

Molecular Peptide Grafting as a Tool to Create Novel Protein Therapeutics


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Functional Peptides
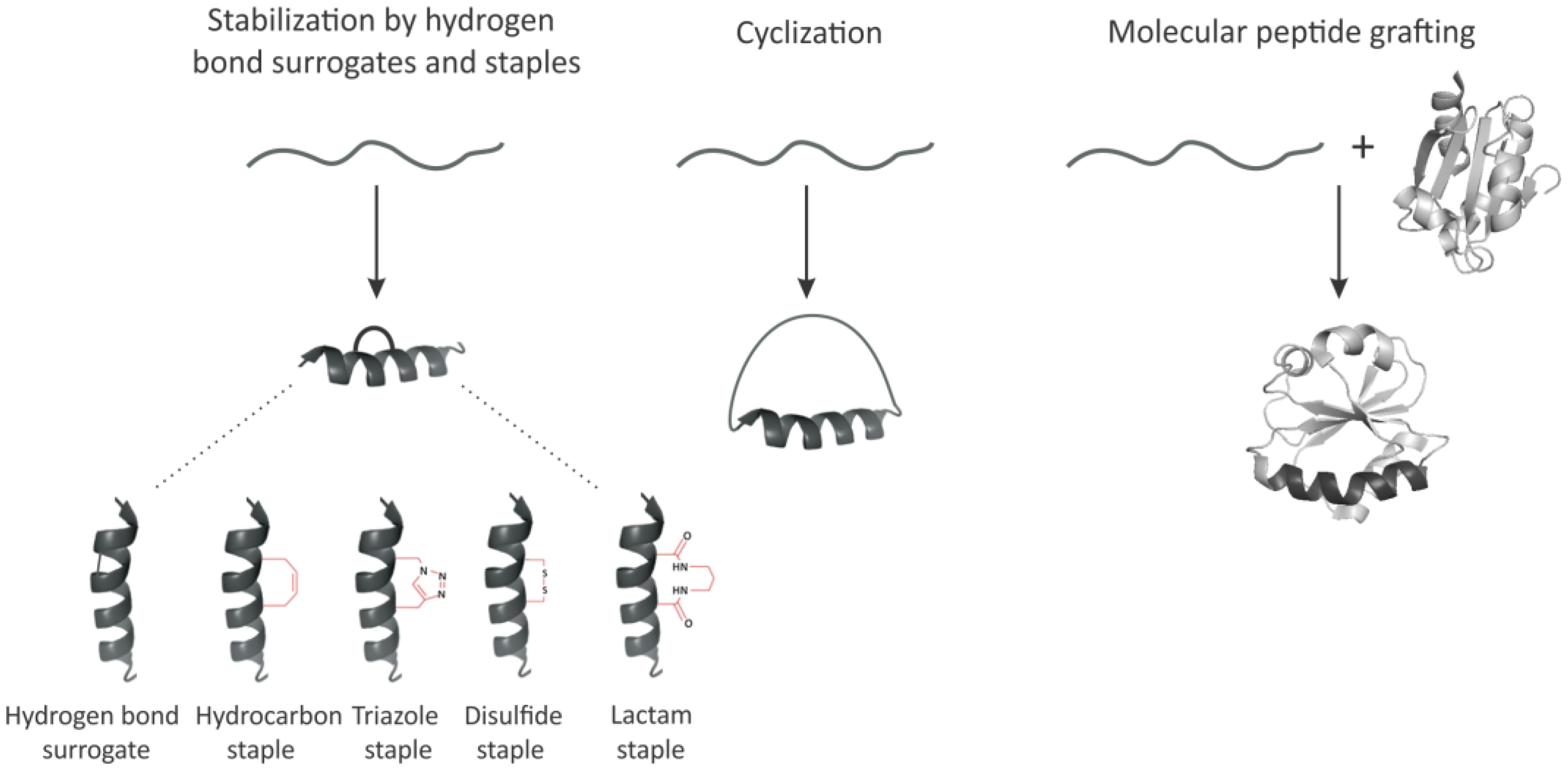
3. Imposing Conformational Constraints in Short Peptides
3.1. Hydrogen Bond Surrogates and Staples
3.2. Peptide Backbone Circularization
3.3. Molecular Peptide Grafting
3.3.1. Chimeric Glutathione S-Transferases (GSTs) Containing Inserts of Kininogen Peptides as Cellular Anti-Proliferating Agents
3.3.2. TNF-α Epitope-Scaffold Immunogen
3.3.3. Grafting Short hDM2/Mdm2 Hydrophobic Epitopes as a Tool to Inhibit Protein–Protein Interactions Critical for Cancer Development and Progression
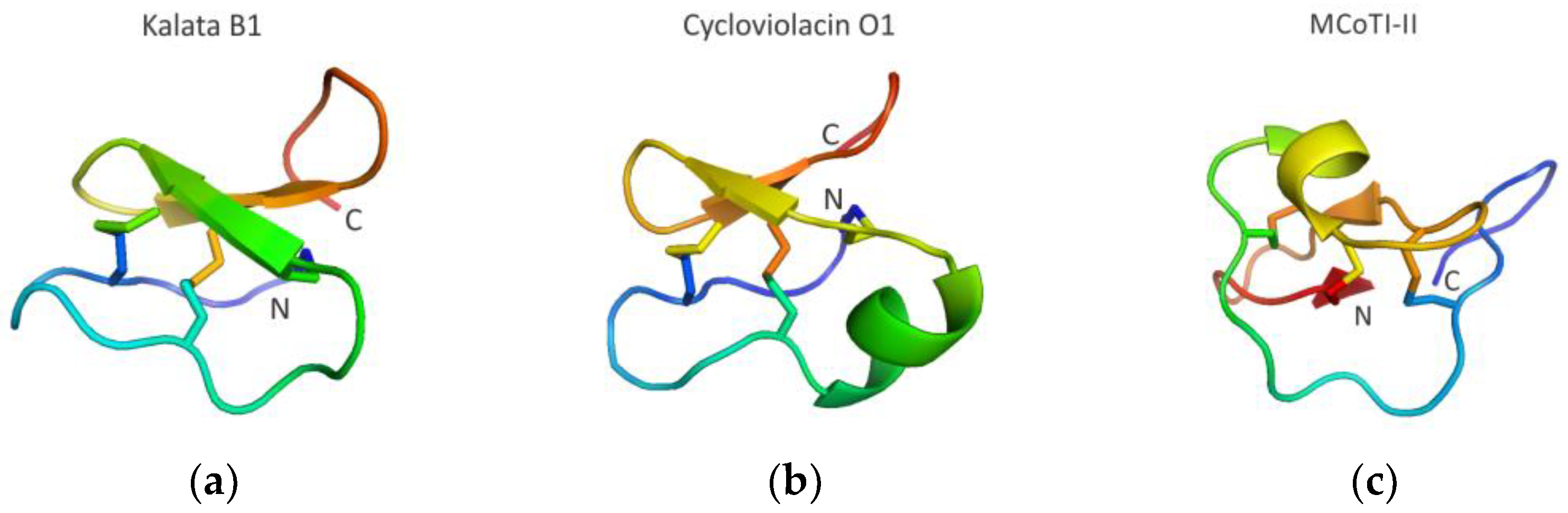
3.3.4. Cyclotide Scaffolds Targeting Biomolecular Interactions
4. Conclusions and Future Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Moult, J.; Melamud, E. From fold to function. Curr. Opin. Struct. Biol. 2000, 3, 384–389. [Google Scholar] [CrossRef]
- Söding, J.; Lupas, A.N. More than the sum of their parts: On the evolution of proteins from peptides. BioEssays 2003, 25, 837–846. [Google Scholar] [CrossRef]
