Chiral 8-Amino-5,6,7,8-tetrahydroquinoline Derivatives in Metal Catalysts for the Asymmetric Transfer Hydrogenation of 1-Aryl Substituted-3,4-dihydroisoquinolines as Alkaloids Precursors



Abstract
1. Introduction
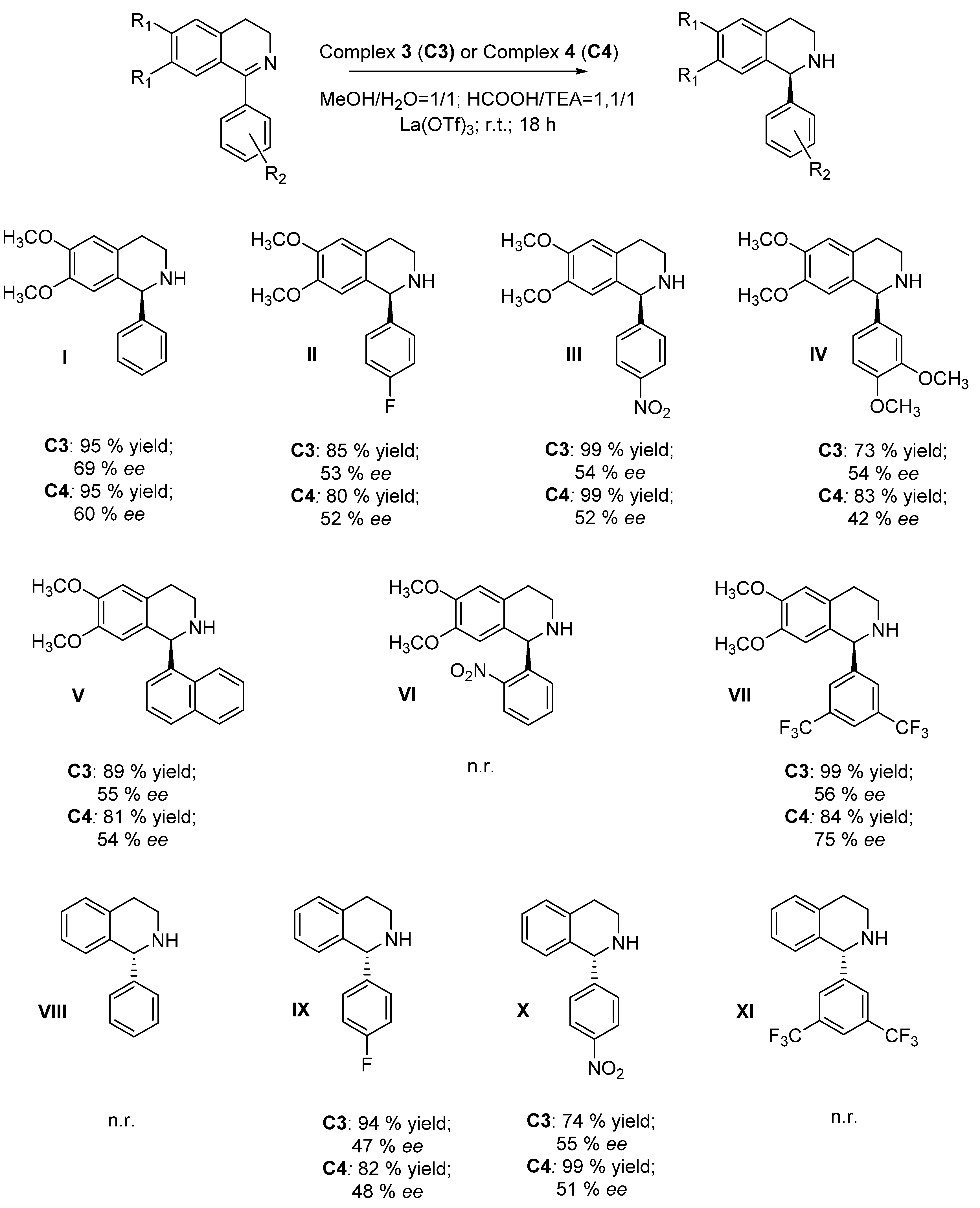
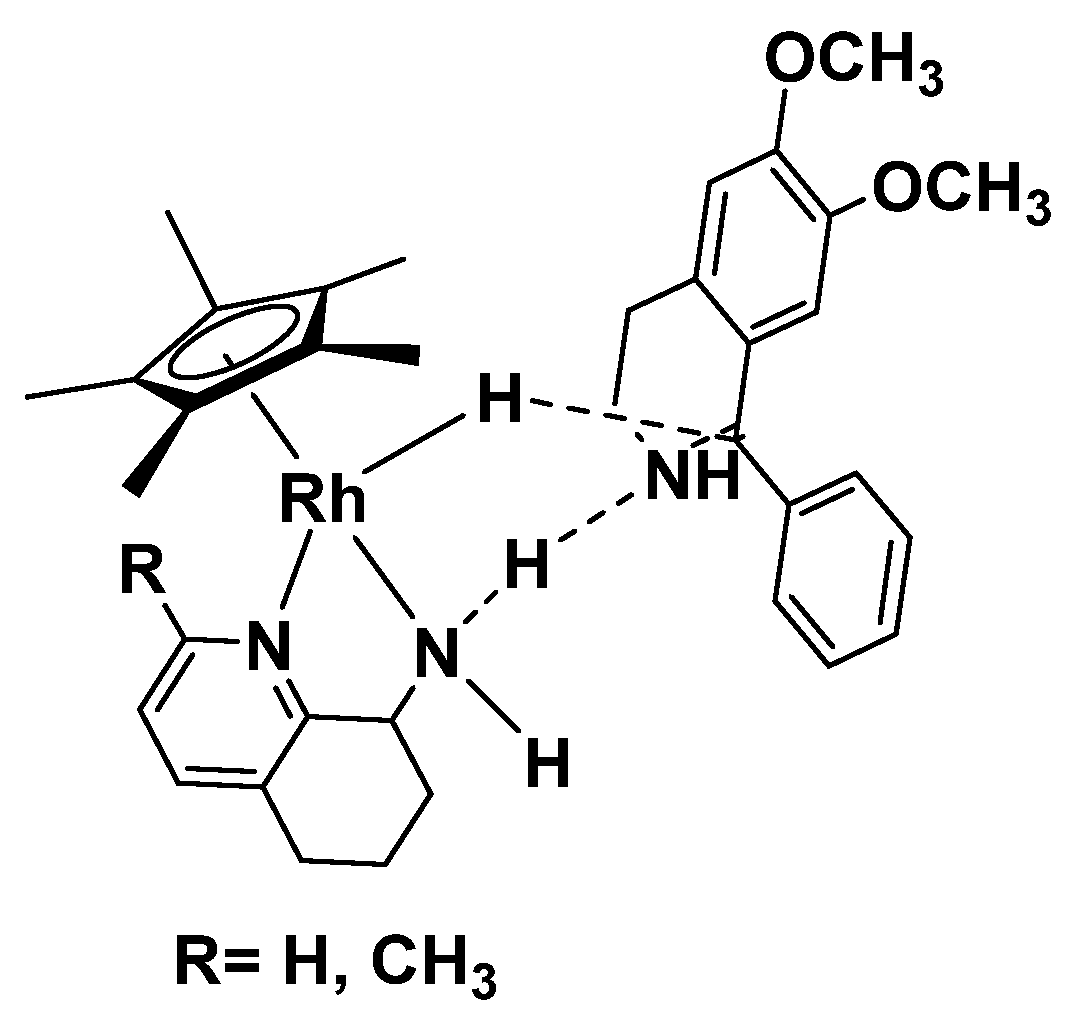
2. Results and Discussion
3. Experimental
3.1. General Procedure for the Synthesis of L1 and L2
- (R)-L1: pale yellow oil (97% yield). 1H NMR (CDCl3, 300 MHz, 25 °C): δ = 1.68–1.75 (m, 2 H), 1.96 (m, 1 H), 2.24 (m, 1 H), 2.68–2.88 (m, 2 H), 3.45 (br, 2 H, NH2), 4.05 (t, J = 6.8 Hz, 1 H, H-8), 7.07 (dd, J = 7.3, 4.4 Hz, 1 H, H-3), 7.37 (d, J = 7.3 Hz, 1 H, H-4), 8.39 (d, J = 4.4 Hz, 1 H, H-2). 