
Comparison of the Determination of Some Antihypertensive Drugs in Clinical Human Plasma Samples by Solvent Front Position Extraction and Precipitation Modes

 , , , and
, , , and
Abstract
1. Introduction
2. Results
2.1. Precipitation Procedure of Sample Preparation
2.2. SFPE Procedure of Sample Preparation
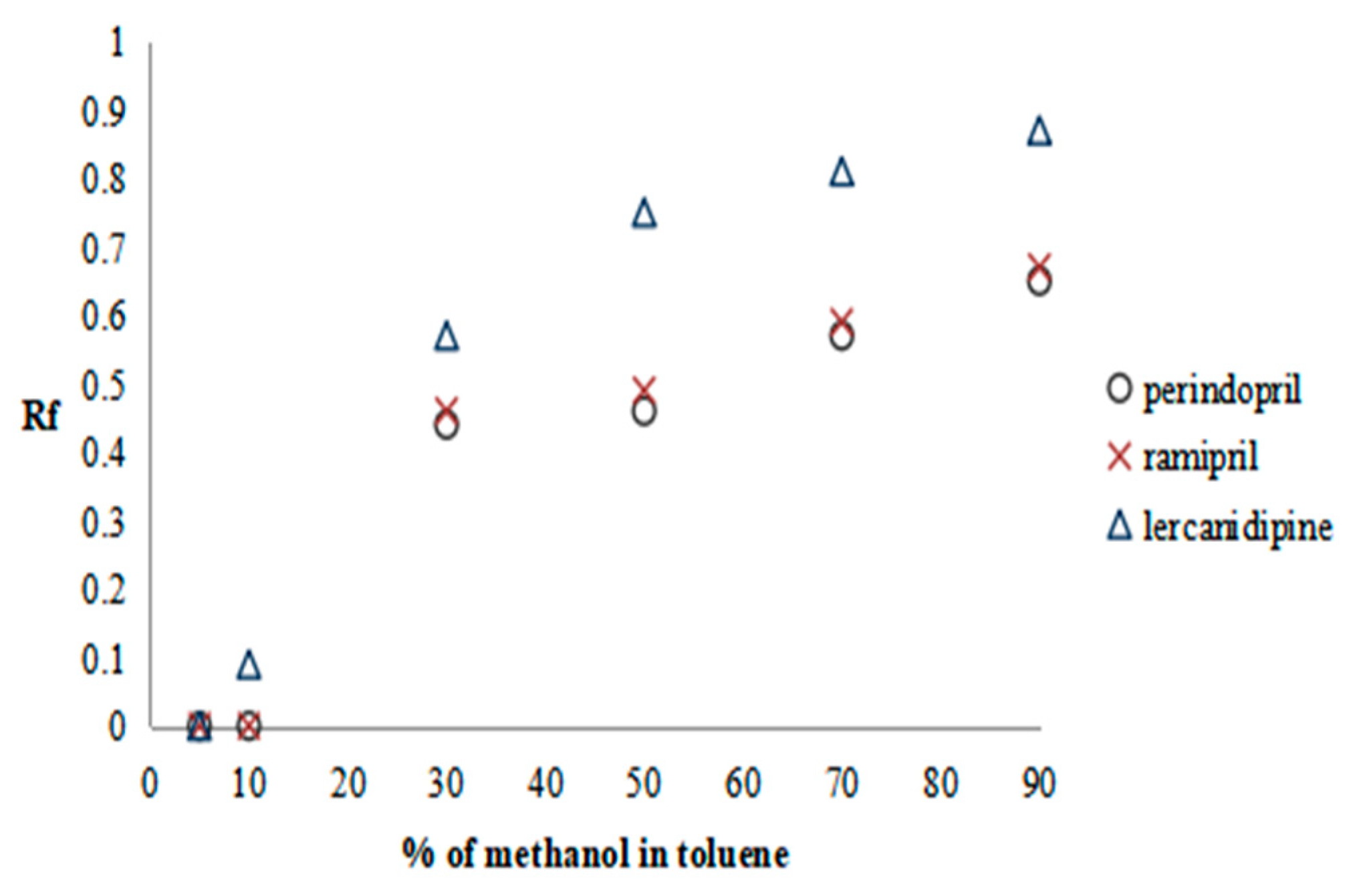
2.2.1. Preliminary Research—SFPE Procedure Optimization
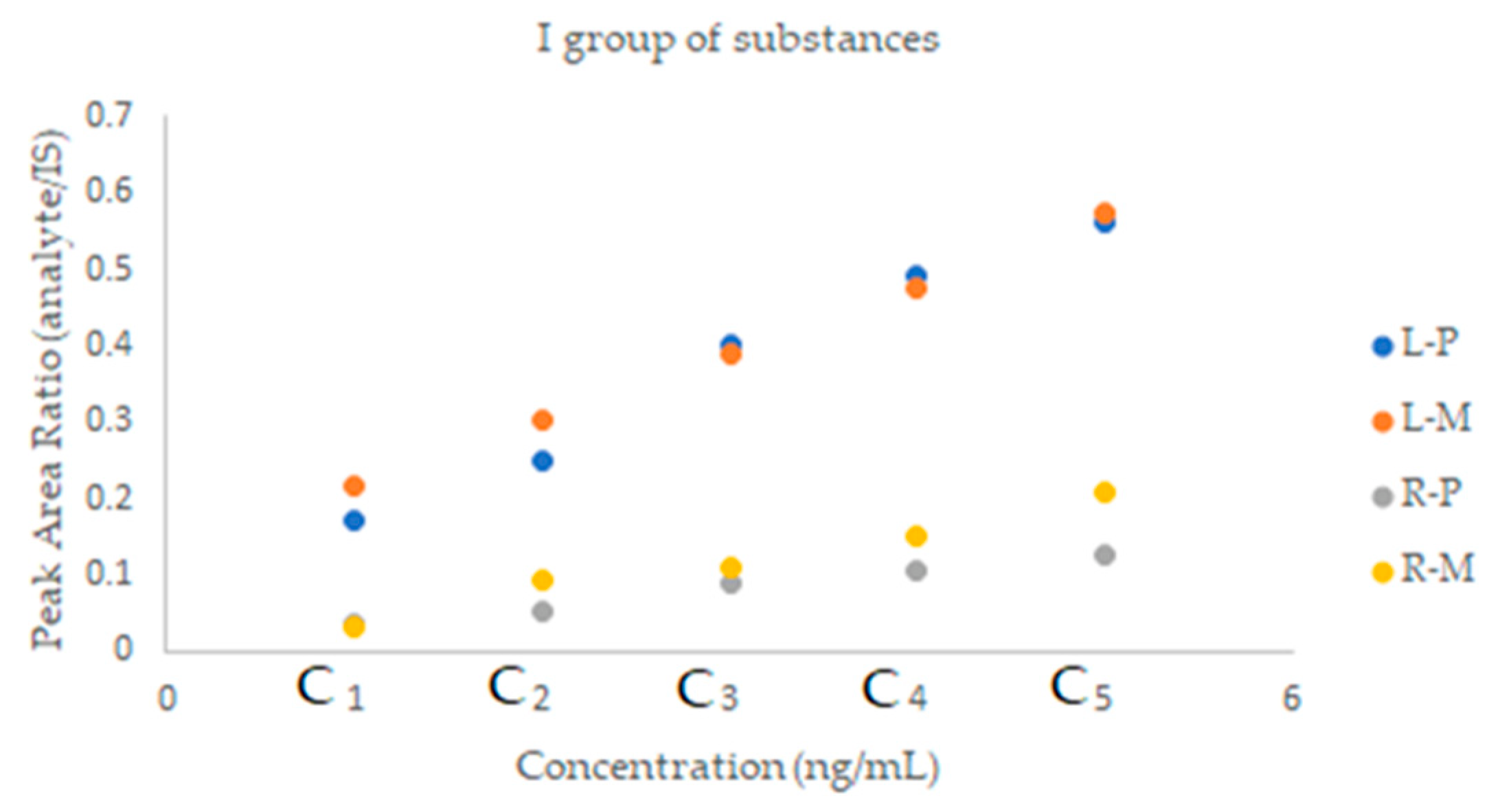
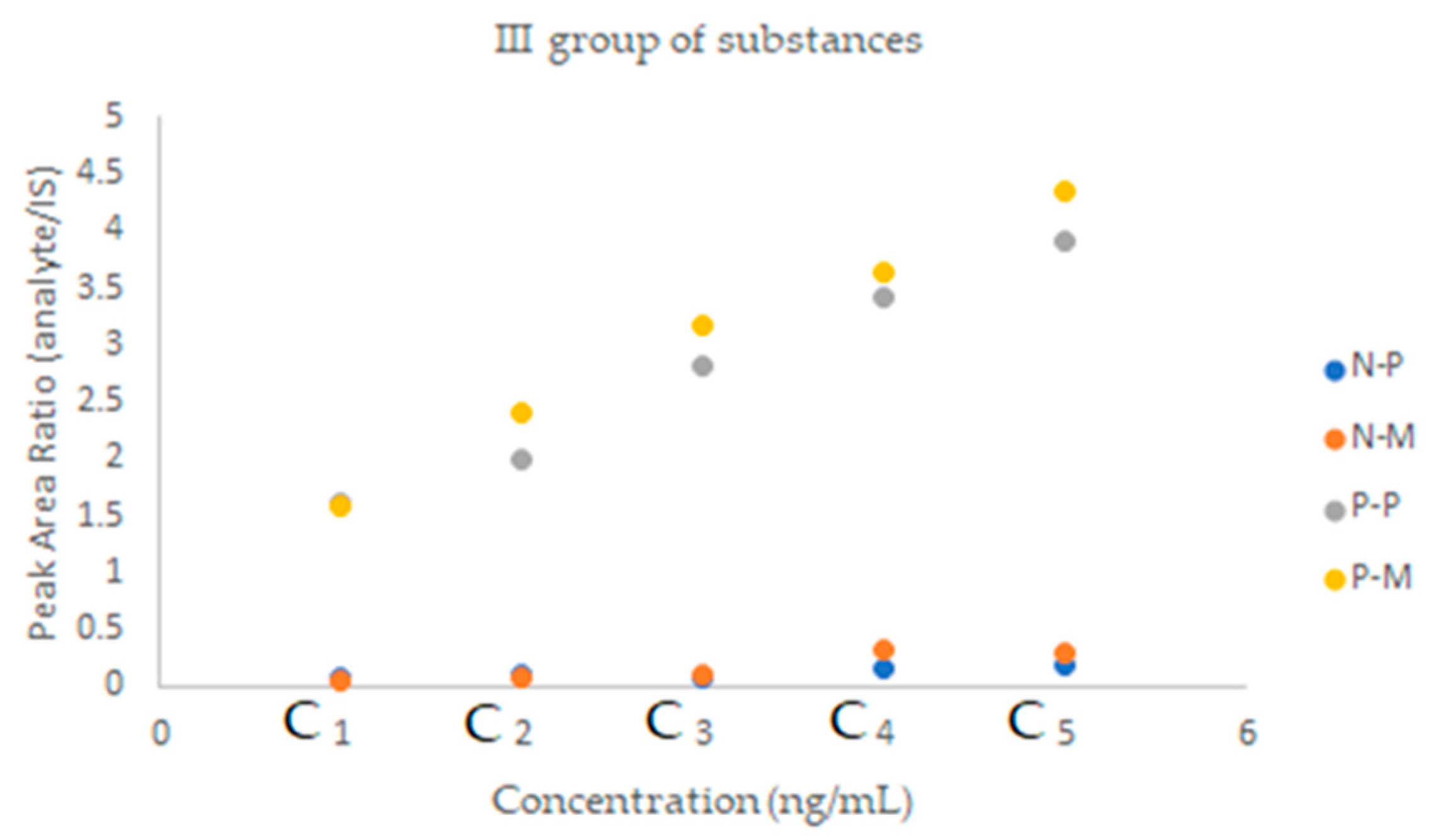
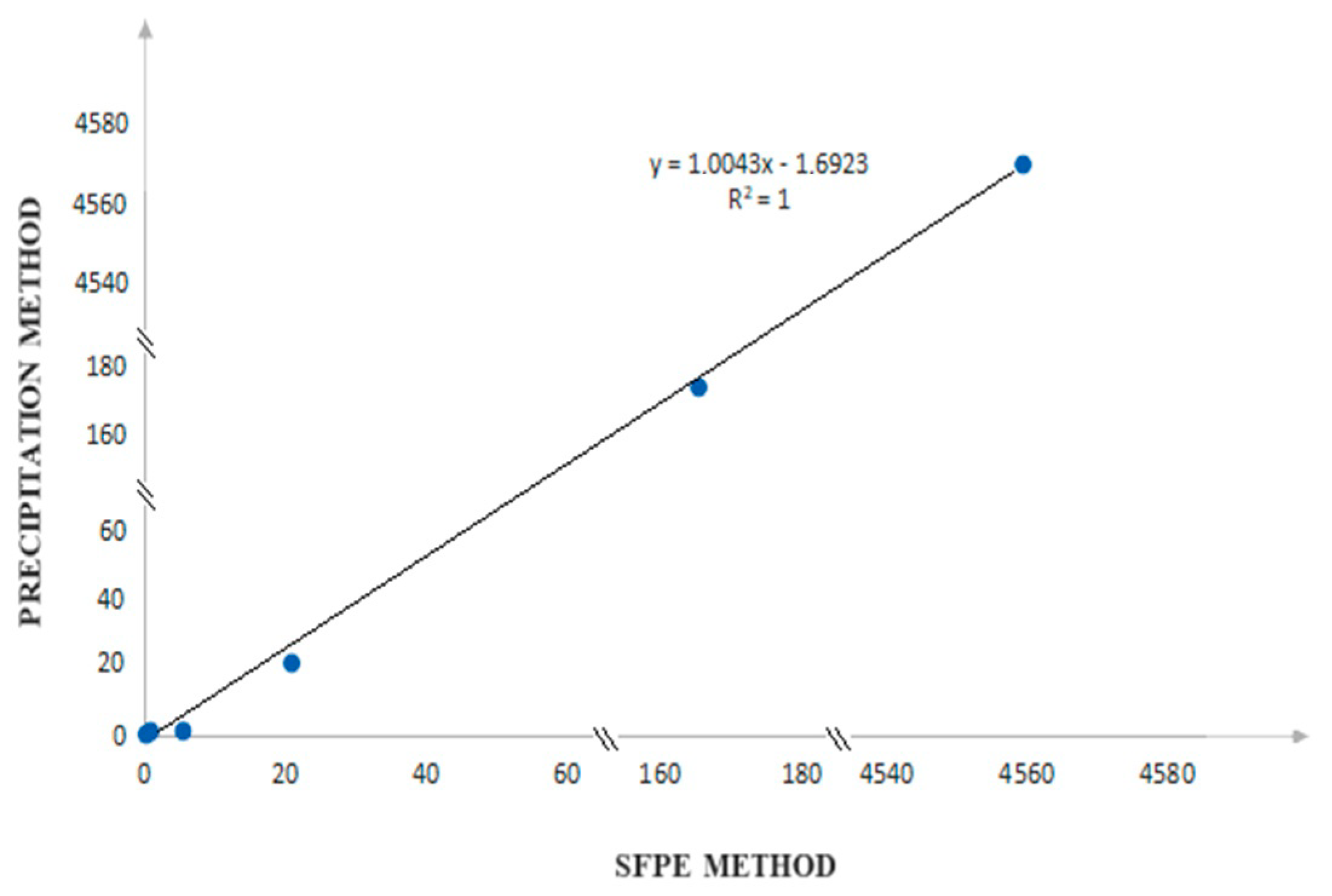
2.2.2. SFPE—Quantitative Analysis
3. Materials and Methods
3.1. Materials and Reagents
3.2. Devices and Instrumentation
3.3. Standard Solutions
3.4. Calibration Curve
3.5. Plasma Sample Preparation
3.5.1. Common Stage of Experiments
3.5.2. Precipitation Method
3.5.3. SFPE Procedure
3.6. LC-MS/MS Analysis Conditions
3.7. Ethics Approval and Consent to Participate
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Lopes, S.; Mesquita-Bastos, J.; Alves, A.J.; Ribeiro, F. Exercise as a tool for hypertension and resistant hypertension management: Current insights. Integr. Blood Press. Control. 2018, 11, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Fagard, R.H. Resistant hypertension. Heart 2012, 98, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Kjeldsen, S.E.; Os, I. Cost-effectiveness of therapeutic drug monitoring in patients with resistant hypertension and improving patients’ adherence. J. Hypertens. 2014, 32, 2357–2358. [Google Scholar] [CrossRef] [PubMed]
- Eskås, P.A.; Heimark, S.; Eek Mariampillai, J.; Larstorp, A.C.K.; Fadl Elmula, F.E.M.; Høieggen, A. Adherence to medication and drug monitoring in apparent treatment-resistant hypertension. Blood Press. 