
Fluorescent Probes cis- and trans-Parinaric Acids in Fluid and Gel Lipid Bilayers: A Molecular Dynamics Study




Abstract

1. Introduction
2. Results and Discussion
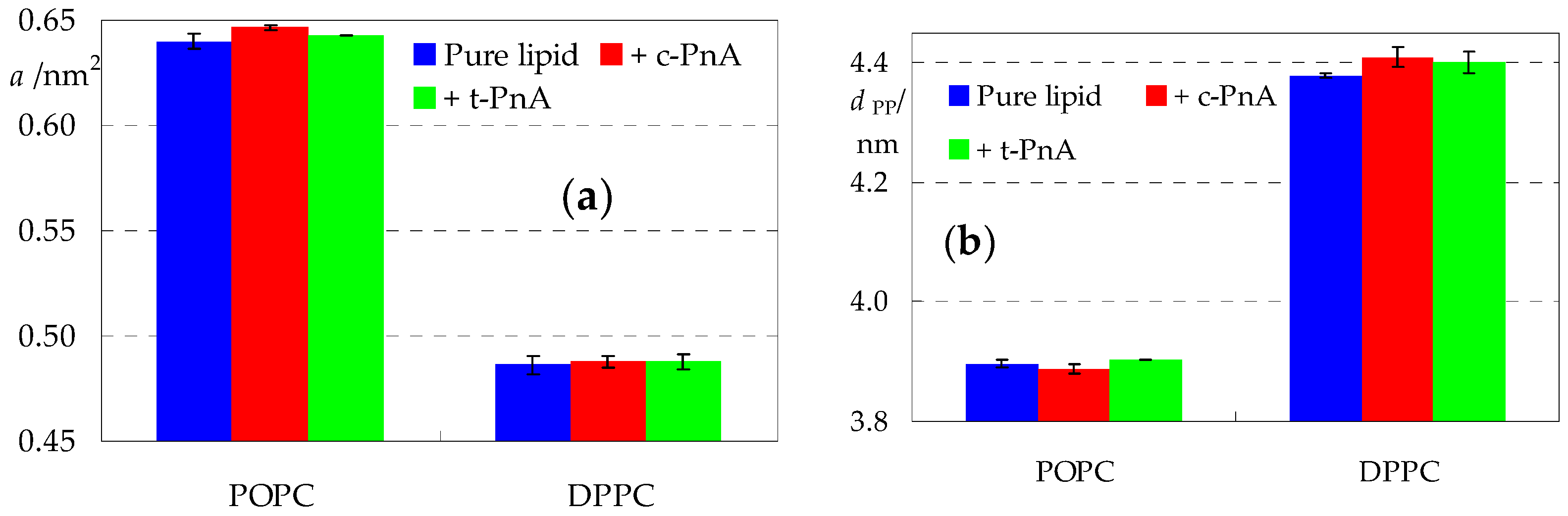
2.1. Bilayer Thickness and Area Per Lipid
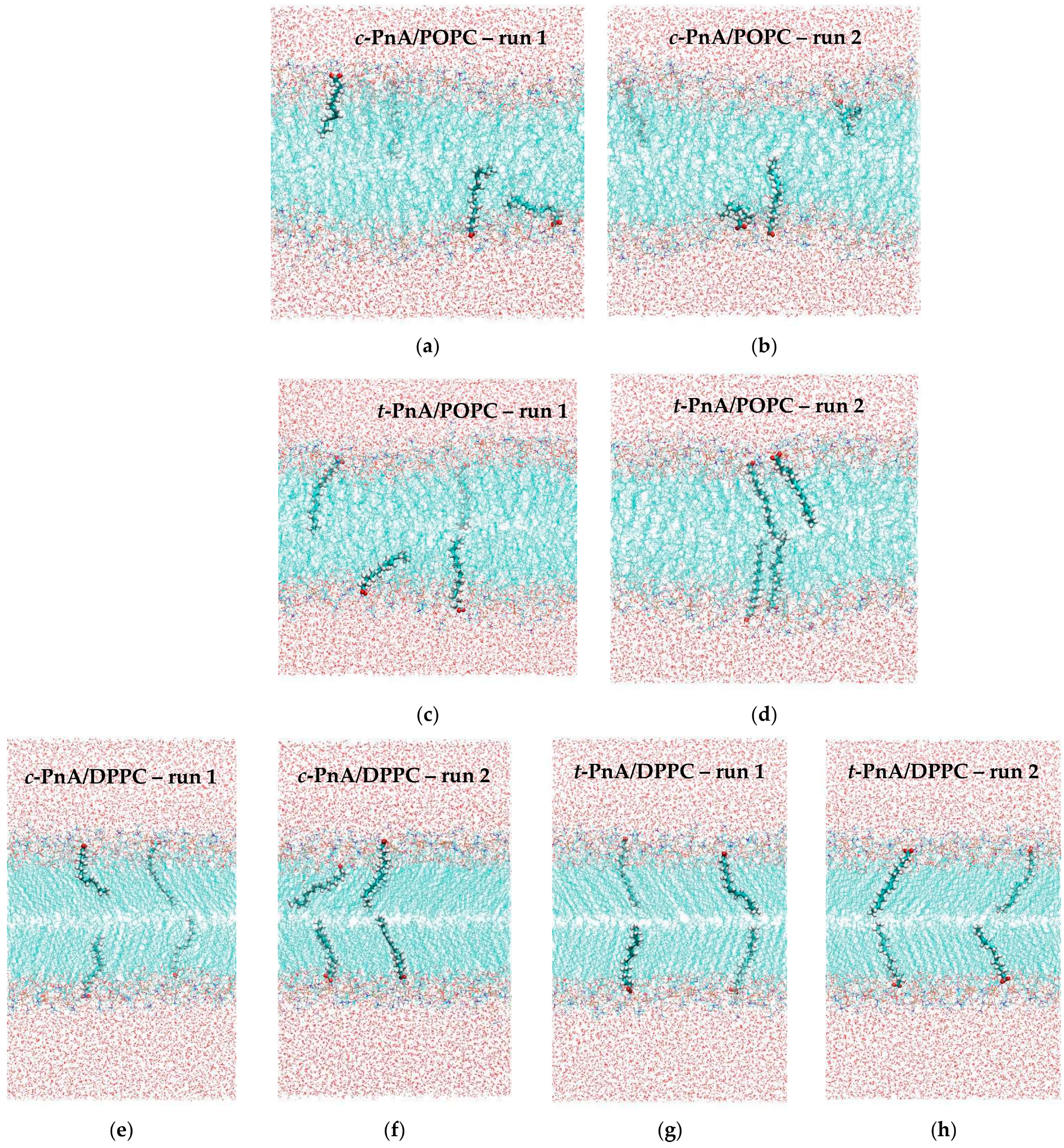
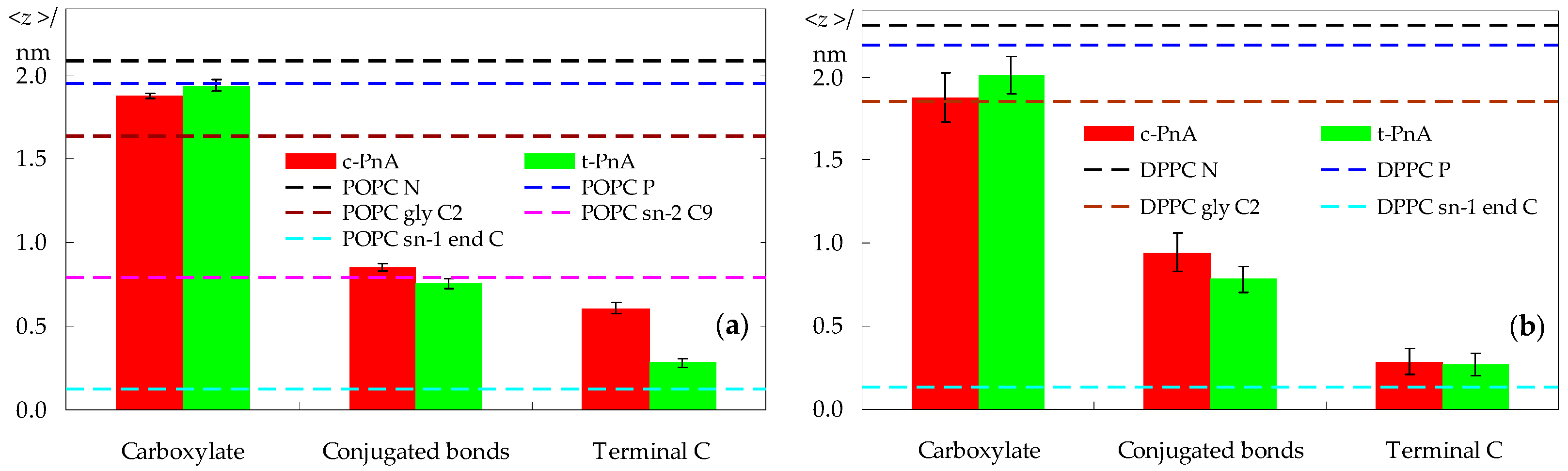
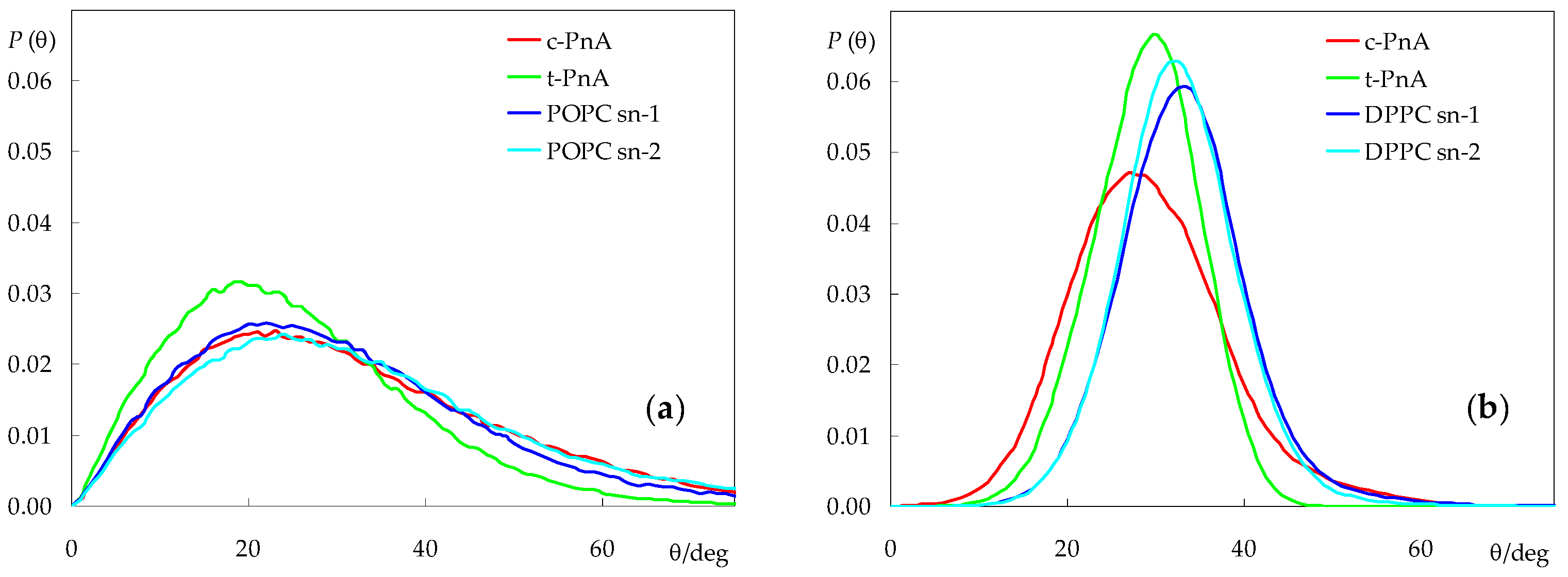
2.2. Probe Location and Orientation
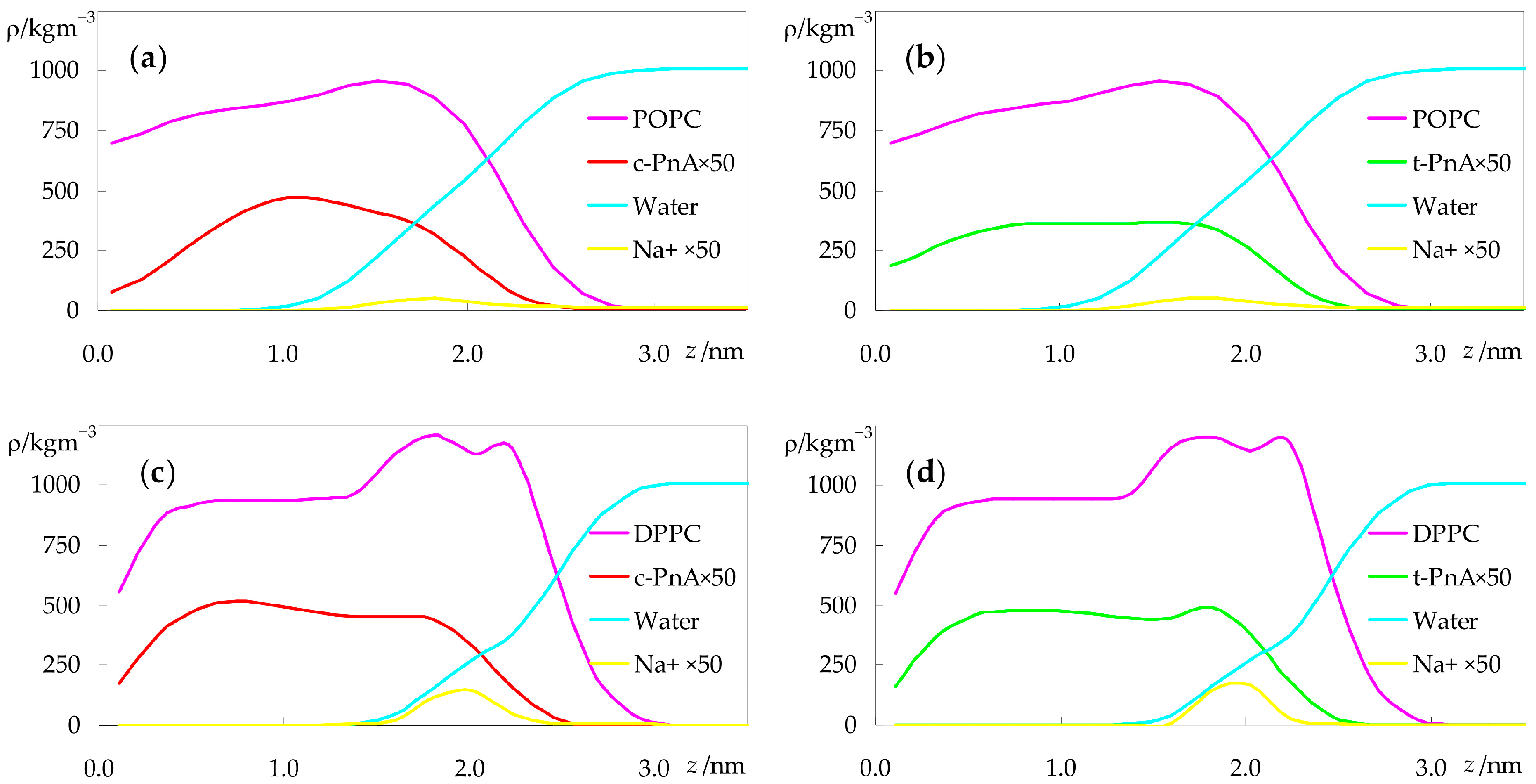
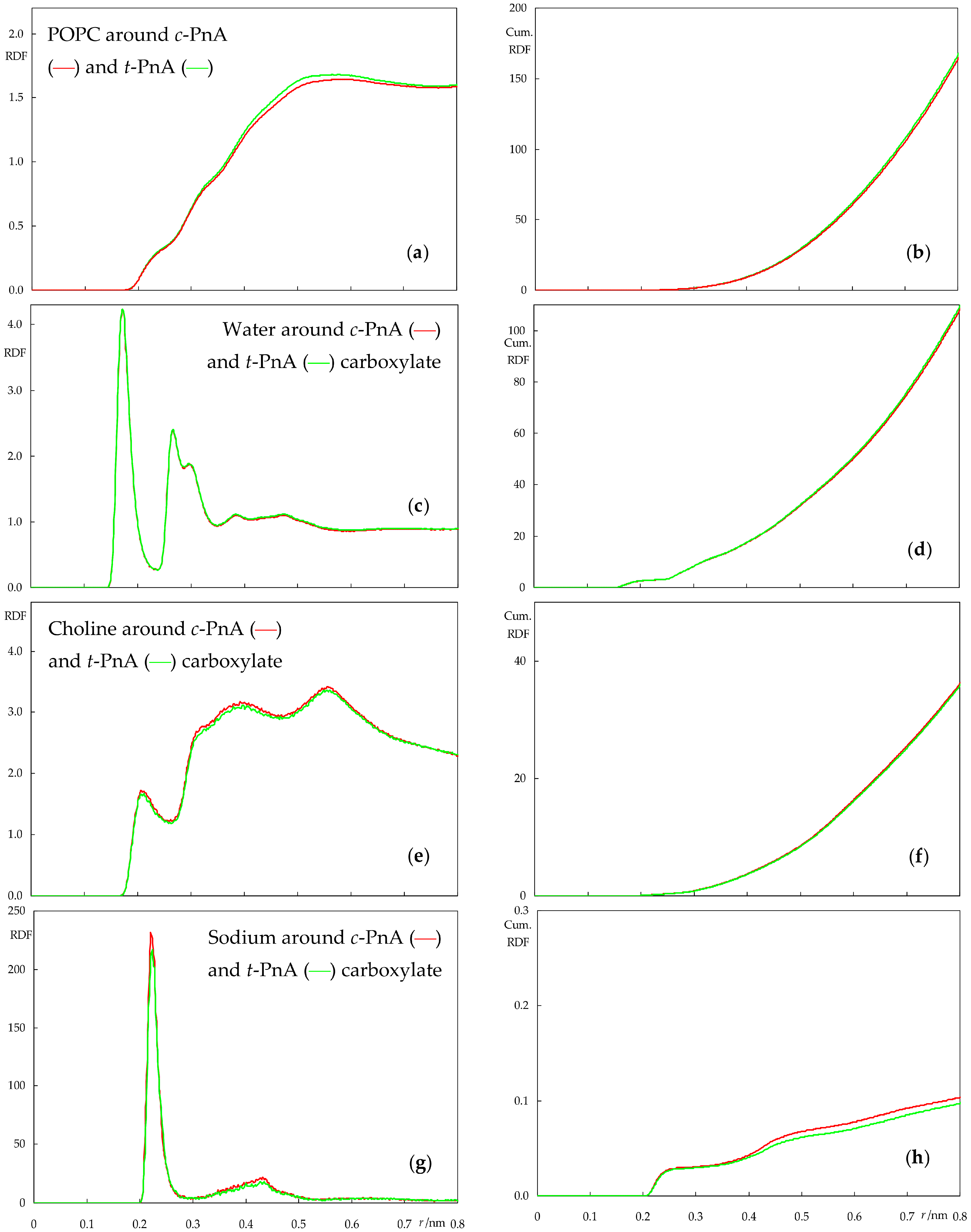
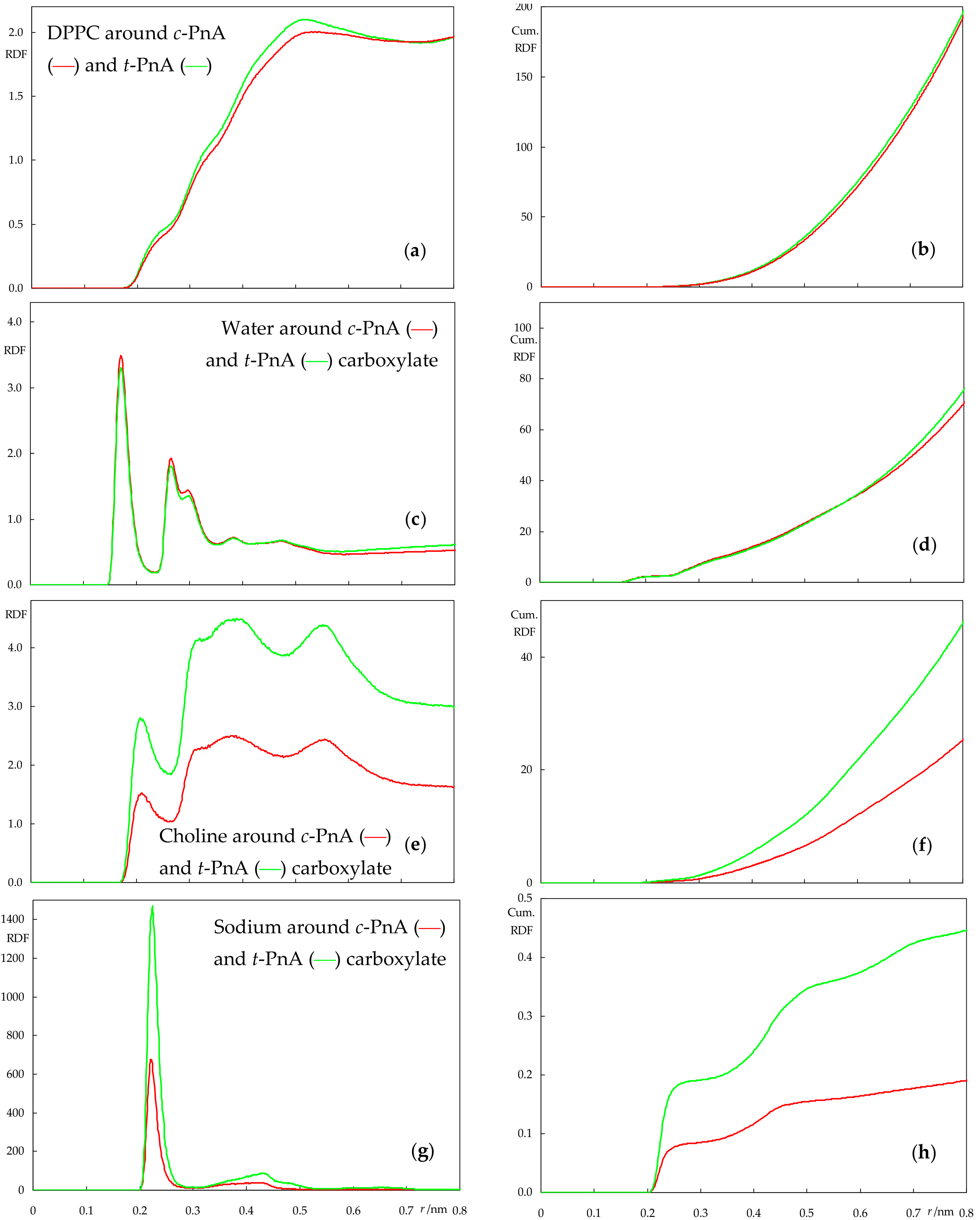
2.3. Interactions between Probes and Lipids or Solvent
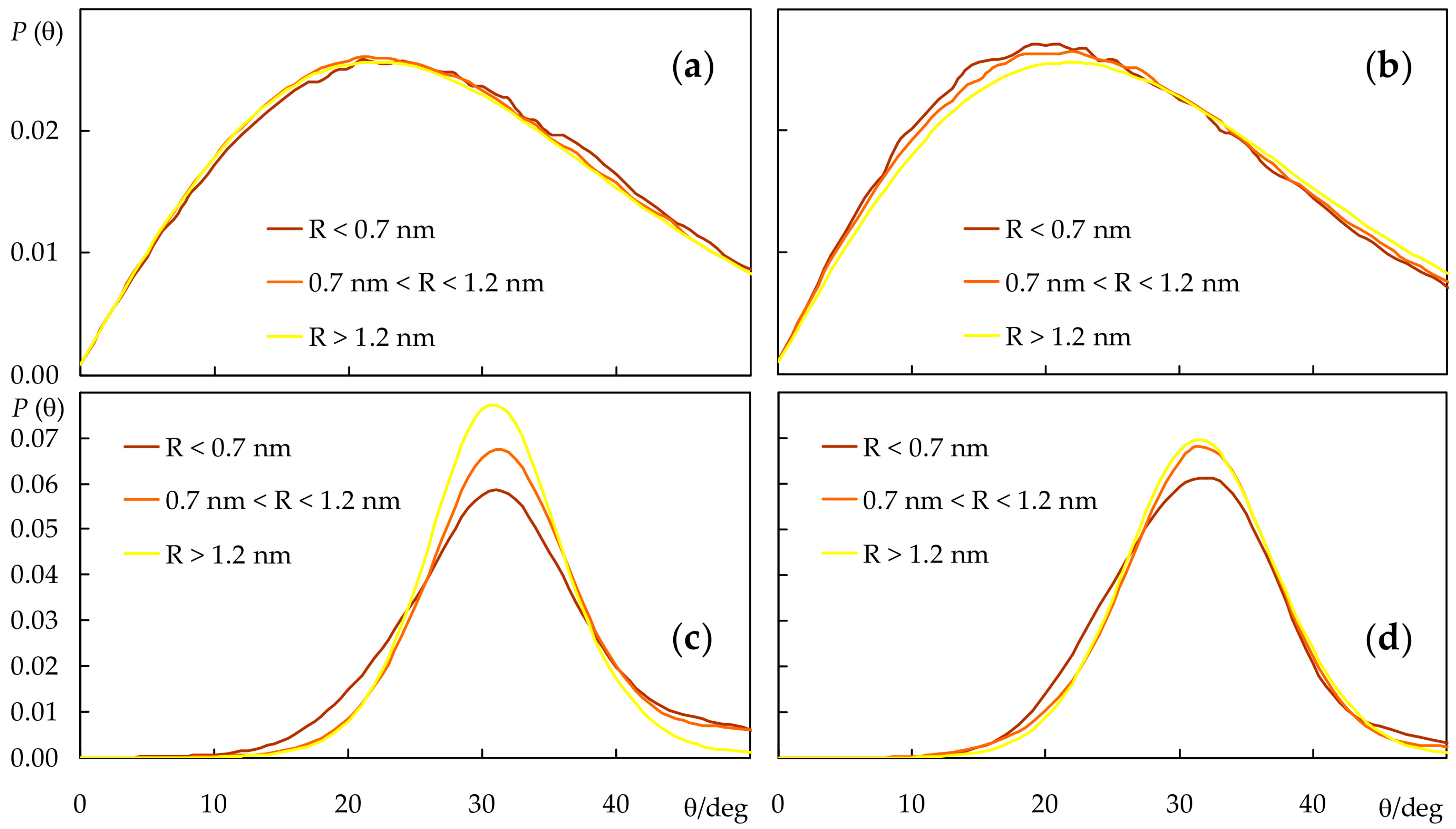
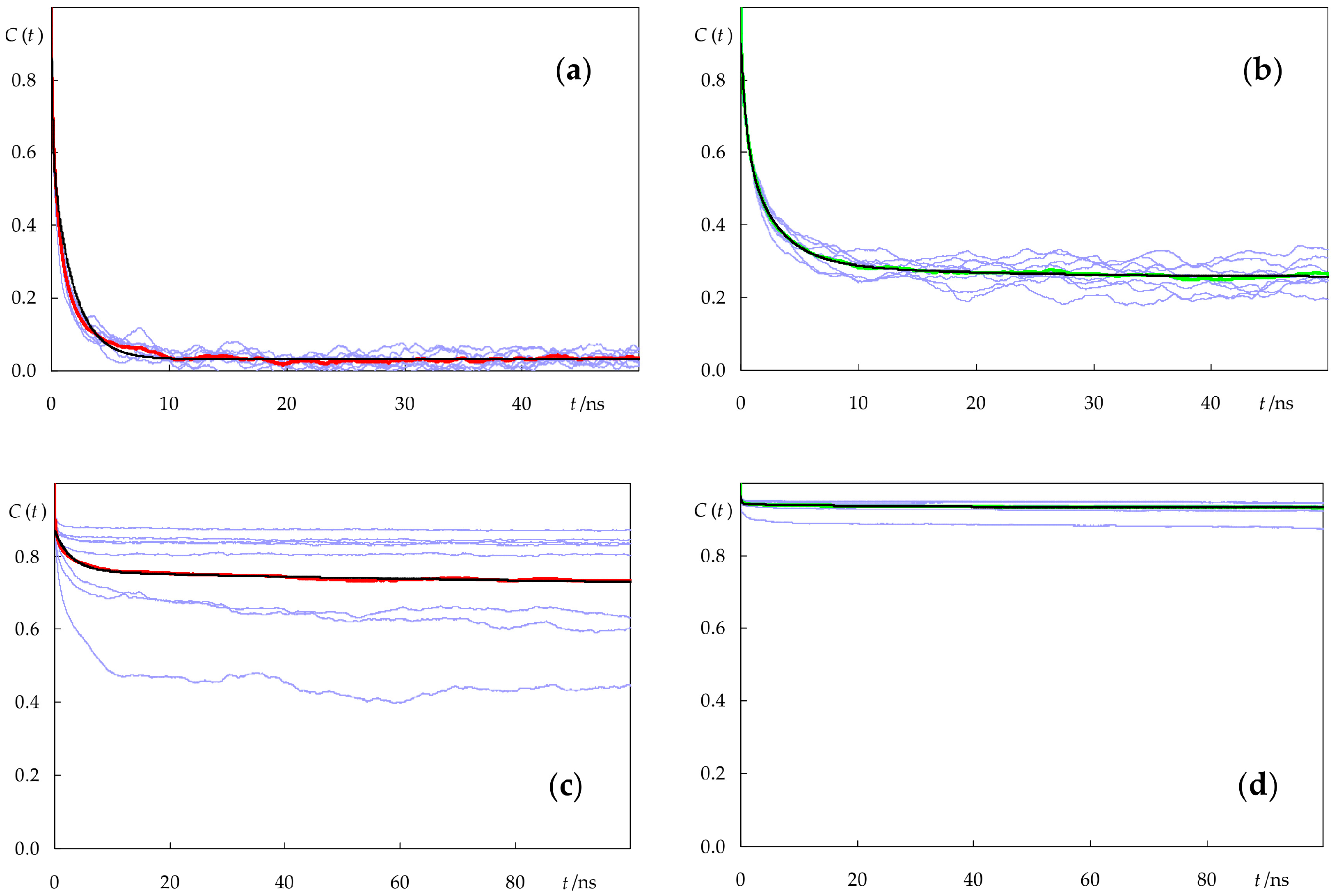
2.4. Rotational and Translational Dynamics
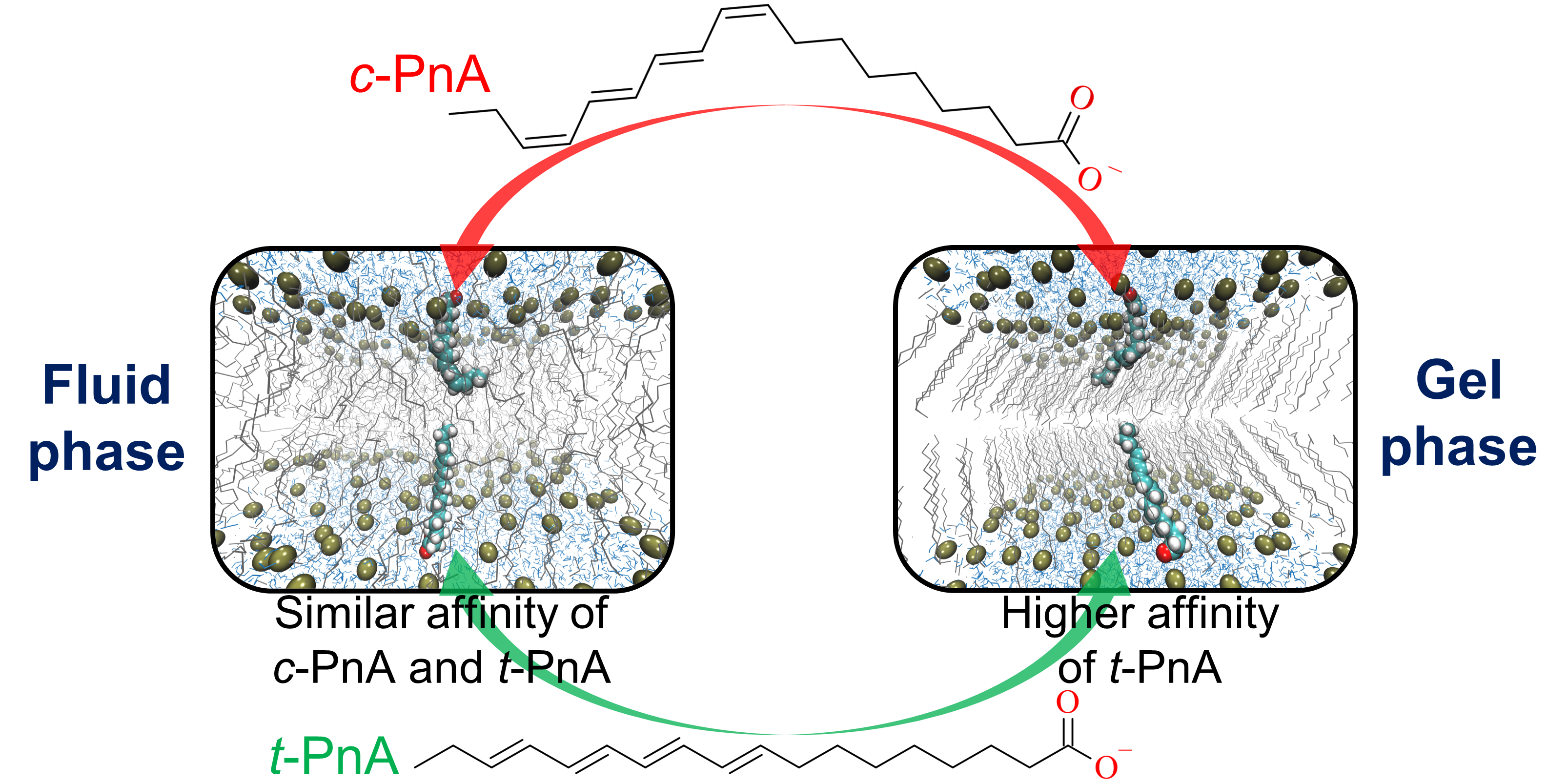
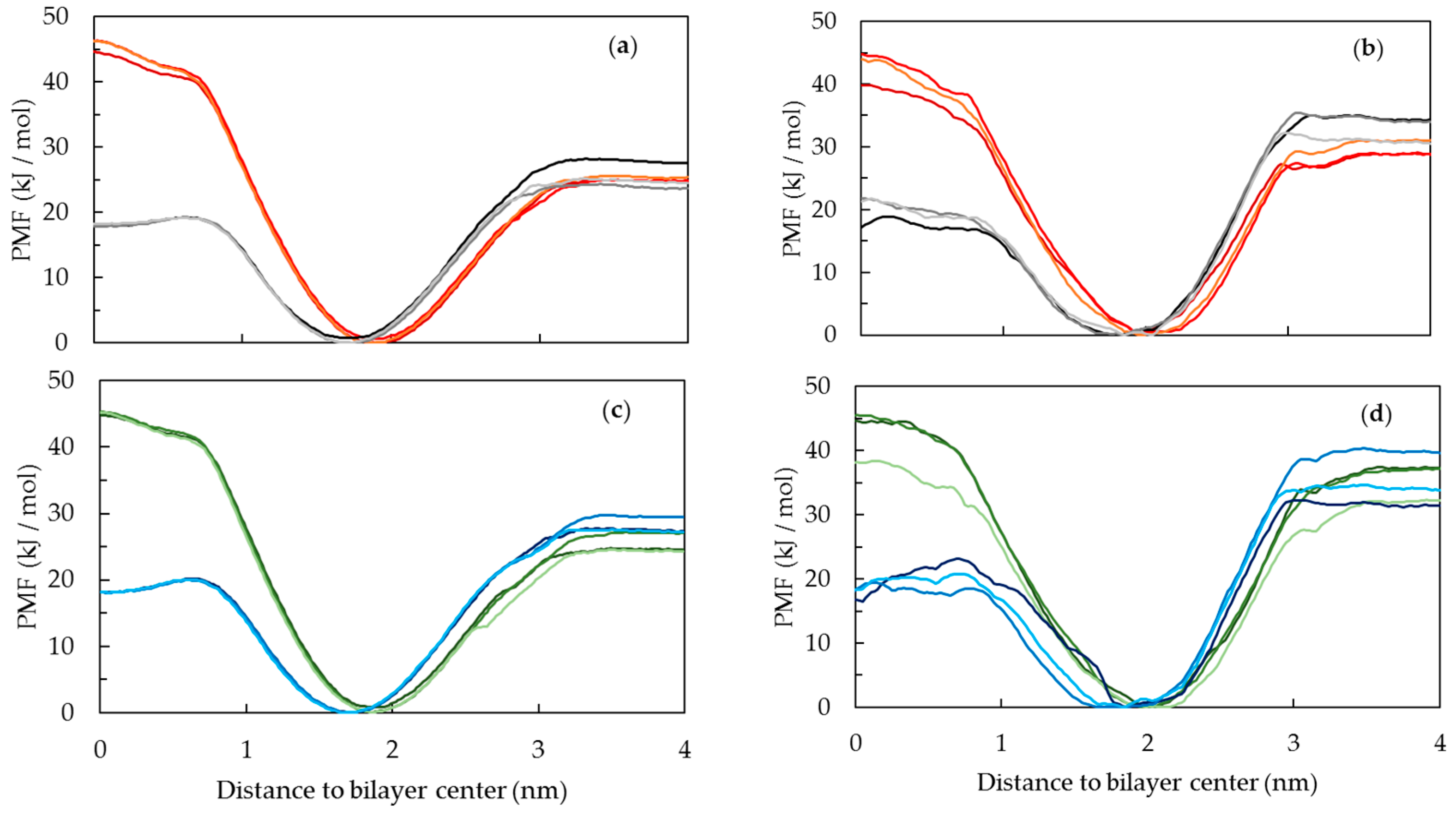
2.5. Free Energy Profile of PnA across DPPC and POPC Bilayers and Its Relation to Probe Partition
3. Methods
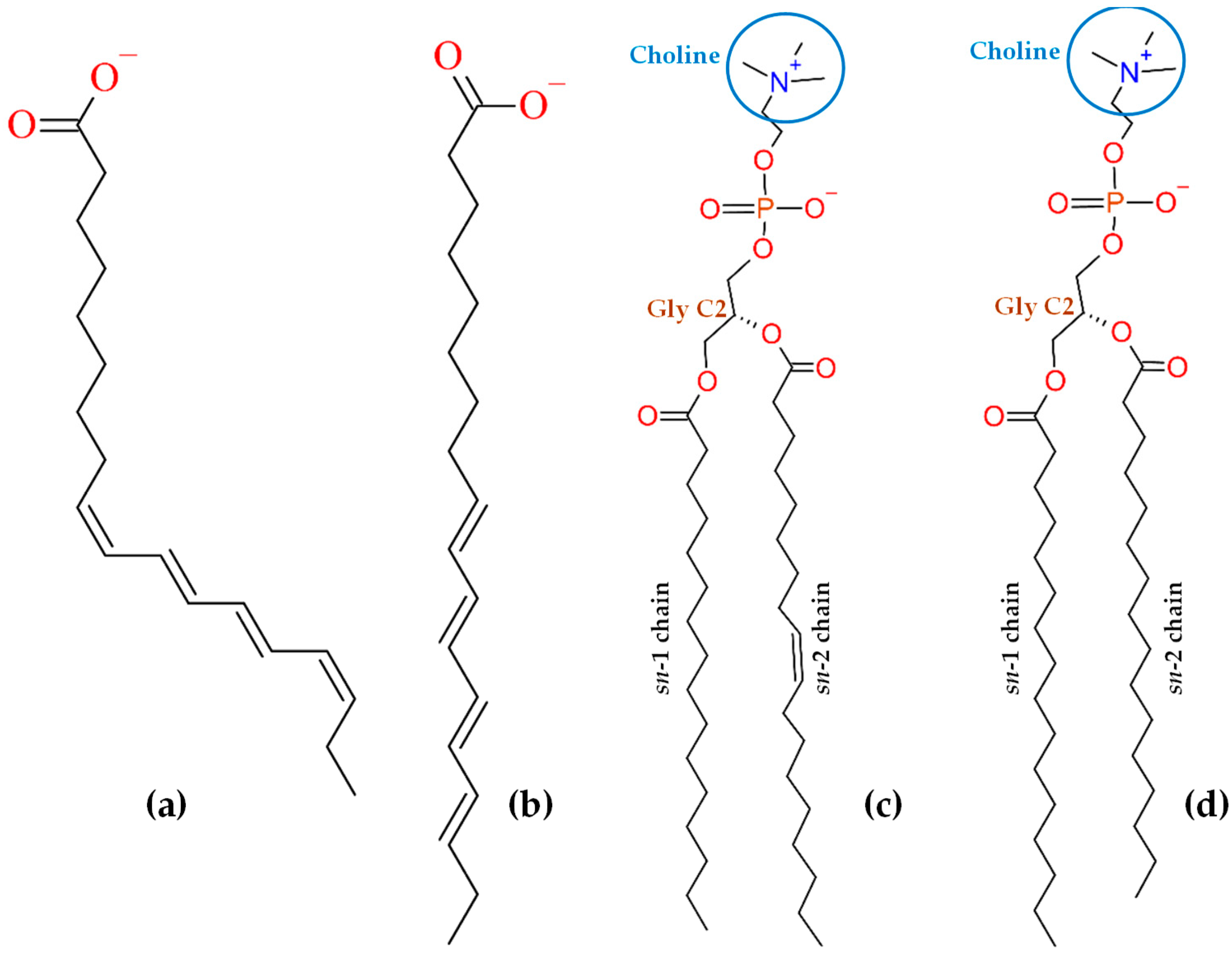
3.1. All-Atom MD Simulation
3.2. CG Simulation and Calculation of PMF Profiles
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 3rd ed.; Lakowicz, J.R., Ed.; Springer: Boston, MA, USA, 2006; ISBN 978-0-387-31278-1. [Google Scholar]
- Demchenko, A.P.; Mély, Y.; Duportail, G.; Klymchenko, A.S. Monitoring Biophysical Properties of Lipid Membranes by Environment-Sensitive Fluorescent Probes. Biophys. J. 2009, 96, 3461–3470. [Google Scholar] [CrossRef] [PubMed]
- Demchenko, A.P.; Duportail, G.; Oncul, S.; Klymchenko, A.S.; Mély, Y. Introduction to Fluorescence Probing of Biological Membranes. In Methods in Membrane Lipids. Methods in Molecular Biology; Humana Press: New York, NY, USA, 2015; Volume 1232, pp. 19–43. ISBN 978-1-4939-1751-8. [Google Scholar]
- Kyrychenko, A. Using Fluorescence for Studies of Biological Membranes: A Review. Methods Appl. Fluoresc. 2015, 3, 042003. [Google Scholar] [CrossRef] [PubMed]
