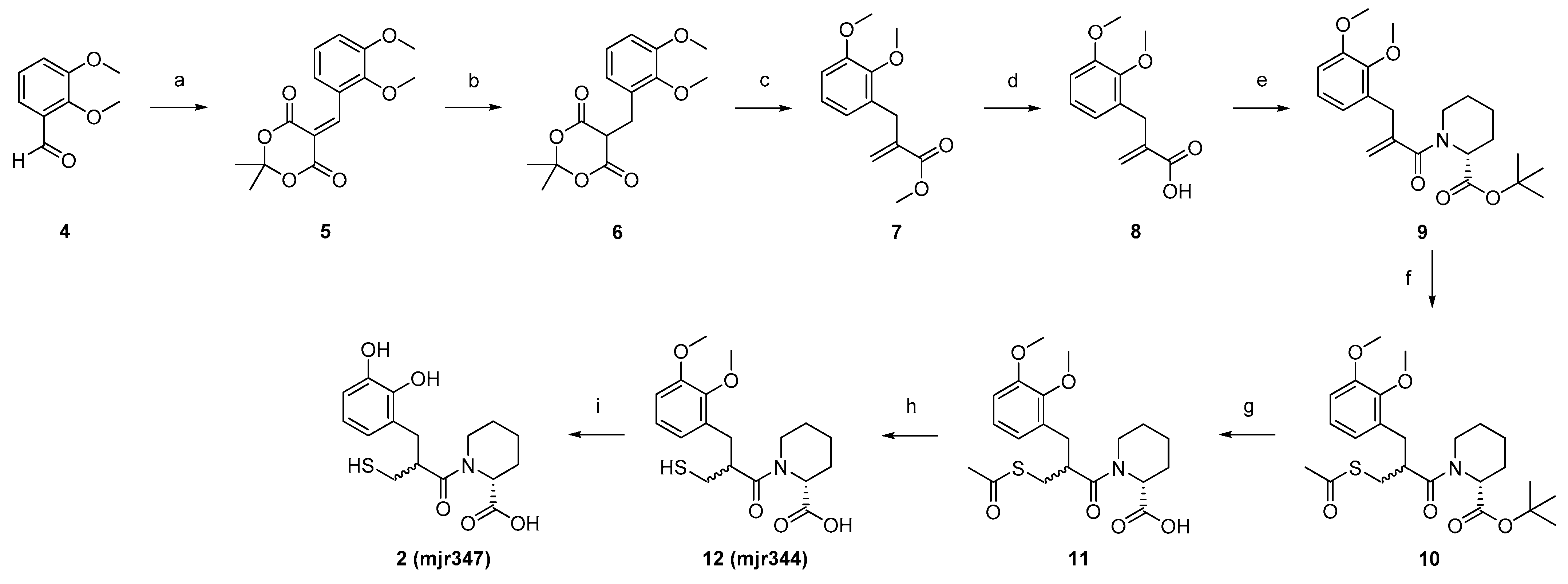
4.2. Synthesis
5-(2,3-Dimethoxybenzylidene)-2,2-dimethyl-1,3-dioxane-4,6-dione (
5). 2,3-Dimethoxybenzaldehyde (
4, 5.1 g, 29.9 mmol, 0.9 eq) and AcOH (0.2 mL, 3.3 mmol, 0.1 eq) were added to 2-propanol (210 mL). Meldrum acid (4.9 g, 33.2 mmol, 1.0 eq) was added portion-wise. The reaction was stirred at 75 °C for 3 h. After cooling to room temperature, the solvent was removed in vacuo. Purification was carried out using flash chromatography, with silica gel as stationary phase and Hexane/EtOAc (gradient 100/0 to 0/100) as mobile phase. Product
5 was obtained as yellow oil (7.3 g, 66%).
1H NMR (400 MHz, DMSO-
d6): δ = 7.32 (d,
J = 8.1 Hz, 1H), 7.25 (d,
J = 8.1 Hz, 1H), 7.13 (t,
J = 8.0 Hz, 1H), 3.85 (s, 3H), 3.79 (s, 3H), 3.31 (s, 1H), 1.77 (s, 6H) ppm. MS (ESI, 70 eV)
m/z (%): 293.85 (100) (M + H).