- Zhu, J.; Avakyan, N.; Kakkis, A.; Hoffnagle, A.M.; Han, K.; Li, Y.; Zhang, Z.; Choi, T.S.; Na, Y.; Yu, C.J.; et al. Protein assembly by design. Chem. Rev. 2021, 121, 13701–13796. [Google Scholar] [CrossRef]
- Wu, L.C.; Grandori, R.; Carey, J. Autonomous subdomains in protein folding. Protein Sci. 1994, 3, 369–371. [Google Scholar] [CrossRef]
- Xu, D.; Nussinov, R. Favorable domain size in proteins. Fold. Des. 1998, 3, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, A.V.; Bogatyreva, N.S.; Ivankov, D.N.; Garbuzynskiy, S.O. Protein folding problem: Enigma, paradox, solution. Biophys. Rev. 2022, 14, 1255–1272. [Google Scholar] [CrossRef]
- Naudin, E.A.; Albanese, K.I.; Smith, A.J.; Mylemans, B.; Baker, E.G.; Weiner, O.D.; Andrews, D.M.; Tigue, N.; Savery, N.J.; Woolfson, D.N. From peptides to proteins: Coiled-coil tetramers to single-chain 4-helix bundles. Chem. Sci. 2022, 13, 11330–11340. [Google Scholar] [CrossRef] [PubMed]
- Kaspar, A.A.; Reichert, J.M. Future directions for peptide therapeutics development. Drug. Discov. Today. 2013, 18, 807–817. [Google Scholar] [CrossRef]