- Sarmento, M.J.; Fernandes, F. Choosing the Right Fluorescent Probe. In Fluorescence Spectroscopy in Biology-Springer Series on Fluorescence; Springer: Cham, Switzerland, 2022; pp. 1–49. [Google Scholar] [CrossRef]
- Filipe, H.A.L.; Moreno, M.J.; Loura, L.M.S. The Secret Lives of Fluorescent Membrane Probes as Revealed by Molecular Dynamics Simulations. Molecules 2020, 25, 3424. [Google Scholar] [CrossRef] [PubMed]
- Faller, R. Molecular Modeling of Lipid Probes and Their Influence on the Membrane. Biochim. Biophys. Acta -Biomembr. 2016, 1858, 2353–2361. [Google Scholar] [CrossRef]
- Loura, L.M.S.; Ramalho, J.P.P. Recent Developments in Molecular Dynamics Simulations of Fluorescent Membrane Probes. Molecules 2011, 16, 5437–5452. [Google Scholar] [CrossRef] [PubMed]
- Loura, L.M.S.; Prates Ramalho, J.P. Fluorescent Membrane Probes’ Behavior in Lipid Bilayers: Insights from Molecular Dynamics Simulations. Biophys. Rev. 2009, 1, 141–148. [Google Scholar] [CrossRef]
- Hoff, B.; Strandberg, E.; Ulrich, A.S.; Tieleman, D.P.; Posten, C. 2H-NMR Study and Molecular Dynamics Simulation of the Location, Alignment, and Mobility of Pyrene in POPC Bilayers. Biophys. J. 2005, 88, 1818–1827. [Google Scholar] [CrossRef] [PubMed]
- Čurdová, J.; Čapková, P.; Plášek, J.; Repáková, J.; Vattulainen, I. Free Pyrene Probes in Gel and Fluid Membranes: Perspective through Atomistic Simulations. J. Phys. Chem. B 2007, 111, 3640–3650. [Google Scholar] [CrossRef]
- Loura, L.M.S.; do Canto, A.M.T.M.; Martins, J. Sensing Hydration and Behavior of Pyrene in POPC and POPC/Cholesterol Bilayers: A Molecular Dynamics Study. Biochim. Biophys. Acta -Biomembr. 2013, 1828, 1094–1101. [Google Scholar] [CrossRef]
- Do Canto, A.M.T.M.; Santos, P.D.; Martins, J.; Loura, L.M.S. Behavior of Pyrene as a Polarity Probe in Palmitoylsphingomyelin and Palmitoylsphingomyelin/Cholesterol Bilayers: A Molecular Dynamics Simulation Study. Colloids Surf. A Physicochem. Eng. Asp. 2015, 480, 296–306. [Google Scholar] [CrossRef]