2016, 25, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Florczak, E.; Tokarczyk, B.; Warchoł-Celiñska, E.; Szwench-Pietrasz, E.; Prejbisz, A.; Gosk, M.; Kabat, M.; Narkiewicz, K.; Januszewicz, A.; Kała, M. Assessment of adherence to treatment in patients with resistant hypertension using toxicological serum analysis. Pol. Arch. Med. Wewn. 2015, 125, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Tomaszewski, M.; White, C.; Patel, P.; Masca, N.; Damani, R.; Hepworth, J.; Samani, N.J.; Gupta, P.; Madira, W.; Stanley, A.; et al. High rates of non-adherence to antihypertensive treatment revealed by high-performance liquid chromatography-tandem mass spectrometry (HP LC-MS/MS) urine analysis. Heart 2014, 100, 855–861. [Google Scholar] [CrossRef]
- Brinker, S.; Pandey, A.; Ayers, C.; Price, A.; Raheja, P.; Arbique, D.; Das, S.R.; Halm, E.A.; Kaplan, N.M.; Vongpatanasin, W. Therapeutic Drug Monitoring Facilitates Blood Pressure Control in Resistant Hypertension. J. Am. Coll. Cardiol. 2014, 63, 834–835. [Google Scholar] [CrossRef] [PubMed]
- Avataneo, V.; De Nicolò, A.; Rabbia, F.; Perlo, E.; Burrello, J.; Berra, E.; Pappaccogli, M.; Cusato, J.; D’Avolio, A.; Di Perri, G.; et al. Therapeutic drug monitoring-guided definition of adherence profiles in resistant hypertension and identification of predictors of poor adherence. Br. J. Clin. Pharmacol. 2018, 84, 2535–2543. [Google Scholar] [CrossRef]
- Varga, A.; Farczádi, L.; Vlase, L.; Primejdie, D.P.; Carasca, E.; Tilea, I. Liquid chromatography tandem mass spectrometry simultaneous determination of amlodipine and telmisartan in human plasma for therapeutic drug monitoring. Rev. De Chim. 2015, 66, 1675–1679. [Google Scholar]
- Johannsen, J.O.; Reuter, H.; Hoffmann, F.; Blaich, C.; Wiesen, M.H.J.; Streichert, T.; Müller, C. Reliable and easy-to-use LC–MS/MS-method for simultaneous determination of the antihypertensives metoprolol, amlodipine, canrenone and hydrochlorothiazide in patients with therapy-refractory arterial hypertension. J. Pharm. Biomed. Anal. 2019, 164, 373–381. [Google Scholar] [CrossRef]