- Agyei, D.; Ahmed, I.; Akram, Z.; Iqbal, H.M.; Danquah, M.K. Protein and peptide biopharmaceuticals: An overview. Protein Pept. Lett. 2017, 24, 94–101. [Google Scholar] [CrossRef]
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorg. Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic peptides: Current applications and future directions. Signal. Transduct. Target. Ther. 2022, 7, 48. [Google Scholar] [CrossRef]
- Sharma, K.; Sharma, K.K.; Sharma, A.; Jain, R. Peptide-based drug discovery: Current status and recent advances. Drug Discov Today 2023, 28, 103464. [Google Scholar] [CrossRef] [PubMed]
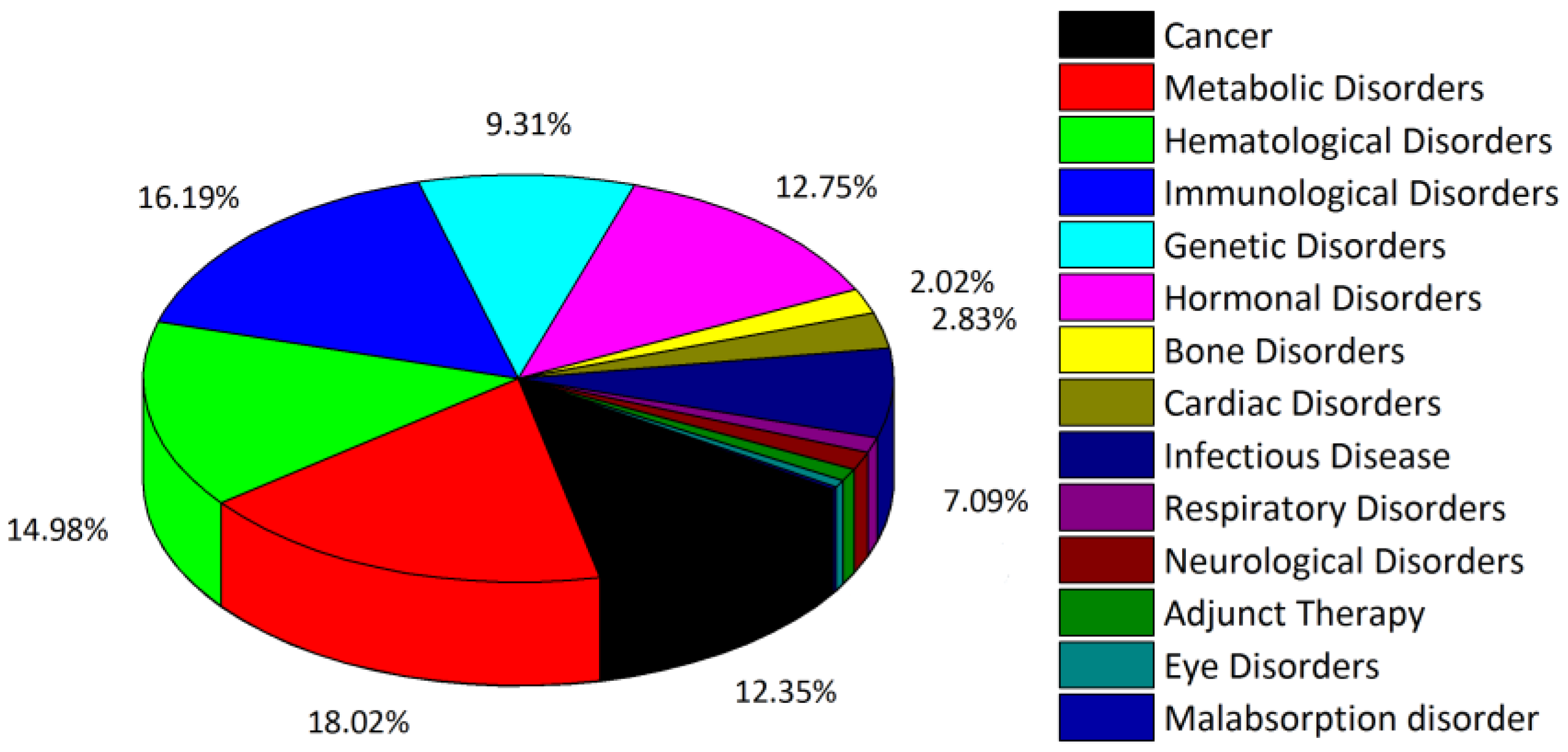
- Usmani, S.S.; Bedi, G.; Samuel, J.S.; Singh, S.; Kalra, S.; Kumar, P.; Ahuja, A.A.; Sharma, M.; Gautam, A.; Raghava, G.P.S. THPdb: Database of FDA-approved peptide and protein therapeutics. PLoS ONE 2017, 12, e0181748. [Google Scholar] [CrossRef]
- THPdb. Browse Disease Area in THPdb. Available online: https://webs.iiitd.edu.in/raghava/thpdb/disease.php (accessed on 24 February 2023).
- THPdb. Browse No. of Amino Acid in THPdb. Available online: https://webs.iiitd.edu.in/raghava/thpdb/length.php (accessed on 24 February 2023).