- Poojari, C.; Wilkosz, N.; Lira, R.B.; Dimova, R.; Jurkiewicz, P.; Petka, R.; Kepczynski, M.; Róg, T. Behavior of the DPH Fluorescence Probe in Membranes Perturbed by Drugs. Chem. Phys. Lipids 2019, 223, 104784. [Google Scholar] [CrossRef] [PubMed]
- Paloncýová, M.; Ameloot, M.; Knippenberg, S. Orientational Distribution of DPH in Lipid Membranes: A Comparison of Molecular Dynamics Calculations and Experimental Time-Resolved Anisotropy Experiments. Phys. Chem. Chem. Phys. 2019, 21, 7594–7604. [Google Scholar] [CrossRef] [PubMed]
- Do Canto, A.M.T.M.; Robalo, J.R.; Santos, P.D.; Carvalho, A.J.P.; Ramalho, J.P.P.; Loura, L.M.S. Diphenylhexatriene Membrane Probes DPH and TMA-DPH: A Comparative Molecular Dynamics Simulation Study. Biochim. Biophys. Acta-Biomembr. 2016, 1858, 2647–2661. [Google Scholar] [CrossRef] [PubMed]
- Repáková, J.; Holopainen, J.M.; Morrow, M.R.; McDonald, M.C.; Čapková, P.; Vattulainen, I. Influence of DPH on the Structure and Dynamics of a DPPC Bilayer. Biophys. J. 2005, 88, 3398–3410. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, M.X.; García de la Torre, J.; Castanho, M.A.R.B. A Brownian Dynamics Simulation of an Acyl Chain and a Trans -Parinaric Acid Molecule Confined in a Phospholipid Bilayer in the Gel and Liquid-Crystal Phases. J. Phys. Chem. B 2000, 104, 11579–11584. [Google Scholar] [CrossRef]
- Sklar, L.A.; Hudson, B.S.; Simoni, R.D. Conjugated Polyene Fatty Acids as Membrane Probes: Preliminary Characterization. Proc. Natl. Acad. Sci. USA 1975, 72, 1649–1653. [Google Scholar] [CrossRef]
- Hudson, B.; Cavalier, S.A. Studies of Membrane Dynamics And Lipid-Protein Interactions With Parinaric Acid. In Spectroscopic Membrane Probes; Loew, L.M., Ed.; CRC Press: Boca Raton, FL, USA, 1988; pp. 43–62. [Google Scholar]