13C NMR (CDCl3, 75 MHz, 25 °C): δ = 19.8, 28.7, 31.3, 51.2, 121.8, 131.7, 136.8, 146.9, 158.3. MS (EI): m/z (%) 148 (M+, 100), 147 (77), 131 (21), 120 (78), 119 (60), 93 (36). MS (ESI) of C9H12N2 (m/z): calcd 148.1 (M+). found 148.1. [α]D22 = −51.0 (c = 0.55, CHCl3)
- (R)-L2: pale yellow oil (82% yield). 1H NMR (CDCl3, 300 MHz, 25 °C): δ = 1.69–1.81 (m, 2H), 1.93–2.01 (m, 1H), 2.10–2.18 (m, 1H), 2.53 (s, 3H), 2.50–2.78 (m, 3H), 3.67 (t, J = 5.2 Hz, 1H), 7.05 (dd, J = 7.7, 4.7 Hz, 1H), 7.39 (d, J = 7.6 Hz, 1H), 8.40 (d, J = 4.6 Hz, 1H) ppm; 13C NMR (CDCl3, 75 MHz, 25 °C): δ = 19.55, 27.82, 28.85, 34.26, 59.56, 121.86, 132.46, 136.89, 146.86, 157,23 ppm. FTIR 3333.9, 3049.6, 2926.7, 2855.2, 2784.1, 1648.1, 1575.3, 1444.5, 1428.1, 1238.7, 