- Kumar, A.; Dwivedi, S.P.; Prasad, T. Method Validation for Simultaneous Quantification of Olmesartan and Hydrochlorothiazide in Human Plasma Using LC-MS/MS and Its Application Through Bioequivalence Study in Healthy Volunteers. Front. Pharmacol. 2019, 10, 810. [Google Scholar] [CrossRef] [PubMed]
- Bylda, C.; Thiele, R.; Kobold, U.; Volmer, D.A. Recent advances in sample preparation techniques to overcome difficulties encountered during quantitative analysis of small molecules from biofluids using LC-MS/MS. Analyst 2014, 139, 2265–2276. [Google Scholar] [CrossRef] [PubMed]
- van der Nagel, B.C.H.; Versmissen, J.; Bahmany, S.; van Gelder, T.; Koch, B.C.P. High-throughput quantification of 8 antihypertensive drugs and active metabolites in human plasma using UPLC–MS/MS. J. Chromatogr. B 2017, 1060, 367–373. [Google Scholar] [CrossRef] [PubMed]
- De Nicolò, A.; Avataneo, V.; Rabbia, F.; Bonifacio, G.; Cusato, J.; Tomasello, C.; Perlo, E.; Mulatero, P.; Veglio, F.; Di Perri, G.; et al. UHPLC–MS/MS method with protein precipitation extraction for the simultaneous quantification of ten antihypertensive drugs in human plasma from resistant hypertensive patients. J. Pharm. Biomed. Anal. 2016, 129, 535–541. [Google Scholar] [CrossRef]
- Thorstensen, C.W.; Clasen, P.-E.; Rognstad, S.; Haldsrud, R.; Føreid, S.; Helstrøm, T.; Bergland, O.U.; Halvorsen, L.V.; Aune, A.; Olsen, E.; et al. Development of UHPLC-MS/MS methods to quantify 25 antihypertensive drugs in serum in a cohort of patients treated for hypertension. J. Pharm. Biomed. Anal. 2022, 219, 114908. [Google Scholar] [CrossRef]
- Klimek-Turek, A.; Chomicki, A.; Fornal, E.; Pradiuch, A.; Hys, M.; Dzido, T.H. Solvent front position extraction with semi-automatic device as a powerful sample preparation procedure to quantitation of tryptophan in human plasma. Sci. Rep. 2020, 10, 15063. [Google Scholar] [CrossRef]