- Reyes Mateo, C.; Brochon, J.C.; Pilar Lillo, M.; Ulises Acuña, A. Lipid Clustering in Bilayers Detected by the Fluorescence Kinetics and Anisotropy of Trans-Parinaric Acid. Biophys. J. 1993, 65, 2237–2247. [Google Scholar] [CrossRef]
- De Almeida, R.F.M.; Fedorov, A.; Prieto, M. Sphingomyelin/Phosphatidylcholine/Cholesterol Phase Diagram: Boundaries and Composition of Lipid Rafts. Biophys. J. 2003, 85, 2406–2416. [Google Scholar] [CrossRef]
- Silva, L.; De Almeida, R.F.M.; Fedorov, A.; Matos, A.P.A.; Prieto, M. Ceramide-Platform Formation and -Induced Biophysical Changes in a Fluid Phospholipid Membrane. Mol. Membr. Biol. 2006, 23, 137–148. [Google Scholar] [CrossRef]
- Castro, B.M.; de Almeida, R.F.M.; Silva, L.C.; Fedorov, A.; Prieto, M. Formation of Ceramide/Sphingomyelin Gel Domains in the Presence of an Unsaturated Phospholipid: A Quantitative Multiprobe Approach. Biophys. J. 2007, 93, 1639–1650. [Google Scholar] [CrossRef]
- Pinto, S.N.; Silva, L.C.; de Almeida, R.F.M.; Prieto, M. Membrane Domain Formation, Interdigitation, and Morphological Alterations Induced by the Very Long Chain Asymmetric C24:1 Ceramide. Biophys. J. 2008, 95, 2867–2879. [Google Scholar] [CrossRef]
- Castro, B.M.; Silva, L.C.; Fedorov, A.; de Almeida, R.F.M.; Prieto, M. Cholesterol-Rich Fluid Membranes Solubilize Ceramide Domains: Implications for the structure and dynamics of mammalian intracellular and plasma membranes. J. Biol. Chem. 2009, 284, 22978–22987. [Google Scholar] [CrossRef] [PubMed]
- Nyholm, T.K.M.; Lindroos, D.; Westerlund, B.; Slotte, J.P. Construction of a DOPC/PSM/Cholesterol Phase Diagram Based on the Fluorescence Properties of Trans -Parinaric Acid. Langmuir 2011, 27, 8339–8350. [Google Scholar] [CrossRef]
- Yapoudjian, S.; Ivanova, M.G.; Brzozowski, A.M.; Patkar, S.A.; Vind, J.; Svendsen, A.; Verger, R. Binding of Thermomyces (Humicola) Lanuginosa Lipase to the Mixed Micelles of Cis -Parinaric Acid/NaTDC. Eur. J. Biochem. 2002, 269, 1613–1621. [Google Scholar] [CrossRef] [PubMed]