- Tibbetts, S.A.; Seetharama Jois, D.; Siahaan, T.J.; Benedict, S.H.; Chan, M.A. Linear and cyclic LFA-1 and ICAM-1 peptides inhibit T cell adhesion and function. Peptides 2000, 21, 1161–1167. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.C.; Claffey, K.; Sakthivel, R.; Darzynkiewicz, Z.; Shaw, D.E.; Leal, J.; Wang, Y.C.; Lu, F.M.; McCrae, K.R. Two-chain high molecular weight kininogen induces apoptosis and inhibits angiogenesis: Partial activity within domain 5. FASEB J. 2000, 14, 2589–2600. [Google Scholar] [CrossRef] [PubMed]
- Church, W.B.; Inglis, A.S.; Tseng, A.; Duell, R.; Lei, P.W.; Bryant, K.J.; Scott, K.F. A novel approach to the design of inhibitors of human secreted phospholipase A2 based on native peptide inhibition. J. Biol. Chem. 2001, 276, 33156–33164. [Google Scholar] [CrossRef]
- Sia, S.K.; Carr, P.A.; Cochran, A.G.; Malashkevich, V.N.; Kim, P.S. Short constrained peptides that inhibit HIV-1 entry. Proc. Natl. Acad. Sci. USA 2002, 99, 14664–14669. [Google Scholar] [CrossRef]
- Kiyota, S.; Franzoni, L.; Nicastro, G.; Benedetti, A.; Oyama, S., Jr.; Viviani, W.; Gambarini, A.G.; Spisni, A.; Miranda, M.T. Introduction of a chemical constraint in a short peptide derived from human acidic fibroblast growth factor elicits mitogenic structural determinants. J. Med. Chem. 2003, 46, 2325–2333. [Google Scholar] [CrossRef] [PubMed]
- Daly, M.E.; Makris, A.; Reed, M.; Lewis, C.E. Hemostatic regulators of tumor angiogenesis: A source of anti-angiogenic agents for cancer treatment? J. Natl. Cancer Inst. 2003, 95, 1660–1673. [Google Scholar] [CrossRef]
- Fowler, S.B.; Poon, S.; Muff, R.; Chiti, F.; Dobson, C.M.; Zurdo, J. Rational design of aggregation-resistant bioactive peptides: Reengineering human calcitonin. Proc. Natl. Acad. Sci. USA 2005, 102, 10105–10110. [Google Scholar] [CrossRef] [PubMed]
- Walport, L.J.; Obexer, R.; Suga, H. Strategies for transitioning macrocyclic peptides to cell-permeable drug leads. Curr. Opin. Biotechnol. 2017, 48, 242–250. [Google Scholar] [CrossRef]
- Qin, L.; Cui, Z.; Wu, Y.; Wang, H.; Zhang, X.; Guan, J.; Mao, S. Challenges and strategies to enhance the systemic absorption of inhaled peptides and proteins. Pharm. Res. 2022, 16, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Deo, S.; Turton, K.L.; Kainth, T.; Kumar, A.; Wieden, H.J. Strategies for improving antimicrobial peptide production. Biotechnol. Adv. 2022, 59, 107968. [Google Scholar] [CrossRef] [PubMed]
- Holub, J.M. Small scaffolds, big potential: Developing miniature proteins as therapeutic agents. Drug. Dev. Res. 2017, 78, 268–282. [Google Scholar] [CrossRef]
- Camarero, J.A.; Campbell, M.J. The potential of the cyclotide scaffold for drug development. Biomedicines 2019, 7, 31. [Google Scholar] [CrossRef]
- Wang, C.K.; Craik, D.J. Linking molecular evolution to molecular grafting. J. Biol. Chem. 2021, 296, 100425. [Google Scholar] [CrossRef]
- Jacob, B.; Vogelaar, A.; Cadenas, E.; Camarero, J.A. Using the cyclotide scaffold for targeting biomolecular interactions in drug development. Molecules 2022, 27, 6430. [Google Scholar] [CrossRef]
- Hillman, R.A.; Nadraws, J.W.; Bertucci, M.A. The hydrocarbon staple and beyond: Recent advances towards stapled peptide therapeutics that target protein-protein interactions. Curr. Top. Med. Chem. 2018, 18, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.M.; Atmaj, J.; Van Oosterwijk, N.; Groves, M.R.; Dömling, A. Stapled peptides inhibitors: A new window for target drug discovery. Comput. Struct. Biotechnol. J. 2019, 17, 263–281. [Google Scholar] [CrossRef]
- Chan, A.M.; Goodis, C.C.; Pommier, E.G.; Fletcher, S. Recent applications of covalent chemistries in protein-protein interaction inhibitors. RSC Med. Chem. 2022, 13, 921–928. [Google Scholar] [CrossRef]
- National Human Genome Research Institute. Peptide. Available online: https://www.genome.gov/genetics-glossary/Peptide (accessed on 3 February 2023).