1H NMR spectra of synthetized compounds can be found in the
Supplementary Materials.
5-(2,3-Dimethoxybenzyl)-2,2-dimethyl-1,3-dioxane-4,6-dione (6). Dione (5, 7.2 g, 17.2 mmol, 1.0 eq) was solved into DCM (200 mL) at 0 °C. AcOH (31 mL, 533 mmol, 31 eq) was added and the mixture was stirred at 0 °C for 15 min. NaBH4 (5.3 g, 0.1 mol, 8.0 eq) was added in portions at 0 °C and the reaction mixture was stirred at room temperature for one hour. After the decolourisation of the reaction mixture, H2O (200 mL) was added and extracted with DCM (3 × 200 mL). The combined organic phases were washed with H2O (200 mL), saturated with NaCl solution (200 mL), and dried over MgSO4. The solvent was removed to obtain the crude product, which was purified by flash chromatography (silica gel as stationary phase, gradient of DCM/MeOH 100/0 to 90/10 as mobile phase) to obtain the product 6 as a colourless solid (3.0 g, 53%). 1H NMR (400 MHz, DMSO-d6): δ = 6.96 (t, J = 7.9 Hz, 1H), 6.89 (d, J = 6.7 Hz, 1H), 6.75 (d, J = 7.0 Hz, 1H), 4.64 (t, J = 5.4 Hz, 1H), 3.78 (s, 3H), 3.72 (s, 3H), 3.29 (d, J = 5.4 Hz, 2H), 1.82 (s, 3H), 1.68 (s, 3H) ppm. MS (ESI, 70 eV) m/z (%): 294.90 (100) (M + H).
Methyl 2-(2,3-dimethoxybenzyl)acrylate (7). Dione (6, 3.0 g, 10.1 mmol, 1.0 eq) and Eschenmoser’s salt (6.7 mg, 35.2 mmol, 3.5 eq) were dissolved in MeOH (160 mL). The reaction mixture was stirred at 70 °C for 18 h. After cooling to room temperature, the solvent was removed in vacuo and the residue was taken up in DCM. The organic phase was washed with saturated NaHCO3 solution (2 × 80 mL) and 10% NaHSO4 solution (2 × 80 mL), and the aqueous phase was extracted with DCM (2 × 80 mL). The combined organic phases were washed with saturated NaCl solution (80 mL) and dried over MgSO4. After removal of the solvent in vacuo, the crude product was purified by flash chromatography (gradient hexane/EtOAc 96/4 to 0/100) to isolate the product 7 as a colourless oil (1.7 g, 67%). 1H NMR (400 MHz, DMSO-d6) δ = 7.01–6.96 (m, 1H), 6.92 (dd, J = 8.2, 1.7 Hz, 1H), 6.69 (dd, J = 7.6, 1.6 Hz, 1H), 6.14 (q, J = 1.1 Hz, 1H), 5.41 (q, J = 1.5 Hz, 1H), 3.79 (s, 3H), 3.68 (s, 3H), 3.67 (s, 3H), 3.56 (t, J = 1.2 Hz, 2H) ppm. MS (ESI, 70 eV) m/z (%): 237.15 (100) (M + H).
2-(2,3-Dimethoxybenzyl)acrylic acid (8). Acrylate (7, 1.7 g, 6.4 mmol, 1.0 eq) was dissolved in NaOH (1 M, 20 mL) and stirred under microwave irradiation at 100 °C for 1 h. The reaction mixture was washed with diethyl ether (2 × 20 mL) and the aqueous phase was adjusted to pH 1 with HCl (2 M, 13 mL). Subsequently, the reaction mixture was extracted with diethyl ether (3 × 20 mL) and the combined organic phases were dried over MgSO4 and in vacuo. The resulting crude product of 8 was obtained as a colourless oil (0.8 mg, 54%) and used without further purification. 1H NMR (400 MHz, DMSO-d6): δ = 12.49 (s, 1H), 7.01–6.96 (m, 1H), 6.91 (dd, J = 8.2, 1.6 Hz, 1H), 6.70 (dd, J = 7.6, 1.6 Hz, 1H), 6.13–6.07 (m, 1H), 5.34 (q, J = 1.6 Hz, 1H), 3.79 (s, 3H), 3.68 (s, 3H), 3.53 (s, 2H) ppm. MS (ESI, 70 eV) m/z (%): 220.75 (100) (M–H).