- Petrescu, A.D.; Hertz, R.; Bar-Tana, J.; Schroeder, F.; Kier, A.B. Role of Regulatory F-Domain in Hepatocyte Nuclear Factor-4α Ligand Specificity. J. Biol. Chem. 2005, 280, 16714–16727. [Google Scholar] [CrossRef]
- De Almeida, R.F.M.; Loura, L.M.S.; Fedorov, A.; Prieto, M. Nonequilibrium Phenomena in the Phase Separation of a Two-Component Lipid Bilayer. Biophys. J. 2002, 82, 823–834. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, T.; Slotte, J.P.; Murata, M.; Hanashima, S. Molecular Dynamics of Glycolipids in Liposomes. In Glycolipids. Methods in Molecular Biology; Kabayama, K., Inokuchi, J., Eds.; Humana: New York, NY, USA, 2023; Volume 2613, pp. 257–270. [Google Scholar] [CrossRef]
- Marsh, D. Handbook of Lipid Bilayers, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2013; ISBN 9780429193125. [Google Scholar]
- Kučerka, N.; Nieh, M.; Katsaras, J. Fluid Phase Lipid Areas and Bilayer Thicknesses of Commonly Used Phosphatidylcholines as a Function of Temperature. Biochim. Biophys. Acta-Biomembr. 2011, 1808, 2761–2771. [Google Scholar] [CrossRef]
- Shahane, G.; Ding, W.; Palaiokostas, M.; Orsi, M. Physical Properties of Model Biological Lipid Bilayers: Insights from All-Atom Molecular Dynamics Simulations. J. Mol. Model. 2019, 25, 76. [Google Scholar] [CrossRef]
- Tjörnhammar, R.; Edholm, O. Reparameterized United Atom Model for Molecular Dynamics Simulations of Gel and Fluid Phosphatidylcholine Bilayers. J. Chem. Theory Comput. 2014, 10, 5706–5715. [Google Scholar] [CrossRef]
- Ferreira, T.M.; Coreta-Gomes, F.; Ollila, O.H.S.; Moreno, M.J.; Vaz, W.L.C.; Topgaard, D. Cholesterol and POPC Segmental Order Parameters in Lipid Membranes: Solid State 1 H– 13 C NMR and MD Simulation Studies. Phys. Chem. Chem. Phys. 2013, 15, 1976–1989. [Google Scholar] [CrossRef]
- Piggot, T.J.; Allison, J.R.; Sessions, R.B.; Essex, J.W. On the Calculation of Acyl Chain Order Parameters from Lipid Simulations. J. Chem. Theory Comput. 2017, 13, 5683–5696. [Google Scholar] [CrossRef] [PubMed]