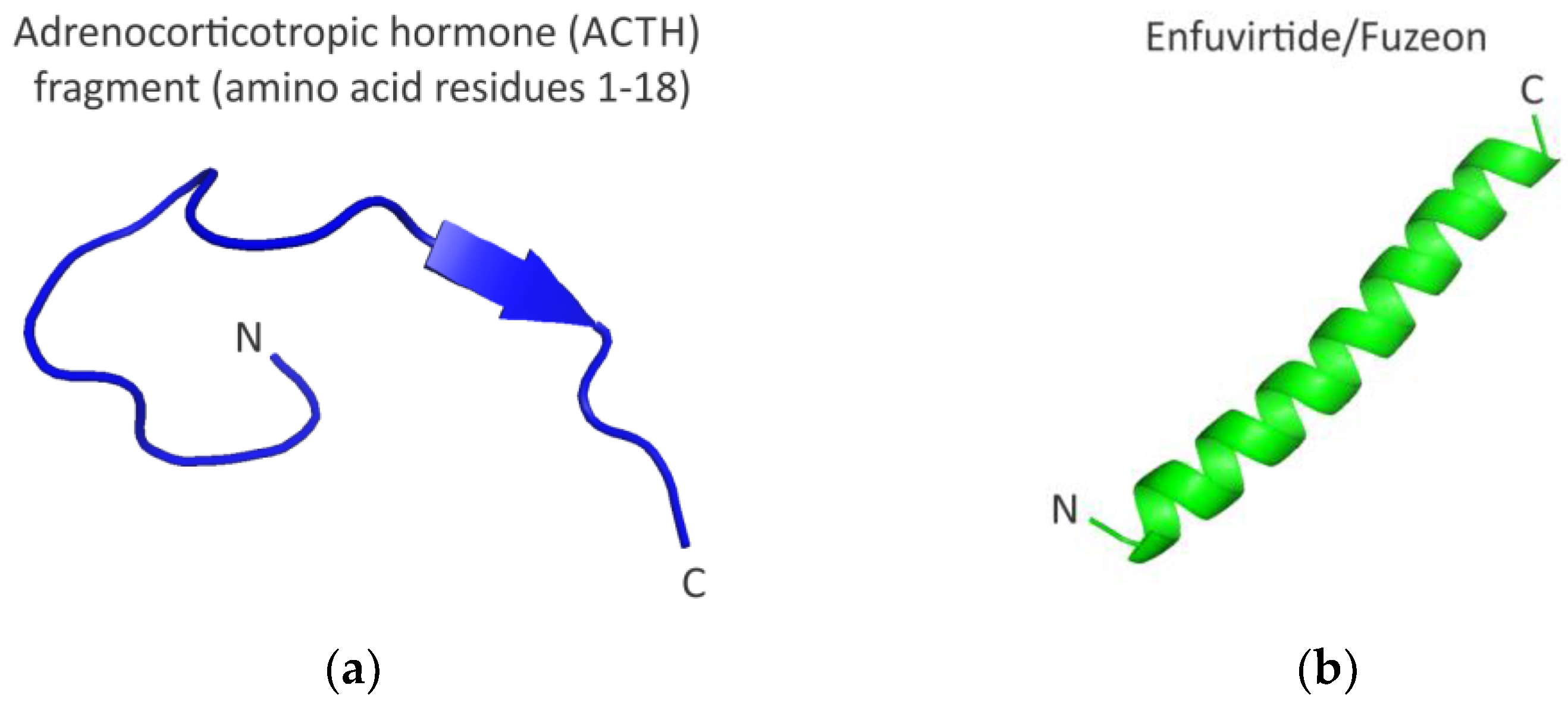
- Elkinton, J.R.; Hunt, A.D., Jr.; Godfrey, L.; McCrory, W.W.; Rogerson, A.G.; Stokes, J. Effects of pituitary adrenocorticotropic hormone ACTH) therapy. J. Am. Med. Assoc. 1949, 141, 1273–1279. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, M.; Cammack, N.; Salgo, M.; Smiley, L. HIV fusion and its inhibition in antiretroviral therapy. Rev. Med. Virol. 2004, 14, 321–337. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, F.; Kodama, E.N. Novel HIV-1 fusion inhibition peptides: Designing the next generation of drugs. Antivir. Chem. Chemother. 2012, 22, 151–158. [Google Scholar] [CrossRef]
- Yi, H.A.; Fochtman, B.C.; Rizzo, R.C.; Jacobs, A. Inhibition of HIV entry by targeting the envelope transmembrane subunit gp41. Curr. HIV Res. 2016, 14, 283–294. [Google Scholar] [CrossRef]
- Ghaddhab, C.; Vuissoz, J.M.; Deladoëy, J. From bioinactive ACTH to ACTH antagonist: The clinical perspective. Front. Endocrinol. 2017, 8, 17. [Google Scholar] [CrossRef]
- Tibbetts, S.A.; Chirathaworn, C.; Nakashima, M.; Jois, D.S.; Siahaan, T.J.; Chan, M.A.; Benedict, S.H. Peptides derived from ICAM-1 and LFA-1 modulate T cell adhesion and immune function in a mixed lymphocyte culture. Transplantation 1999, 68, 685–692. [Google Scholar] [CrossRef]
- Steward-Tharp, S.M.; Song, Y.J.; Siegel, R.M.; O'Shea, J.J. New insights into T cell biology and T cell-directed therapy for autoimmunity, inflammation, and immunosuppression. Ann. N. Y. Acad. Sci. 2010, 1183, 123–148. [Google Scholar] [CrossRef]
- Anderson, M.E.; Siahaan, T.J. Targeting ICAM-1/LFA-1 interaction for controlling autoimmune diseases: Designing peptide and small molecule inhibitors. Peptides 2003, 24, 487–501. [Google Scholar] [CrossRef] [PubMed]
- Azzarito, V.; Long, K.; Murphy, N.S.; Wilson, A.J. Inhibition of α-helix-mediated protein–protein interactions using designed molecules. Nat. Chem. 2013, 5, 161–173. [Google Scholar] [CrossRef]
- Araghi, R.R.; Keating, A.E. Designing helical peptide inhibitors of protein–protein interactions. Curr. Opin. Struct. Biol. 2016, 39, 27–38. [Google Scholar] [CrossRef]
- Ohtake, S.; Kita, Y.; Payne, R.; Manning, M.; Arakawa, T. Structural characteristics of short peptides in solution. Protein Pept. Lett. 2013, 20, 1308–1323. [Google Scholar] [CrossRef] [PubMed]
- Patgiri, A.; Jochim, A.L.; Arora, P.S. A hydrogen bond surrogate approach for stabilization of short peptide sequences in alpha-helical conformation. Acc. Chem. Res. 2008, 41, 1289–1300. [Google Scholar] [CrossRef]
- Henchey, L.K.; Jochim, A.L.; Arora, P.S. Contemporary strategies for the stabilization of peptides in the alpha-helical conformation. Curr. Opin. Chem. Biol. 2008, 12, 692–697. [Google Scholar] [CrossRef] [PubMed]
- Kritzer, J.A. Stapled peptides: Magic bullets in nature’s arsenal. Nat. Chem. Biol. 2010, 6, 566–567. [Google Scholar] [CrossRef] [PubMed]
- Verdine, G.L.; Hilinski, G.J. Stapled peptides for intracellular drug targets. Methods Enzymol. 2012, 503, 3–33. [Google Scholar]
- Walensky, L.D.; Bird, G.H. Hydrocarbon-stapled peptides: Principles, practice, and progress. J. Med. Chem. 2014, 57, 6275–6288. [Google Scholar] [CrossRef]
- Roy, S.; Ghosh, P.; Ahmed, I.; Chakraborty, M.; Naiya, G.; Ghosh, B. Constrained α-helical peptides as inhibitors of protein-protein and protein-DNA interactions. Biomedicines 2018, 6, 118. [Google Scholar] [CrossRef]
- Bechtler, C.; Lamers, C. Macrocyclization strategies for cyclic peptides and peptidomimetics. RSC Med. Chem. 2021, 12, 1325–1351. [Google Scholar] [CrossRef]