1104.1, 782.2 cm−1. MS (ESI) of C10H14N2 (m/z): calcd 162.1, found 163.2 [M+1]+. [α]D22 = −20.8 (c 0.5, CH2Cl2).
3.2. General Procedure for ATH
3.3. General Procedure for the Synthesis of Cp* Metal Complexes
- [Ir(Cp*)(R)-CAMPY(Cl)]Cl (C1): 1H NMR (300 MHz, CDCl3) δ 8.36 (d, J = 8.21 Hz, 1H), 7.59 (d, J = 8.69 Hz, 1H), 7.38–7.23 (m, 1H), 4.38–4.22 (m, 1H), 3.37–3.21 (m, 2H), 2.78–2.67 (m, 2H), 2.11–2.00 (m, 2H), 1.97 (15H) ppm. 13C NMR (75 MHz, CDCl3) δ 159.89, 148.87, 139.30, 136.22, 125.90, 87.54, 61.87, 31.83, 27.33, 21.42, 9.40 ppm. MS (ESI+) for C19H27ClN2Ir m/z: calculated 511.15, found 511.00 [M]+. Elemental analysis for C19H27Cl2N2Ir: calcd. C, 41.75; H, 4.98; N, 5.13; found C, 41.26; H, 4.47; N, 5.08.