- Castanho, M.; Prieto, M.; Ulises Acuña, A. The Transverse Location of the Fluorescent Probe Trans-Parinaric Acid in Lipid Bilayers. Biochim. Biophys. Acta-Biomembr. 1996, 1279, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Tristram-Nagle, S.; Zhang, R.; Suter, R.M.; Worthington, C.R.; Sun, W.J.; Nagle, J.F. Measurement of Chain Tilt Angle in Fully Hydrated Bilayers of Gel Phase Lecithins. Biophys. J. 1993, 64, 1097–1109. [Google Scholar] [CrossRef]
- Lopes, S.; Fernandes, M.X.; Prieto, M.; Castanho, M.A.R.B. Orientational Order of the Polyene Fatty Acid Membrane Probe Trans -Parinaric Acid in Langmuir−Blodgett Multilayer Films. J. Phys. Chem. B 2001, 105, 562–568. [Google Scholar] [CrossRef]
- Shang, Q.; Dou, X.; Hudson, B.S. Off-Axis Orientation of the Electronic Transition Moment for a Linear Conjugated Polyene. Nature 1991, 352, 703–705. [Google Scholar] [CrossRef]
- Kinosita, K.; Kawato, S.; Ikegami, A. A Theory of Fluorescence Polarization Decay in Membranes. Biophys. J. 1977, 20, 289–305. [Google Scholar] [CrossRef] [PubMed]
- Tao, T. Time-Dependent Fluorescence Depolarization and Brownian Rotational Diffusion Coefficients of Macromolecules. Biopolymers 1969, 8, 609–632. [Google Scholar] [CrossRef]
- Loura, L.M.S.; Ramalho, J.P.P. Location and Dynamics of Acyl Chain NBD-Labeled Phosphatidylcholine (NBD-PC) in DPPC Bilayers. A Molecular Dynamics and Time-Resolved Fluorescence Anisotropy Study. Biochim. Biophys. Acta -Biomembr. 2007, 1768, 467–478. [Google Scholar] [CrossRef]
- Reyes Mateo, C.; Ulises Acuña, A.; Brochon, J.C. Liquid-Crystalline Phases of Cholesterol/Lipid Bilayers as Revealed by the Fluorescence of Trans-Parinaric Acid. Biophys. J. 1995, 68, 978–987. [Google Scholar] [CrossRef][Green Version]