- Li, X.; Craven, T.W.; Levine, P.M. Cyclic peptide screening methods for preclinical drug discovery. J. Med. Chem. 2022, 65, 11913–11926. [Google Scholar] [CrossRef]
- Bentley, A.A.; Merkulov, S.M.; Peng, Y.; Rozmarynowycz, R.; Qi, X.; Pusztai-Carey, M.; Merrick, W.C.; Yee, V.C.; McCrae, K.R.; Komar, A.A. Chimeric glutathione S-transferases containing inserts of kininogen peptides: Potential novel protein therapeutics. J. Biol. Chem. 2012, 287, 22142–22150. [Google Scholar] [CrossRef]
- Colman, R.W.; Jameson, B.A.; Lin, Y.; Johnson, D.; Mousa, S.A. Domain 5 of high molecular weight kininogen (kininostatin) downregulates endothelial cell proliferation and migration and inhibits angiogenesis. Blood 2000, 95, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Kamiyama, F.; Maeda, T.; Yamane, T.; Li, Y.H.; Ogukubo, O.; Otsuka, T.; Ueyama, H.; Takahashi, S.; Ohkubo, I.; Matsui, N. Inhibition of vitronectin-mediated haptotaxis and haptoinvasion of MG-63 cells by domain 5 (D5H) of human high molecular weight kininogen and identification of a minimal amino acid sequence. Biochem. Biophys. Res. Commun. 2001, 288, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.C.; Qi, X.; Juarez, J.; Plunkett, M.; Donaté, F.; Sakthivel, R.; Mazar, A.P.; McCrae, K.R. Inhibition of angiogenesis by two-chain high molecular weight kininogen (HKa) and kininogen-derived polypeptides. Can. J. Physiol. Pharmacol. 2002, 80, 85–90. [Google Scholar] [CrossRef]
- Zhang, J.C.; Donate, F.; Qi, X.; Ziats, N.P.; Juarez, J.C.; Mazar, A.P.; Pang, Y.P.; McCrae, K.R. The anti-angiogenic activity of cleaved high molecular weight kininogen is mediated through binding to endothelial tropomyosin. Proc. Natl. Acad. Sci. USA 2002, 99, 12224–12229. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, M.; Maeda, T.; Hanasawa, K.; Ohkubo, I.; Tani, T. Effect of His-Gly-Lys motif derived from domain 5 of high molecular weight kininogen on suppression of cancer metastasis both in vitro and in vivo. J. Biol. Chem. 2003, 278, 49301–49307. [Google Scholar] [CrossRef]
- Sheehan, D.; Meade, G.; Foley, V.M.; Dowd, C.A. Structure, function, and evolution of glutathione transferases: Implications for classification of non-mammalian members of an ancient enzyme superfamily. Biochem. J. 2001, 360, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Tracey, K.J.; Cerami, A. Tumor necrosis factor: A pleiotropic cytokine and therapeutic target. Annu. Rev. Med. 1994, 45, 491–503. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, M.; Maini, R.N. TNF defined as a therapeutic target for rheumatoid arthritis and other autoimmune diseases. Nat. Med. 2003, 9, 1245–1250. [Google Scholar] [CrossRef]
- Feldmann, M.; Steinman, L. Design of effective immunotherapy for human autoimmunity. Nature 2005, 435, 612–619. [Google Scholar] [CrossRef]
- Tracey, D.; Klareskog, L.; Sasso, E.H.; Salfeld, J.G.; Tak, P.P. Tumor necrosis factor antagonist mechanisms of action: A comprehensive review. Pharmacol. Ther. 2008, 117, 244–279. [Google Scholar] [CrossRef]
- Vincent, F.B.; Morand, E.F.; Murphy, K.; Mackay, F.; Mariette, X.; Marcelli, C. Antidrug antibodies (ADAb) to tumour necrosis factor (TNF)-specific neutralising agents in chronic inflammatory diseases: A real issue, a clinical perspective. Ann. Rheum. Dis. 2013, 72, 165–178. [Google Scholar] [CrossRef]
- van Schie, K.A.; Hart, M.H.; de Groot, E.R.; Kruithof, S.; Aarden, L.A.; Wolbink, G.J.; Rispens, T. The antibody response against human and chimeric anti-TNF therapeutic antibodies primarily targets the TNF binding region. Ann. Rheum. Dis. 2015, 74, 311–314. [Google Scholar] [CrossRef]