tert-Butyl (R)-piperidine-2-carboxylate (19). D-Pipecolic acid (2.0 g, 15.0 mmol, 1.0 eq) was suspended in tert-butyl acetate (0.1 L, 0.7 mol, 47.0 eq). Under ice bath cooling, 60% perchloric acid (2.3 mL, 24.0 mmol, 1.6 eq) was added dropwise and the reaction mixture was stirred overnight at room temperature. The reaction mixture was extracted with water (3 × 90 mL) and saturated NaHCO3 solution was added to the combined aqueous phases until gas development stopped. The aqueous phase was extracted with EtOAc (5 × 45 mL), the combined organic phases were washed with NaCl solution, dried over Na2SO4, and the solvent was removed in vacuo. Product 19 was obtained as a yellowish solid (1.0 g, 48%) and used without further purification. 1H NMR (400 MHz, DMSO-d6): δ = 3.24 (dd, J = 9.7, 3.4 Hz, 1H), 2.95 (dtd, J = 12.0, 3.8, 1.3 Hz, 1H), 2.58–2.53 (m, 1H), 2.53–2.51 (m, 1H), 1.82–1.75 (m, 1H), 1.70–1.64 (m, 1H), 1.50–1.43 (m, 1H), 1.41 (s, 9H), 1.40–1.28 (m, 3H) ppm. MS (ESI, 70 eV) m/z (%): 185.90 (100) (M + H).
tert-Butyl (R)-1-(2-(2,3-dimethoxybenzyl)acryloyl)piperidine-2-carboxylate (9). SOCl2 (0.6 mL, 8.3 mmol, 2.5 eq) was solved in DCM (30 mL), together with a drop of DMF. Acrylic acid (8, 0.8 g, 3.3 mmol, 1.0 eq) was added and the resulting mixture was stirred for 3 h at 50 °C. After the removal of the excess SOCl2, the resulting acid chloride was suspended in 1,2-dichloroethane (19 mL) and protected D-pipecolic acid (19, 0.8 mg, 3.6 mmol, 1.1 eq). DIPEA (1.7 mL, 9.9 mmol, 3.0 eq) was added slowly to the suspension. The reaction was stirred under microwave irradiation for 1 h at 90 °C. The mixture was diluted with water (40 mL), the aqueous phase was acidified with HCl (2 M), and extracted with EtOAc (3 × 20 mL). The combined organic phases were washed with HCl (2 M, 3 × 20 mL) and saturated NaCl solution (17 mL), and dried over MgSO4. After the removal of all solvents in vacuo, the crude product was obtained. Purification was performed using flash chromatography with silica gel as stationary phase and a mixture of hexane/EtOAc (gradient 100/0 to 0/100) as mobile phase, to isolate product 9 as a yellow oil (1.1 g, 63%). 1H NMR (400 MHz, DMSO-d6) δ = 7.01 (t, J = 7.8 Hz, 1H), 6.94 (d, J = 8.4 Hz, 1H), 6.74 (t, J = 7.8 Hz, 1H), 5.05–4.80 (m, 3H), 3.79 (s, 3H), 3.70 (s, 3H), 3.51–3.43 (m, 2H), 3.03 (t, J = 13.0 Hz, 1H), 2.61 (t, J = 12.5 Hz, 1H), 2.02 (dd, J = 54.8, 13.2 Hz, 2H), 1.64–1.56 (m, 1H), 1.41 (s, 9H), 1.27–0.95 (m, 3H) ppm. MS (ESI, 70 eV) m z (%): 390.05 (100) (M + H).