- Jaglíńska, K.; Polak, B.; Klimek-Turek, A.; Błaszczyk, R.; Wysokíński, A.; Dzido, T.H. Preparation of Antihypertensive Drugs in Biological Matrix with Solvent Front Position Extraction for LC-MS/MS Analysis. Molecules 2022, 27, 205. [Google Scholar] [CrossRef]
- Klimek-Turek, A.; Sikora, E.; Dzido, T.H. Solvent front position extraction procedure with thin-layer chromatography as a mode of multicomponent sample preparation for quantitative analysis by instrumental technique. J. Chromatogr. A 2017, 1530, 204–210. [Google Scholar] [CrossRef]
- Klimek-Turek, A.; Sikora, M.; Rybicki, M.; Dzido, T.H. Frontally eluted components procedure with thin layer chromatography as a mode of sample preparation for high performance liquid chromatography quantitation of acetaminophen in biological matrix. J. Chromatogr. A 2016, 1436, 19–27. [Google Scholar] [CrossRef]
- Punt, A.M.; Stienstra, N.A.; van Kleef, M.E.A.; Lafeber, M.; Spiering, W.; Blankestijn, P.J.; Bots, M.L.; van Maarseveen, E.M. Screening of cardiovascular agents in plasma with LC-MS/MS: A valuable tool for objective drug adherence assessment. J. Chromatogr. B 2019, 1121, 103–110. [Google Scholar] [CrossRef]
- Sengupta, P.; Chatterjee, B.; Mandal, U.K.; Gorain, B.; Pal, T.K. Development and validation of a high throughput LC–MS/MS method for simultaneous quantitation of pioglitazone and telmisartan in rat plasma and its application to a pharmacokinetic study. J. Pharm. Anal. 2017, 7, 381–387. [Google Scholar] [CrossRef]
- Lwin, E.M.P.; Gerber, C.; Song, Y.; Leggett, C.; Ritchie, U.; Turner, S.; Garg, S. A new LC–MS/MS bioanalytical method for atenolol in human plasma and milk. Bioanalysis 2017, 9, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Chunduri, R.H.B.; Dannana, G.S. Development and validation of a reliable andrapid LC-MS/MS method for simultaneousquantification of sacubitril and valsartan in rat plasma and its application to apharmacokinetic study. Biomed. Chromatogr. 2016, 30, 1467–1475. [Google Scholar] [CrossRef] [PubMed]