- Jia, T.; Pan, Y.; Li, J.; Wang, L. Strategies for active TNF-alpha vaccination in rheumatoid arthritis treatment. Vaccine 2013, 31, 4063–4068. [Google Scholar] [CrossRef] [PubMed]
- Le Buanec, H.; Delavallée, L.; Bessis, N.; Paturance, S.; Bizzini, B.; Gallo, R.; Zagury, D.; Boissier, M.C. TNFalpha kinoid vaccination-induced neutralizing antibodies to TNFalpha protect mice from autologous TNFalpha-driven chronic and acute inflammation. Proc. Natl. Acad. Sci. USA 2006, 103, 19442–19447. [Google Scholar] [CrossRef] [PubMed]
- Grünewald, J.; Tsao, M.L.; Perera, R.; Dong, L.; Niessen, F.; Wen, B.G.; Kubitz, D.M.; Smider, V.V.; Ruf, W.; Nasoff, M.; et al. Immunochemical termination of self-tolerance. Proc. Natl. Acad. Sci. USA 2008, 105, 11276–11280. [Google Scholar] [CrossRef]
- Capini, C.J.; Bertin-Maghit, S.M.; Bessis, N.; Haumont, P.M.; Bernier, E.M.; Muel, E.G.; Laborie, M.A.; Autin, L.; Paturance, S.; Chomilier, J.; et al. Active immunization against murine TNFalpha peptides in mice: Generation of endogenous antibodies cross-reacting with the native cytokine and in vivo protection. Vaccine 2004, 22, 3144–3153. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, J.; Xu, A.; Zhong, C.; Lu, W.; Deng, L.; Li, R. A rationally designed TNF-α epitope-scaffold immunogen induces sustained antibody response and alleviates collagen-induced arthritis in mice. PLoS ONE 2016, 11, e0163080. [Google Scholar] [CrossRef] [PubMed]
- Young, L.; Jernigan, R.L.; Covell, D.G. A role for surface hydrophobicity in protein-protein recognition. Protein Sci. 1994, 3, 717–729. [Google Scholar] [CrossRef]
- Yan, C.; Wu, F.; Jernigan, R.L.; Dobbs, D.; Honavar, V. Characterization of protein-protein interfaces. Protein J. 2008, 27, 59–70. [Google Scholar] [CrossRef]
- Chen, J.; Sawyer, N.; Regan, L. Protein-protein interactions: General trends in the relationship between binding affinity and interfacial buried surface area. Protein Sci. 2013, 22, 510–515. [Google Scholar] [CrossRef]
- Wójcik, P.; Berlicki, Ł. Peptide-based inhibitors of protein-protein interactions. Bioorg. Med. Chem. Lett. 2016, 26, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Kritzer, J.A.; Zutshi, R.; Cheah, M.; Ran, F.A.; Webman, R.; Wongjirad, T.M.; Schepartz, A. Miniature protein inhibitors of the P53-HDM2 Interaction. ChemBioChem 2006, 7, 29–31. [Google Scholar] [CrossRef]
- Ji, Y.; Majumder, S.; Millard, M.; Borra, R.; Bi, T.; Elnagar, A.Y.; Neamati, N.; Shekhtman, A.; Camarero, J.A. In vivo activation of the P53 tumor suppressor pathway by an engineered cyclotide. J. Am. Chem. Soc. 2013, 135, 11623–11633. [Google Scholar] [CrossRef]
- Fujiwara, D.; Kitada, H.; Oguri, M.; Nishihara, T.; Michigami, M.; Shiraishi, K.; Yuba, E.; Nakase, I.; Im, H.; Cho, S.; et al. A cyclized helix-loop-helix peptide as a molecular scaffold for the design of inhibitors of intracellular protein-protein interactions by epitope and arginine grafting. Angew. Chem. Int. Ed. 2016, 55, 10612–10615. [Google Scholar] [CrossRef]
- Li, C.; Liu, M.; Monbo, J.; Zou, G.; Li, C.; Yuan, W.; Zella, D.; Lu, W.Y.; Lu, W. Turning a scorpion toxin into an antitumor miniprotein. J. Am. Chem. Soc. 2008, 130, 13546–13548. [Google Scholar] [CrossRef] [PubMed]
- Ricardo, M.G.; Ali, A.M.; Plewka, J.; Surmiak, E.; Labuzek, B.; Neochoritis, C.G.; Atmaj, J.; Skalniak, L.; Zhang, R.; Holak, T.A.; et al. multicomponent peptide stapling as a diversity-driven tool for the development of Inhibitors of protein−protein interactions. Angew. Chem. Int. Ed. 2020, 59, 5235–5241. [Google Scholar] [CrossRef]