tert-Butyl (2R)-1-(3-(acetylthio)-2-(2,3-dimethoxybenzyl)propanoyl)piperidine-2-carboxylate (10). Carboxylate (9, 1.0 g, 2.7 mmol, 1.0 eq) was dissolved in thioacetic acid (40 mL, 535 mmol, 200 eq) and the mixture was stirred at room temperature for 70 h. The mixture was then diluted with water (70 mL) and extracted with EtOAc (3 × 50 mL). The combined organic phases were washed with saturated NaCl solution (60 mL) and dried over MgSO4. The solvents were removed in vacuo and the crude product was purified using flash chromatography with silica gel as stationary phase and a mixture of hexane/EtOAC (gradient) as mobile phase, to isolate product 10 as a dark red oil (1.0 g, 72%). 1H NMR (400 MHz, DMSO-d6): δ = 6.98–6.91 (m, 2H), 6.69 (td, J = 6.7, 2.4 Hz, 1H), 5.10–4.98 (m, 1H), 3.79 (s, 3H), 3.74 (s, 3H), 3.27–3.08 (m, 2H), 3.02–2.87 (m, 2H), 2.74 (d, J = 7.4 Hz, 1H), 2.27 (s, 3H), 2.01 (s, 2H), 1.72–1.62 (m, 2H), 1.55–1.45 (m, 2H), 1.41 (s, 9H), 1.32–1.22 (m, 2H) ppm. MS (ESI, 70 eV) m/z (%): 466.40 (100) (M + H).
(2R)-1-(3-(acetylthio)-2-(2,3-dimethoxybenzyl)propanoyl)piperidine-2-carboxylic acid (11). Carboxylate (10, 1.0 g, 2.1 mmol, 1.0 eq) was dissolved in DCM (35 mL), and TFA (8.8 mL, 0.1 mol, 57.0 eq) was slowly added by stirring. The reaction mixture was stirred overnight at 50 °C and the solvent was removed under reduced pressure afterwards. After co-evaporation with toluene, 11 was obtained as a crude product. After washing the crude product with hexane (8 × 15 mL), 11 was obtained as a red, highly viscous oil (0.9 g, 83%). 1H NMR (400 MHz, DMSO-d6): δ = 12.73 (s, 1H), 7.00–6.90 (m, 2H), 6.81–6.65 (m, 1H), 5.10 (dd, J = 25.7, 5.6 Hz, 1H), 3.79 (d, J = 4.4 Hz, 3H), 3.76–3.72 (m, 3H), 3.31–3.05 (m, 2H), 2.97–2.83 (m, 2H), 2.74 (d, J = 7.0 Hz, 1H), 2.31–2.21 (m, 3H), 2.07 (dd, J = 42.8, 13.9 Hz, 2H), 1.76–1.61 (m, 2H), 1.57–1.43 (m, 2H), 1.39–1.13 (m, 2H) ppm. MS (ESI, 70 eV) m/z (%): 409.90 (100) (M + H).
(2R)-1-(2-(2,3-dimethoxybenzyl)-3-mercaptopropanoyl)piperidine-2-carboxylic acid (12, mjr344). Acid (11, 0.9 mg, 1.7 mmol, 1.0 eq) was added to water (40 mL) and degassed under argon in an ultrasonic bath for 30 min. The solution was cooled to 0 °C and NH3 solution (25% in water, 6.5 mL, 45.0 eq) was added. The mixture was stirred for 2 h at room temperature. The reaction was terminated by adding HCl (2 M) and adjusted to pH < 3, extracted with EtOAc (3 × 20 mL), and the combined organic phases were washed with saturated NaCl solution (20 mL) and dried over MgSO4. All volatile components were removed in vacuo to obtain 15 as crude product. Purification was carried out using preparative HPLC (C18 column, mobile phase: Iso 42% ACN) to isolate product 12 as a colourless solid (0.2 g, 34%). 1H NMR (500 MHz, DMSO-d6): δ = 12.80 (s, 1H), 7.00–6.88 (m, 2H), 6.79–6.68 (m, 1H), 5.14 (ddd, J = 35.7, 6.1, 2.2 Hz, 1H), 4.04 (d, J = 13.5 Hz, 1H), 3.79 (d, J = 3.2 Hz, 3H), 3.76–3.73 (m, 3H), 3.30–3.15 (m, 2H), 3.14–3.02 (m, 2H), 2.85 (dd, J = 13.6, 4.9 Hz, 1H), 2.12 (dd, J = 10.1, 6.9 Hz, 2H), 1.71–1.61 (m, 2H), 1.53–1.45 (m, 2H), 1.32–1.23 (m, 2H) ppm. 13C NMR (126 MHz, DMSO): δ = 172.66, 172.45, 146.83, 146.71, 132.01, 123.69, 122.54, 111.55, 60.07, 55.67, 51.38, 44.93, 42.90, 32.45, 26.54, 25.18, 20.78, 20.57 ppm. MS (ESI, 70 eV) m/z (%): 367.95 (100) (M + H). HRMS calculated m/z 368.15262 andfound m/z 368.15317.