- [Ir(Cp*)(R)-Me-CAMPY (Cl)]Cl (C2): 1H NMR (300 MHz, CDCl3) δ 7.55 (d, J = 8.57 Hz, 1H), 7.28-7.21 (m, 1H), 4.23–4.09 (m, 1H), 3.57–3.42 (m, 2H), 2.96 (s, 3H), 2.82–2.66 (m, 2H), 2.16–1.98 (m, 2H), 1.88 (15H) ppm. 13C NMR (75 MHz, CDCl3) δ 159.12, 140.07, 139.51, 134.05, 126.31, 88.86, 87.94, 62.87, 35.78, 28.98, 26.91, 22.37, 9.81 ppm. MS (ESI+) for C20H29ClN2Ir m/z: calculated 525.16, found 525.19 [M]+. Elemental analysis for C20H29Cl2N2Ir: calcd. C, 42.85; H, 5.21; N, 5.00; found C, 43.26; H, 5.33; N, 5.06.
- [RhCp*(R)-CAMPY (Cl)]Cl (C3): 1H NMR (300 MHz, CDCl3) δ 8.48 (d, J = 8.00 Hz, 1H), 7.57 (d, J = 8.10 Hz, 1H), 7.39–7.24 (m, 1H), 4.48–4.27 (m, 1H), 3.32–3.09 (m, 2H), 2.91–2.78 (m, 2H), 2.18–2.02 (m, 2H), 1.88 (s, 15H) ppm. 13C NMR (75 MHz, CDCl3) δ 159.23, 148.99, 139.62, 135.85, 125.77, 95.88, 95.78, 60.30, 32.11, 27.23, 21.19, 9.63 ppm. MS (ESI+) for C19H27ClN2Rh m/z: calculated 421.09, found 422.95 [M + H]+. Elemental analysis for C19H27Cl2N2Rh: calcd. C, 49.91; H, 5.95; N, 6.13; found C, 49.26; H, 6.01; N, 6.08.
- [RhCp*(R)-Me-CAMPY (Cl)]Cl (C4): 1H NMR (300 MHz, CDCl3) δ 7.46 (d, J = 8.19 Hz, 1H), 7.24 (d, J = 6.41 Hz, 1H), 4.59–4.36 (m, 1H), 3.58–3.48 (m, 2H), 2.96 (s, 3H), 2.89–2.71 (m, 2H), 2.16–1.94 (m, 2H), 1.89 (s, 15H) ppm. 13C NMR (75 MHz, CDCl3) δ 159.77, 157.92, 139.34, 131.86, 125.11, 106.66, 105.33, 102.02, 99.13, 95.90, 58.41, 33.28, 28.01, 21.44, 19.55, 9.83 ppm. MS (ESI+) for C20H29ClN2Rh m/z: calculated 435.11, found 436.05 [M + H]+. Elemental analysis for C20H29Cl2N2Rh: calcd. C, 50.97; H, 6.20; N, 5.94; found C, 50.98; H, 6.11; N, 5.88.
- RuCp*(R)-CAMPY (Cl) (C5): 1H NMR (300 MHz, CDCl3) δ 8.34 (d, J = 7.89 Hz, 1H), 7.62 (d, J = 8.21 Hz, 1H), 7.39–7.28 (m, 1H), 4.45–4.14 (m, 1H), 3.33–3.18 (m, 2H), 2.91–2.63 (m, 2H), 2.19–2.00 (m, 2H), 1.91 (s, 15H) ppm. 13C NMR (75 MHz, CDCl3) δ 159.67, 157.86, 133.31, 132.01, 125.45, 107.00, 105.27, 101.97, 99.15, 96.12, 58.21, 33.38, 28.13, 21.33, 10.11 ppm. MS (ESI+) for C19H27ClN2Ru m/z: calculated 420.09, found 421.11 [M + H]+. Elemental analysis for C19H27ClN2Ru: calcd. C, 54.34; H, 6.48; N, 6.67; found C, 53.78; H, 6.13; N, 6.56.