- Nagano, Y.; Arafiles, J.V.V.; Kuwata, K.; Kawaguchi, Y.; Imanishi, M.; Hirose, H.; Futaki, S. Grafting hydrophobic amino acids critical for inhibition of protein-protein interactions on a cell-penetrating peptide scaffold. Mol. Pharm. 2022, 19, 558–567. [Google Scholar] [CrossRef]
- Klein, C.; Vassilev, L.T. Targeting the P53-MDM2 interaction to treat cancer. Br. J. Cancer 2004, 91, 1415–1419. [Google Scholar] [CrossRef]
- Kussie, P.H.; Gorina, S.; Marechal, V.; Elenbaas, B.; Moreau, J.; Levine, A.J.; Pavletich, N.P. Structure of the MDM2 oncoprotein bound to the P53 tumor suppressor transactivation domain. Science 1996, 274, 948–953. [Google Scholar] [CrossRef]
- Böttger, A.; Böttger, V.; Sparks, A.; Liu, W.L.; Howard, S.F.; Lane, D.P. Design of a synthetic Mdm2-binding mini protein that activates the p53 response in vivo. Curr. Biol. 1997, 7, 860–869. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.K.; Kaas, Q.; Chiche, L.; Craik, D.J. Cybase: A database of cyclic protein sequences and structures, with applications in protein discovery and engineering. Nucleic Acids Res. 2008, 36, D206–D210. [Google Scholar] [CrossRef] [PubMed]
- Daly, N.L.; Wilson, D.T. Plant derived cyclic peptides. Biochem. Soc. Trans. 2021, 49, 1279–1285. [Google Scholar] [CrossRef] [PubMed]
- Aboye, T.L.; Ha, H.; Majumder, S.; Christ, F.; Debyser, Z.; Shekhtman, A.; Neamati, N.; Camarero, J.A. Design of a novel cyclotide-based cxcr4 antagonist with anti-human immunodeficiency virus (hiv)-1 activity. J. Med. Chem. 2012, 55, 10729–10734. [Google Scholar] [CrossRef] [PubMed]
- Saether, O.; Craik, D.J.; Campbell, I.D.; Sletten, K.; Juul, J.; Norman, D.G. Elucidation of the primary and three-dimensional structure of the uterotonic polypeptide kalata b1. Biochemistry 1995, 34, 4147–4158. [Google Scholar] [CrossRef]
- Grover, T.; Mishra, R.; Bushra; Gulati, P.; Mohanty, A. An insight into biological activities of native cyclotides for potential applications in agriculture and pharmaceutics. Peptides 2021, 135, 170430. [Google Scholar] [CrossRef] [PubMed]
- Gunasekera, S.; Foley, F.M.; Clark, R.J.; Sando, L.; Fabri, L.J.; Craik, D.J.; Daly, N.L. Engineering stabilized vascular endothelial growth factor-A antagonists: Synthesis, structural characterization, and bioactivity of grafted analogues of cyclotides. J. Med. Chem. 2008, 51, 7697–7704. [Google Scholar] [CrossRef]
- González-Castro, R.; Gómez-Lim, M.A.; Plisson, F. Cysteine-rich peptides: Hyperstable scaffolds for protein engineering. Chembiochem 2021, 22, 961–973. [Google Scholar] [CrossRef]
- Lagassé, H.A.D.; McCormick, Q.; Sauna, Z.E. Secondary failure: Immune responses to approved protein therapeutics. Trends Mol. Med. 2021, 27, 1074–1083. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Komar, A.A. Molecular Peptide Grafting as a Tool to Create Novel Protein Therapeutics. Molecules 2023, 28, 2383. https://doi.org/10.3390/molecules28052383
Komar AA. Molecular Peptide Grafting as a Tool to Create Novel Protein Therapeutics. Molecules. 2023; 28(5):2383. https://doi.org/10.3390/molecules28052383
Chicago/Turabian StyleKomar, Anton A. 2023. "Molecular Peptide Grafting as a Tool to Create Novel Protein Therapeutics" Molecules 28, no. 5: 2383. https://doi.org/10.3390/molecules28052383
APA StyleKomar, A. A. (2023). Molecular Peptide Grafting as a Tool to Create Novel Protein Therapeutics. Molecules, 28(5), 2383. https://doi.org/10.3390/molecules28052383