- Elgawish, M.S.; Soltan, M.K.; Sebaiy, M.M. An LC–MS/MS spectrometry method for the simultaneous determination of rosuvastatin and irbesartan in rat plasma: Insight into pharmacokinetic and drug-drug interaction studies. J. Pharm. Biomed. Anal. 2019, 174, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Das, D.; Halder, D.; Maji, H.S.; De, P.K.; Pal, T.K. Special emphasis on bioanalytical method development and validation of an anti-hypertensive drug azelnidipine by lc-esi-ms/ms in healthy human volunteer’s blood plasma. Res. J. Pharm. Technol. 2021, 14, 3571–3577. [Google Scholar] [CrossRef]
- Caudron, E.; Laurent, S.; Billaud, E.M.; Prognon, P. Simultaneous determination of the acid/base antihypertensive drugs celiprolol, bisoprolol and irbesartan in human plasma by liquid chromatography. J. Chromatogr. B 2004, 801, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Shah, H.J.; Kundlik, M.L.; Patel, N.K.; Subbaiah, G.; Patel, D.M.; Suhagia, B.N.; Patel, C.N. Rapid determination of losartan and losartan acid in human plasma by multiplexed LC-MS/MS. J. Sep. Sci. 2009, 32, 3388–3394. [Google Scholar] [CrossRef]
- Lee, J.; Son, J.; Lee, M.; Lee, K.T.; Kim, D.H. Simultaneous quantitation of enalapril and enalaprilat in human plasma by 96-well solid-phase extraction and liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. RCM 2003, 17, 1157–1162. [Google Scholar] [CrossRef]
- Williams, B.; Mancia, G.; Spiering, W.; Rosei, E.A.; Azizi, M.; Burnier, M.; Clement, D.L.; Coca, A.; Simone, G.; Dominiczak, A.; et al. 2018 ESC/ESH Guidelines for the management of arterial hypertension. Kardiol. Pol. 2019, 77, 71–159. [Google Scholar] [CrossRef]
- Tykarski, A.; Filipiak, K.J.; Januszewicz, A.; Litwin, M.; Narkiewicz, K.; Prejbisz, A.; Ostalska-Nowicka, D.; Widecka, K.; Kostka-Jeziorny, K.; Adamczak, M.; et al. Zasady postępowania w nadciśnieniu tętniczym—2019 rok. Nadciśnienie Tętnicze W Prakt. 2019, 5, 99–151. [Google Scholar]