(2R)-1-(2-(2,3-dihydroxybenzyl)-3-mercaptopropanoyl)piperidine-2-carboxylic acid (2, mjr347). Acid (12, 0.16 mg, 0.42 mmol, 1.00 eq) was dissolved in DCM (3 mL) at −78 °C. BBr3 (1 M in DCM, 2.09 mL, 2.09 mmol, 5.00 eq) was added, stirred for 2 h at −78 °C and then overnight at room temperature. The resulting mixture was cooled to 0 °C and water was added dropwise until the remaining BBr3 was consumed (no more smoke was produced). The aqueous phase was extracted with DCM (3 × 6 mL), the organic phases were combined and all volatile components were removed under reduced pressure to obtain the crude product. The purification was done using preparative HPLC (C18 column, mobile phase: Iso 34% ACN) to isolate product 2/mjr347 as a colourless glass (0.04 g, 28%). 1H NMR (500 MHz, DMSO-d6): δ = 12.72 (s, 1H), 9.22 (d, J = 11.8 Hz, 1H), 8.43–8.22 (m, 1H), 6.68–6.62 (m, 1H), 6.55–6.51 (m, 1H), 6.50 (d, J = 4.4 Hz, 1H), 5.14 (ddd, J = 37.3, 6.0, 2.3 Hz, 1H), 4.11 (d, J = 13.7 Hz, 1H), 3.29–3.11 (m, 2H), 3.03 (td, J = 13.1, 3.0 Hz, 2H), 2.87–2.71 (m, 1H), 2.34–2.10 (m,2H), 1.72–1.59 (m, 2H), 1.58–1.38 (m, 2H), 1.35–1.07 (m, 2H) ppm. 13C NMR (126 MHz, DMSO): δ = 172.97, 172.42, 145.03, 143.33, 125.56, 121.43, 118.56, 113.75, 51.33, 44.56, 42.82, 34.29, 26.57, 25.21, 20.86, 20.54 ppm. MS (ESI, 70 eV) m/z (%): 339.95 (100) (M + H).HRMS calculated m/z 362.10326 and found m/z 362.10352. HPLC: tR: 11.56 min (method 1), purity >95%.