- Calafut, T.M.; Dix, J.A.; Verkman, A.S. Fluorescence Depolarization of Cis- and Trans-Parinaric Acids in Artificial and Red Cell Membranes Resolved by a Double Hindered Rotational Model. Biochemistry 1989, 28, 5051–5058. [Google Scholar] [CrossRef] [PubMed]
- Wolber, P.K.; Hudson, B.S. Bilayer Acyl Chain Dynamics and Lipid-Protein Interaction: The Effect of the M13 Bacteriophage Coat Protein on the Decay of the Fluorescence Anisotropy of Parinaric Acid. Biophys. J. 1982, 37, 253–262. [Google Scholar] [CrossRef][Green Version]
- Lyubartsev, A.P.; Rabinovich, A.L. Recent Development in Computer Simulations of Lipid Bilayers. Soft Matter 2011, 7, 25–39. [Google Scholar] [CrossRef]
- Filippov, A.; Orädd, G.; Lindblom, G. The Effect of Cholesterol on the Lateral Diffusion of Phospholipids in Oriented Bilayers. Biophys. J. 2003, 84, 3079–3086. [Google Scholar] [CrossRef]
- Harb, F.; Simon, A.; Tinland, B. Ripple Formation in Unilamellar-Supported Lipid Bilayer Revealed by FRAPP. Eur. Phys. J. E 2013, 36, 140. [Google Scholar] [CrossRef] [PubMed]
- Filipe, H.A.L.; Moreno, M.J.; Róg, T.; Vattulainen, I.; Loura, L.M.S. How To Tackle the Issues in Free Energy Simulations of Long Amphiphiles Interacting with Lipid Membranes: Convergence and Local Membrane Deformations. J. Phys. Chem. B 2014, 118, 3572–3581. [Google Scholar] [CrossRef]
- Kamp, F.; Zakim, D.; Zhang, F.; Noy, N.; Hamilton, J.A. Fatty Acid Flip-Flop in Phospholipid Bilayers Is Extremely Fast. Biochemistry 1995, 34, 11928–11937. [Google Scholar] [CrossRef] [PubMed]
- Filipe, H.A.L.; Javanainen, M.; Salvador, A.; Galvão, A.M.; Vattulainen, I.; Loura, L.M.S.; Moreno, M.J. Quantitative Assessment of Methods Used To Obtain Rate Constants from Molecular Dynamics Simulations—Translocation of Cholesterol across Lipid Bilayers. J. Chem. Theory Comput. 2018, 14, 3840–3848. [Google Scholar] [CrossRef] [PubMed]
- GROMACS development team. GROMACS 2019.3 Source Code. Available online: https://doi.org/10.5281/zenodo.3243833 (accessed on 30 January 2023).