- Carr, G.P.; Wahlich, J.C. A practical approach to method validation in pharmaceutical analysis. J. Pharm. Biomed. Anal. S 1990, 8, 613–618. [Google Scholar] [CrossRef]
- Lawson, G.; Cocks, E.; Tanna, S. Bisoprolol, ramipril and simvastatin determination in dried blood spot samples using LC-HRMS for assessing medication adherence. J. Pharm. Biomed. Anal. 2013, 81–82, 99–107. [Google Scholar] [CrossRef]
- Beermann, B.; Groschinsky-Grind, M. Pharmacokinetics of hydrochlorothiazide in man. Eur. J. Clin. Pharmacol. 1977, 12, 297–303. [Google Scholar] [CrossRef]
- Ramakrishna, N.V.S.; Vishwottam, K.N.; Koteshwara, M.; Manoj, S.; Santosh, M.; Varma, D.P. Rapid quantification of nebivolol in human plasma by liquid chromatography coupled with electrospray ionization tandem mass spectrometry. J. Pharm. Biomed. Anal. 2005, 39, 1006–1013. [Google Scholar] [CrossRef]
- Barchielli, M.; Dolfini, E.; Farina, P.; Leoni, B.; Targa, G.; Vinaccia, V.; Tajana, A. Clinical Pharmacokinetics of Lercanidipine. J. Cardiovasc. Pharmacol. 1997, 29, 1–15. [Google Scholar] [CrossRef]
- Prasad, P.P.; Yeh, C.M.; Gurrieri, P.; Glazer, R.J. McLeod, Pharmacokinetics of multiple doses of valsartan in patients with heart failure. J. Cardiovasc. Pharmacol. 2002, 40, 801–807. [Google Scholar] [CrossRef]
- Stangier, J.; Su, C.; Roth, W. Pharmacokinetics of orally and intravenously administered telmisartan in healthy young and elderly volunteers and in hypertensive patients. J. Int. Med. Res. 2000, 28, 149–167. [Google Scholar] [CrossRef]
- Macfadyen, R.J.; Lees, K.R.; Reid, J.L. A review of its pharmacokinetics and clinical pharmacology. Drugs 1990, 39, 49–63. [Google Scholar] [CrossRef]
- PubChem, National Center for Biotechnology Information. Available online: https://pubchem.ncbi.nlm.nih.gov (accessed on 14 February 2022).