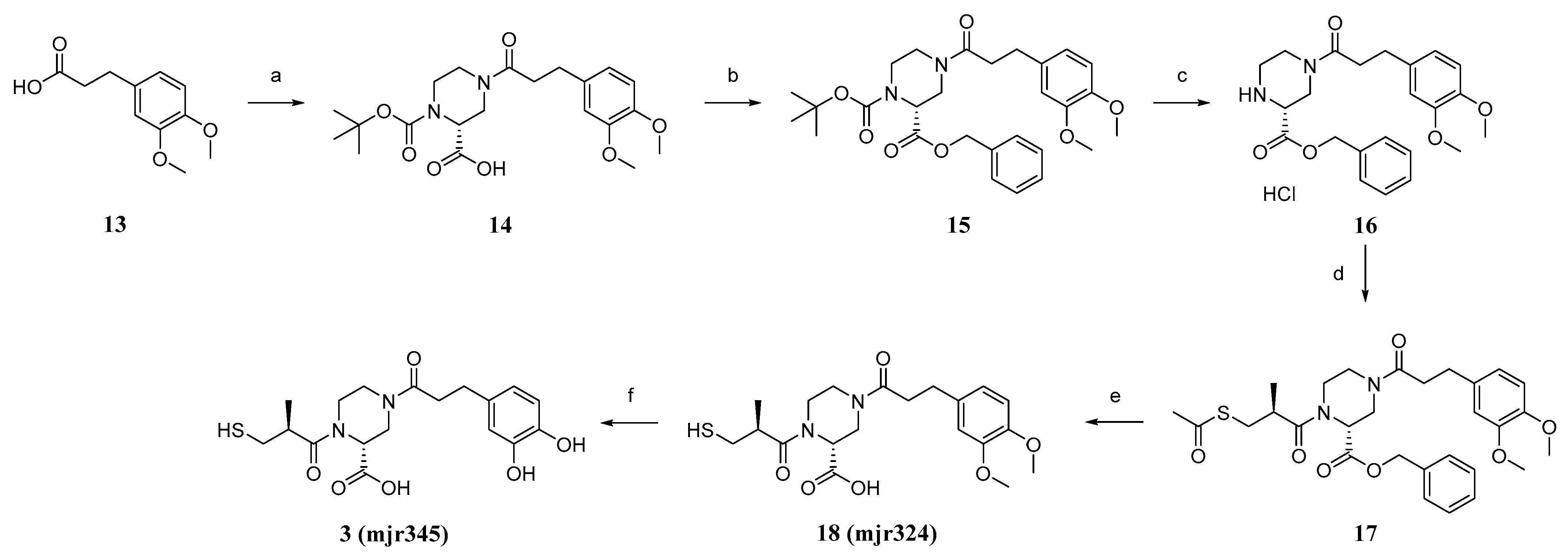
(R)-1-(tert-butoxycarbonyl)-4-(3-(3,4-dimethoxyphenyl)propanoyl)piperazine-2-carboxylic acid (14). 3-(3,4-dimethoxyphenyl)-propionic acid (13, 3.4 g, 16.2 mmol, 1.0 eq) was dissolved in SOCl2 (50 mL) and stirred for 3 h at 90 °C. After removing the excess SOCl2 in vacuo, the obtained acid chloride was mixed with (R)-1-boc-piperazine-2-carboxylic acid (5.0 g, 21.0 mmol, 1.3 eq) in DCM (160 mL), pyridine (20 mL), and DMF (2 mL), and the suspension was stirred for 19 h at room temperature. HCl (10%, 200 mL) was added and the aqueous phase was extracted with EtOAc (3 × 80 mL). The purified organic phases were dried over MgSO4 and in vacuo to obtain the crude product. The crude product was purified using flash column chromatography with silica gel as stationary phase and DCM/MeOH +1%AcOH (gradient DCM/MeOH 100/0 to 90/10) as mobile phase, to obtain product 14 as a beige foamy solid (2.5 g, 29%) after drying. 1H NMR (400 MHz, DMSO-d6): δ = 12.94 (s, 1H), 6.85–6.80 (m, 2H), 6.71 (dd, J = 8.2, 1.9 Hz, 1H), 4.63–4.40 (m, 1H), 3.91–3.78 (m, 2H), 3.73 (s, 3H), 3.70 (s, 3H), 3.19–2.79 (m, 4H), 2.75–2.67 (m, 2H), 2.66–2.54 (m, 2H), 1.39 (d, J = 15.8 Hz, 9H) ppm. MS (ESI, 70 eV) m/z (%): 422.95 (100) (M + H).