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O’Connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; MacKerell, A.D.; Pastor, R.W. Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J. Phys. Chem. B 2010, 114, 7830–7843. [Google Scholar] [CrossRef]
- Beglov, D.; Roux, B. Finite Representation of an Infinite Bulk System: Solvent Boundary Potential for Computer Simulations. J. Chem. Phys. 1994, 100, 9050–9063. [Google Scholar] [CrossRef]
- Noskov, S.Y.; Roux, B. Control of Ion Selectivity in LeuT: Two Na+ Binding Sites with Two Different Mechanisms. J. Mol. Biol. 2008, 377, 804–818. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM General Force Field: A Force Field for Drug-like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. J. Comput. Chem. 2009, 31, 21367. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L. Quantum and Statistical Mechanical Studies of Liquids. 10. Transferable Intermolecular Potential Functions for Water, Alcohols, and Ethers. Application to Liquid Water. J. Am. Chem. Soc. 1981, 103, 335–340. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef]
- Vega, C.; Abascal, J.L.F. Simulating Water with Rigid Non-Polarizable Models: A General Perspective. Phys. Chem. Chem. Phys. 2011, 13, 19663. [Google Scholar] [CrossRef]
- Kim, S.; Lee, J.; Jo, S.; Brooks, C.L.; Lee, H.S.; Im, W. CHARMM-GUI Ligand Reader and Modeler for CHARMM Force Field Generation of Small Molecules. J. Comput. Chem. 2017, 38, 1879–1886. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A Web-Based Graphical User Interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Wishart, D.S.; Sayeeda, Z.; Budinski, Z.; Guo, A.; Lee, B.L.; Berjanskii, M.; Rout, M.; Peters, H.; Dizon, R.; Mah, R.; et al. NP-MRD: The Natural Products Magnetic Resonance Database. Nucleic Acids Res. 2022, 50, D665–D677. [Google Scholar] [CrossRef]
- Wu, E.L.; Cheng, X.; Jo, S.; Rui, H.; Song, K.C.; Dávila-Contreras, E.M.; Qi, Y.; Lee, J.; Monje-Galvan, V.; Venable, R.M.; et al. CHARMM-GUI Membrane Builder toward Realistic Biological Membrane Simulations. J. Comput. Chem. 2014, 35, 1997–2004. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Nosé, S. A Molecular Dynamics Method for Simulations in the Canonical Ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical Dynamics: Equilibrium Phase-Space Distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Marrink, S.J.; Risselada, H.J.; Yefimov, S.; Tieleman, D.P.; de Vries, A.H. The MARTINI Force Field: Coarse Grained Model for Biomolecular Simulations. J. Phys. Chem. B 2007, 111, 7812–7824. [Google Scholar] [CrossRef] [PubMed]
- GROMACS Development Team. GROMACS 2022 Source Code. Available online: https://doi.org/10.5281/zenodo.6103835 (accessed on 30 January 2023).
- Carpenter, T.S.; López, C.A.; Neale, C.; Montour, C.; Ingólfsson, H.I.; Di Natale, F.; Lightstone, F.C.; Gnanakaran, S. Capturing Phase Behavior of Ternary Lipid Mixtures with a Refined Martini Coarse-Grained Force Field. J. Chem. Theory Comput. 2018, 14, 6050–6062. [Google Scholar] [CrossRef]
- Wassenaar, T.A.; Ingólfsson, H.I.; Böckmann, R.A.; Tieleman, D.P.; Marrink, S.J. Computational Lipidomics with Insane: A Versatile Tool for Generating Custom Membranes for Molecular Simulations. J. Chem. Theory Comput. 2015, 11, 2144–2155. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Huang, H.; Starodub, O.; McIntosh, A.; Kier, A.B.; Schroeder, F. Liver Fatty Acid-Binding Protein Targets Fatty Acids to the Nucleus. J. Biol. Chem. 2002, 277, 29139–29151. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Host Lipid | POPC | DPPC | ||
|---|---|---|---|---|
| Probe | c-PnA | t-PnA | c-PnA | t-PnA |
| a1 | 0.42 | 0.27 | 0.11 | 0.02 |
| Φ1/ns | 0.10 | 0.46 | 2.9 | 0.16 |
| a2 | 0.58 | 0.33 | 0.050 | 0.01 |
| Φ2/ns | 1.8 | 2.6 | 120 | 22 |
| a3 | - | 0.047 | - | 0.01 |
| Φ3/ns | - | 15 | - | 850 |
| a∞ | 0.032 | 0.26 | 0.71 | 0.92 |
| <Φ>/ns * | 1.1 | 1.7 | 5.2 | 2.3 |
| Lipid Phase | Fluid | Gel | ||
|---|---|---|---|---|
| Probe | c-PnA | t-PnA | c-PnA | t-PnA |
| Experimental | 9 × 105 a | 1.7 × 106 a | 5.3 × 105 b | 5 × 106 b |
| Estimate from simulation | 2.3 × 104 | 2.4 × 104 | 1.3 × 105 | 1.2 × 106 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oliveira, A.C.; Filipe, H.A.L.; Loura, L.M.S. Fluorescent Probes cis- and trans-Parinaric Acids in Fluid and Gel Lipid Bilayers: A Molecular Dynamics Study. Molecules 2023, 28, 2241. https://doi.org/10.3390/molecules28052241
Oliveira AC, Filipe HAL, Loura LMS. Fluorescent Probes cis- and trans-Parinaric Acids in Fluid and Gel Lipid Bilayers: A Molecular Dynamics Study. Molecules. 2023; 28(5):2241. https://doi.org/10.3390/molecules28052241
Chicago/Turabian StyleOliveira, Alexandre C., Hugo A. L. Filipe, and Luís M. S. Loura. 2023. "Fluorescent Probes cis- and trans-Parinaric Acids in Fluid and Gel Lipid Bilayers: A Molecular Dynamics Study" Molecules 28, no. 5: 2241. https://doi.org/10.3390/molecules28052241
APA StyleOliveira, A. C., Filipe, H. A. L., & Loura, L. M. S. (2023). Fluorescent Probes cis- and trans-Parinaric Acids in Fluid and Gel Lipid Bilayers: A Molecular Dynamics Study. Molecules, 28(5), 2241. https://doi.org/10.3390/molecules28052241