- DrugBank, Database for Drug and Drug Target Info. Available online: https://go.drugbank.com (accessed on 14 February 2022).
- Wawrzyniak, R.; Kosnowska, A.; Macioszek, S.; Bartoszewski, R.; Markuszewski, M.J. New plasma preparation approach to enrich metabolome coverage in untargeted metabolomics: Plasma protein bound hydrophobic metabolite release with proteinase K. Sci. Rep. 2018, 8, 9541. [Google Scholar] [CrossRef]
- Sitnikov, D.G.; Monnin, C.S.; Vuckovic, D. Systematic assessment of seven solvent and solid-phase extraction methods for metabolomics analysis of human plasma by LC-MS. Sci. Rep. 2016, 6, 38885. [Google Scholar] [CrossRef]
- Bruce, S.J.; Tavazzi, I.; Parisod, V.; Rezzi, S.; Kochhar, S.; Guy, P.A. Investigation of human blood plasma sample preparation for performing metabolomics using ultrahigh Performance Liquid Chromatography/Mass Spectrometry. Anal. Chem. 2009, 81, 3285–3296. [Google Scholar] [CrossRef]
- Kim, S.B.; Palmer, J.C.; Debenedetti, P.G. Computational investigation of cold denaturation in the Trp-cage miniprotein. PNAS 2016, 113, 8991–8996. [Google Scholar] [CrossRef]
- Davidovic, M.; Mattea, C.; Qvist, J.; Halle, B. Protein cold denaturation as seen from the solvent. J. Am. Chem. Soc. 2009, 131, 1025–1106. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substance of Interest | Linearity | R2 |
|---|---|---|
| Lercanidipine | y = 1.111x − 0.004 | 0.999 |
| Ramipril | y = 1.226x + 0.022 | 0.989 |
| Hydrochlorothiazide | y = 0.01x + 2.821 | 0.998 |
| Valsartan | y = 0.833x + 0.907 | 0.998 |
| Nebivolol | y = 0.169x − 0.002 | 0.997 |
| Perindopril | y = 0.840x − 0.018 | 0.998 |
| I Group of Substances * | ||||
|---|---|---|---|---|
| Concentration (ng/mL) | %RSD | LOD (ng/mL) | LOQ (ng/mL) | |
| Lercanidipine | 0.16 | 0.44 | 0.02 | 0.05 |
| Ramipril | 1.11 | 6.48 | 0.32 | 0.97 |
| II Group of Substances ** | ||||
| Hydrochlorothiazide | 165.56 | 2.05 | 0.63 | 1.90 |
| Valsartan | 4577.62 | 0.52 | 4.42 | 13.39 |
| III Group of Substances *** | ||||
| Nebivolol | 1.00 | 4.84 | 0.12 | 0.36 |
| Perindopril | 19.49 | 2.50 | 0.47 | 1.44 |
| Substance of Interest | Linearity | R2 |
|---|---|---|
| Lercanidipine | y = 1.286x − 0.008 | 0.981 |
| Ramipril | y = 1.109x − 0.012 | 0.998 |
| Hydrochlorothiazide | y = 0.063x + 0.014 | 0.984 |
| Valsartan | y = 3.275x − 2.508 | 0.994 |
| Nebivolol | y = 0.241x − 0.002 | 0.993 |
| Perindopril | y = 1.543x − 0.673 | 0.981 |
| I Group of Substances * | |||||||
|---|---|---|---|---|---|---|---|
| Conc. (ng/mL) | %RSD | LOD (ng/mL) | LOQ (ng/mL) | %Recovery | %CV Intra-day | %CV Inter-day | |
| Lercanidipine | 0.19 | 5.41 | 0.06 | 0.17 | 98.44–105.01 | 6.61–1.49 | 5.22–1.21 |
| Ramipril | 5.43 | 0.92 | 0.16 | 0.47 | 117.63–84.36 | 6.04–2.61 | 5.30–2.11 |
| II Group of Substances ** | |||||||
| Hydrochloro thiazide | 166.41 | 5.84 | 1.46 | 4.41 | 88.90–79.88 | 7.45–4.58 | 9.74–7.97 |
| Valsartan | 4559.76 | 4.97 | 9.78 | 29.64 | 107.20–115.40 | 5.04–1.87 | 5.60–2.40 |
| III Group of Substances *** | |||||||
| Nebivolol | 0.83 | 4.75 | 0.23 | 0.70 | 120.36–80.92 | 7.70–1.85 | 6.44–3.13 |
| Perindopril | 22.10 | 1.73 | 1.82 | 5.52 | 108.26–85.42 | 4.14–1.34 | 3.64–1.10 |
| Name of the Substance | Formula | Value pKa * | Concentration Ranges Therapeutic (ng/mL) | Drug Classes |
|---|---|---|---|---|