2-Benzyl 1-(tert-butyl) (R)-4-(3-(3,4-dimethoxyphenyl)propanoyl)piperazine-1,2-dicarboxylate (15). Acid (14, 2.4 g, 4.9 mmol, 1.0 eq), benzyl bromide (0.7 mL, 5.9 mmol, 1.2 eq), and KHCO3 (0.7 g, 7.3 mmol,1.5 eq) were suspended in acetone (40 mL) and refluxed for 18 h at 65 °C. Acetone was removed in vacuo and the reaction was diluted with DCM (60 mL) and water (60 mL). The phases were separated and the aqueous phase was extracted with DCM (2 × 60 mL), acidified, and extracted again with EtOAc (2 × 60 mL). The combined organic phases were dried over MgSO4 and in vacuo to obtain the crude product. Purification was performed using flash chromatography with silica gel as stationary phase and a mixture of hexane/EtOAc (gradient 87/13 to 0/100) as mobile phase. After drying, product 15 was isolated as an orange oil (2.2 g, 67%). 1H NMR (600 MHz, DMSO-d6): δ = 7.37–7.27 (m, 5H), 6.84–6.78 (m, 2H), 6.73–6.63 (m, 1H), 5.17–5.03 (m, 2H), 4.82–4.58 (m, 1H), 3.86–3.75 (m, 2H), 3.71 (dd, J = 13.6, 10.6 Hz, 6H), 3.19–2.83 (m, 4H), 2.74–2.64 (m, 2H), 2.63–2.54 (m, 2H), 1.41–1.28 (m, 9H) ppm. MS (ESI, 70 eV) m/z (%): 512.80 (100) (M + H).
Benzyl (R)-4-(3-(3,4-dimethoxyphenyl)propanoyl)piperazine-2-carboxylate (16). Carboxylate (15, 2.1 g, 3.3 mmol, 1.0 eq) was dissolved in DCM (50 mL). HCl in dioxane (4 M, 3.3 mL, 13.3 mmol, 4.0 eq) was added, and the mixture was stirred at room temperature overnight. All volatiles were removed in vacuo to obtain product 16 as a colourless foamy solid (1.8 g., quant.), which was used without further purification. 1H NMR (600 MHz, DMSO-d6): δ = 7.46–7.35 (m, 5H), 6.85–6.80 (m, 2H), 6.71 (dd, J = 24.6, 8.2 Hz, 1H), 5.25 (s, 2H), 4.50–4.23 (m, 1H), 3.74–3.70 (m, 6H), 3.56 (s, 1H), 3.25 (td, J = 10.5, 9.7, 5.1 Hz, 2H), 3.05–2.92 (m, 4H), 2.72 (q, J = 7.6 Hz, 2H), 2.65 (dd, J = 9.8, 6.8 Hz, 2H) ppm. MS (ESI, 70 eV) m/z (%): 412.85 (100) (M + H).
Benzyl (R)-1-((S)-3-(acetylthio)-2-methylpropanoyl)-4-(3-(3,4-dimethoxyphenyl)propanoyl)piperazine-2-carboxylate (17). (S)-(-)-3-acethylthio)-2-methylpropionic acid (4.1 g, 25.1 mmol, 1.0 eq) was dissolved in DCM (100 mL + 0.1 mL DMF), and SOCl2 (2.4 mL, 32.6 mmol, 1.3 eq) was added at 0 °C and the mixture was stirred at room temperature overnight. The solvent was removed in vacuo to obtain the acid chloride. Amine (16, 1.2 g, 2.7 mmol, 1.0 eq) was dissolved in DCM (40 mL), and DIPEA (0.7 mL, 4.0 mmol, 1.5 eq) was added and the mixture was stirred for 30 min at room temperature. Acid chloride (0.5 g, 2.7 mmol, 1.0 eq) was added and the mixture stirred overnight at room temperature. The reaction mixture was washed with HCl (2 M, 40 mL), as well as NaOH (1 M, 40 mL) and saturated sodium chloride solution (40 mL). The organic phase was dried in vacuo to obtain 17 as a crude product. The purification was performed using flash chromatography with silica gel as stationary phase and a mixture of DCM/EtOAc (gradient 100/0 to 0/100) as mobile phase. After drying, product 17 was isolated as a yellow oil (1.4 g, 75%). 1H NMR (600 MHz, DMSO-d6): δ = 7.38–7.27 (m, 5H), 6.85–6.78 (m, 2H), 6.68 (dd, J = 34.6, 8.2 Hz, 1H), 5.18–4.99 (m, 2H), 4.38–4.21 (m, 1H), 3.94–3.78 (m, 2H), 3.75–3.67 (m, 6H), 3.33–3.14 (m, 4H), 3.05–2.93 (m, 3H), 2.85–2.78 (m, 2H), 2.62–2.53 (m, 2H), 2.33–2.29 (m, 3H), 1.13–1.07 (m, 3H) ppm. MS (ESI, 70 eV) m/z (%): 557.00 (100) (M + H).
