) | T (°C) | Time (h) | Ratio (%) a 4/5 |
| 1 | n-Bu | DCM | ZnCl2 (1.5) [41] | - | 40 | 3 | 0/100 b |
| 2 | Ph | DCM | ZnCl2 (1.5) | - | 40 | 48 | 60/40 |
| 3 | Ph | DCM | ZnCl2 (1.5) | - | 60 | 48 | 50/50 |
| 4 | Ph | DCM | Ag2CO3 (0.1) | TFA (2) | 60 | 24 | 15/85 |
| 5 | Ph | DCE | Ag2CO3 (0.1) [40] | TFA (2) | 60 | 6 | 0/100 c |
| 6 | Ph | DCE | Ag2CO3 (0.1) | TFA (2) | 40 | 24 | 5/95 |
 | |||||
|---|---|---|---|---|---|
| Entry | Comp. | Charge | Entry | Comp. | Charge |
| 1 | 4a | α = −0.100 β = +0.125 | 9 | 4i | α = −0.012 β = +0.064 |
| 2 | 4b | α = −0.041 β = +0.078 | 10 | 4j | α = −0.020 β = +0.069 |
| 3 | 4c | α = −0.057 β = +0.082 | 11 | 4k | α = −0.018 β = +0.038 |
| 4 | 4d | α = −0.045 β = +0.081 | 12 | 4l | α = −0.017 β = +0.059 |
| 5 | 4e | α = −0.049 β = +0.080 | 13 | 4m | α = −0.023 β = +0.070 |
| 6 | 4f | α = −0.018 β = +0.071 | 14 | 4n | α = −0.038 β = +0.075 |
| 7 | 4g | α = −0.026 β = +0.070 | 15 | 4o | α = −0.095 β = +0.130 |
| 8 | 4h | α = −0.032 β = +0.073 | -- | -- | -- |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Qami, A.; Jismy, B.; Akssira, M.; Jacquemin, J.; Tikad, A.; Abarbri, M. Efficient Synthesis of 1H-Benzo[4,5]imidazo[1,2-c][1,3]oxazin-1-one Derivatives Using Ag2CO3/TFA-Catalyzed 6-endo-dig Cyclization: Reaction Scope and Mechanistic Study. Molecules 2023, 28, 2403. https://doi.org/10.3390/molecules28052403
El Qami A, Jismy B, Akssira M, Jacquemin J, Tikad A, Abarbri M. Efficient Synthesis of 1H-Benzo[4,5]imidazo[1,2-c][1,3]oxazin-1-one Derivatives Using Ag2CO3/TFA-Catalyzed 6-endo-dig Cyclization: Reaction Scope and Mechanistic Study. Molecules. 2023; 28(5):2403. https://doi.org/10.3390/molecules28052403
Chicago/Turabian StyleEl Qami, Abdelkarim, Badr Jismy, Mohamed Akssira, Johan Jacquemin, Abdellatif Tikad, and Mohamed Abarbri. 2023. "Efficient Synthesis of 1H-Benzo[4,5]imidazo[1,2-c][1,3]oxazin-1-one Derivatives Using Ag2CO3/TFA-Catalyzed 6-endo-dig Cyclization: Reaction Scope and Mechanistic Study" Molecules 28, no. 5: 2403. https://doi.org/10.3390/molecules28052403
APA StyleEl Qami, A., Jismy, B., Akssira, M., Jacquemin, J., Tikad, A., & Abarbri, M. (2023). Efficient Synthesis of 1H-Benzo[4,5]imidazo[1,2-c][1,3]oxazin-1-one Derivatives Using Ag2CO3/TFA-Catalyzed 6-endo-dig Cyclization: Reaction Scope and Mechanistic Study. Molecules, 28(5), 2403. https://doi.org/10.3390/molecules28052403