| lercanidipine |  | 9.36 (ionogenic basic group) | 1.23–10.23 | calcium channel blockers (CCB) |
| ramipril |  | 3.75 (ionogenic acid group) 5.2 (ionogenic basic group) | 11–35 | angiotensin converting enzyme (ACE) inhibitors |
| hydrochlorothiazide |  | 9.09 (ionogenic acid group) | 70–376 | thiazide diuretic |
| valsartan |  | 4.35 (ionogenic acid group) | 1940–6400 | angiotensin II receptor blockers (ARB) |
| telmisartan |  | 3.62 (ionogenic acid group) 5.86 (ionogenic basic group) | 8.9–366 | angiotensin II receptor blockers (ARB) |
| nebivolol |  | 8.9 (ionogenic basic group) | 1.48–26.1 | β-blockers |
| perindopril |  | 3.79 (ionogenic acid group) 5.48 (ionogenic basic group) | 20 – 83 | angiotensin converting enzyme (ACE) inhibitors |
| Calibration Level | |||||
|---|---|---|---|---|---|
| Substance | 1 | 2 | 3 | 4 | 5 |
| Group I | |||||
| ramipril | 10 | 20 | 30 | 40 | 50 |
| lercanidipine | 1 | 2 | 5 | 7.5 | 10 |
| perindopril (IS) * | 40 | 40 | 40 | 40 | 40 |
| Group II | |||||
| valsartan | 1500 | 3000 | 4500 | 5500 | 6500 |
| hydrochlorothiazide | 60 | 140 | 220 | 300 | 380 |
| telmisartan (IS) ** | 200 | 200 | 200 | 200 | 200 |
| Group III | |||||
| perindopril | 15 | 30 | 50 | 70 | 90 |
| nebivolol | 1 | 5 | 10 | 20 | 30 |
| ramipril (IS) *** | 20 | 20 | 20 | 20 | 20 |
| Substance | Prec. Ion (m/z) | Prod. Ion (m/z) | Frag. (V) | CE (eV) | Polarity | Ret. Time (min) |
|---|---|---|---|---|---|---|
| Lercanidipine | 612.3 | 280.1/100 | 145 | 26/40 | Positive | 4.94 |
| Ramipril | 417.2 | 253.8/234 | 128 | 26/22 | Positive | 3.75 */6.03 ** |
| Perindopril | 369.2 | 172/98 | 103 | 22/35 | Positive | 3.33 */5.19 ** |
| Hydrochlorothiazide | 298 | 268.8/204.9 | 96 | 10/18 | Positive | 2.1 |
| Valsartan | 436.2 | 235/207 | 83 | 18/30 | Positive | 5.39 |
| Telmisartan | 515.2 | 497/276 | 155 | 35 | Positive | 3.98 |
| Nebivolol | 406.2 | 150.9/123 | 112 | 35 | Positive | 6.44 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jaglińska, K.; Polak, B.; Klimek-Turek, A.; Fornal, E.; Stachniuk, A.; Trzpil, A.; Błaszczyk, R.; Wysokiński, A. Comparison of the Determination of Some Antihypertensive Drugs in Clinical Human Plasma Samples by Solvent Front Position Extraction and Precipitation Modes. Molecules 2023, 28, 2213. https://doi.org/10.3390/molecules28052213
Jaglińska K, Polak B, Klimek-Turek A, Fornal E, Stachniuk A, Trzpil A, Błaszczyk R, Wysokiński A. Comparison of the Determination of Some Antihypertensive Drugs in Clinical Human Plasma Samples by Solvent Front Position Extraction and Precipitation Modes. Molecules. 2023; 28(5):2213. https://doi.org/10.3390/molecules28052213
Chicago/Turabian StyleJaglińska, Kamila, Beata Polak, Anna Klimek-Turek, Emilia Fornal, Anna Stachniuk, Alicja Trzpil, Robert Błaszczyk, and Andrzej Wysokiński. 2023. "Comparison of the Determination of Some Antihypertensive Drugs in Clinical Human Plasma Samples by Solvent Front Position Extraction and Precipitation Modes" Molecules 28, no. 5: 2213. https://doi.org/10.3390/molecules28052213
APA StyleJaglińska, K., Polak, B., Klimek-Turek, A., Fornal, E., Stachniuk, A., Trzpil, A., Błaszczyk, R., & Wysokiński, A. (2023). Comparison of the Determination of Some Antihypertensive Drugs in Clinical Human Plasma Samples by Solvent Front Position Extraction and Precipitation Modes. Molecules, 28(5), 2213. https://doi.org/10.3390/molecules28052213