(R)-4-(3-(3,4-dimethoxyphenyl)propanoyl)-1-((S)-3-mercapto-2-methylpropanoyl)piperazine-2-carboxylic acid (18, mjr324). LiOH (0.2 g, 7.9 mmol, 4.5 eq) was dissolved in water (120 mL) and THF (60 mL) and carboxylate (17, 1.0 g, 1.8 mmol, 1 eq) were added. The suspension was stirred for 4.5 h at room temperature. HCl (2 M, 200 mL) was added and the mixture was extracted with DCM (3 × 60 mL). Purification was performed using preparative HPLC (C18 column, mobile phase: Iso 42% ACN) to obtain the product (18, mjr324) as a colourless foam (0.4 g, 47%). 1H NMR (500 MHz, DMSO-d6): δ = 12.96 (s, 1H), 6.86–6.81 (m, 2H), 6.72 (dd, J = 8.1, 2.3 Hz, 1H), 4.87 (dd, J = 5.0, 2.3 Hz, 1H), 4.37–4.24 (m, 1H), 3.97–3.79 (m, 2H), 3.73 (s, 3H), 3.70 (s, 3H), 3.32–3.10 (m, 4H), 3.05–2.87 (m, 2H), 2.79–2.68 (m, 3H), 2.66–2.56 (m, 2H), 1.13–0.97 (m, 3H) ppm. 13C NMR (126 MHz, DMSO): δ = 174.14, 171.08, 170.13, 148.63, 147.09, 133.74, 120.00, 112.34, 111.94, 55.57, 45.98, 45.57, 45.40, 42.24, 41.22, 38.75, 33.94, 30.18, 16.96 ppm. MS (ESI, 70 eV) m/z (%): 425.30 (100) (M + H). HRMS calculated m/z 447.15603 and found m/z 447.15561.
(R)-4-(3-(3,4-dihydroxyphenyl)propanoyl)-1-((S)-3-mercapto-2-methylpropanoyl)piperazine-2-carboxylic acid (3, mjr345). Acid (3, 0.3 g, 0.7 mmol, 1.0 eq) was dissolved in DCM (3 mL) at −78 °C. BBr3 (1 M in DCM, 3.5 mL, 3.5 mmol, 5.0 eq) was added. The resulting mixture was stirred for 2 h at −78 °C and then overnight at room temperature. The reaction mixture was cooled to 0 °C and water was added dropwise until the remaining BBr3 was consumed (no more smoke was produced). The aqueous phase was extracted with DCM (3 × 6 mL), the organic phases were combined, and all volatile components were removed under reduced pressure to obtain the crude product. Purification was performed using preparative HPLC (C18 column, mobile phase: Iso 26% ACN) to obtain the product 3, mjr345 as a colourless solid (20.0 mg, 7%). 1H NMR (500 MHz, DMSO-d6): δ = 13.18 (s, 1H), 8.63 (s, 2H), 6.61 (dd, J = 6.6, 3.9 Hz, 1H), 6.59 (d, J = 2.0 Hz, 1H), 6.44 (dd, J = 8.0, 2.1 Hz, 1H), 5.07–4.66 (m, 1H), 4.28 (q, J = 12.0, 11.2 Hz, 1H), 3.98–3.76 (m, 2H), 3.34–3.13 (m, 4H), 3.04–2.88 (m, 2H), 2.77–2.68 (m, 1H), 2.65–2.56 (m, 4H), 1.13–0.96 (m, 3H) ppm. 12C NMR (126 MHz, DMSO): δ = 174.09, 171.27, 171.04, 144.96, 143.32, 131.99, 118.75, 115.74, 115.42, 45.94, 45.37, 42.72, 42.21, 41.92, 38.79, 34.11, 29.97, 16.90 ppm. MS (ESI, 70 eV) m/z (%): 396.95 (100) (M + H). HRMS calculated m/z 419.12473 and found m/z 419.12439.


 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}