- RuCp*(R)-Me-CAMPY(Cl) (C6): 1H NMR (300 MHz, CDCl3) δ 7.66 (d, J = 7.12 Hz, 1H), 7.48–7.39 (m, 1H), 4.44–4.19 (m, 1H),3.31–3.18 (m, 2H), 3.11 (s, 3H), 2.87–2.64 (m, 2H), 2.18–1.98 (m, 2H), 1.95 (s, 15H) ppm. 13C NMR (75 MHz, CDCl3) δ 159.61, 158.52, 139.28, 132.46, 125.01, 105.21, 104.72, 101.96, 99.17, 96.00, 58.33, 33.36, 27.58, 21.32, 18.46, 10.31 ppm. MS (ESI+) for C20H29ClN2Ru m/z: calculated 434.11, found 435.12 [M + H]+. Elemental analysis for C20H29ClN2Ru: calcd. C, 55.35; H, 6.74; N, 6.46; found C, 53.78; H, 6.13; N, 6.56.
- [RhCp*-AMPY (Cl)]Cl (C7): 1H NMR (300 MHz, CDCl3) δ 8.58 (d, J = 7.63 Hz, 1H), 7.96–7.70 (m, 1H), 7.52–7.34 (m, 2H), 4.44 (dd, J = 8.23, 4.81 Hz, 2H), 1.89 (s, 15H) ppm. 13C NMR (75 MHz, CDCl3) δ 161.99, 150.68, 139.25, 125.51, 122.16, 95.88, 95.77, 51.68, 9.50 ppm. MS (ESI+) for C16H23ClN2Rh m/z: calculated 381.06, found 404.04 [M + Na]+. Elemental analysis for C20H29Cl2N2Rh: calcd. C, 46.07; H, 5.56; N, 6.72; found C, 46.44; H, 6.01; N, 6.78.
- RhCp*-Ts-AMPY(Cl) (C8): 1H NMR (300 MHz, CDCl3) δ 9.19 (d, J = 5.64 Hz, 1H), 7.81 (t, J = 6.2 Hz, 1H), 7.65 (d, J = 8.01 Hz, 2H), 7.35 (d, J = 7.63 Hz, 2H), 7.15 (d, J = 7.89 Hz, 2H), 4.03 (q, J = 17.12 Hz, 2H), 2.46 (s, 15H), 2.28 (s, 3H) ppm. 13C NMR (75 MHz, CDCl3) δ 162.04, 151.12, 142.32, 139.89, 139.02, 129.64, 127.83, 126.77, 121.85, 96.32, 95.53, 50.87, 21.44, 9.76 ppm. MS (ESI+) for C23H28ClN2O2RhS m/z: calculated 534.06, found 535.07 [M + H]+. Elemental analysis for C23H28ClN2O2RhS: calcd. C, 51.65; H, 5.28; N, 5.24; found C, 51.37; H, 5.17; N, 5.23.
3.4. Analytical Conditions
- 6,7-dimethoxy-1-phenyl-1,2,3,4-tetrahydroisoquinoline (I): R-isomer: 11.1 min (min); S-isomer: 15.4 min (maj); column: Chiralcel OD-H, eluent: 2-propanol/hexane = 30/70 (0.01% DEA), flow = 0.7 mL/min, λ = 285 nm.
- 1-(4-fluorophenyl)-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline (II): R-isomer: 21.0 min (min); S-isomer: 23.9 min (maj); column: Chiralpak AD-H, eluent: 2-propanol/hexane = 10/90, flow = 0.8 mL/min, λ = 220 nm.
- 6,7-dimethoxy-1-(4-nitrophenyl)-1,2,3,4-tetrahydroisoquinoline (III): R-isomer: 27.9 min (min); S-isomer: 35.7 min (maj); column: Chiralcel OD-H, eluent: 2-propanol/hexane = 30/70 (0.01% DEA), flow = 0.7 mL/min, λ = 285 nm.
- 1-(3,4-dimethoxyphenyl)-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline (IV): R-isomer: 23.7 min (min); S-isomer: 35.0 min (maj); column: Chiralcel OD-H, eluent: 2-propanol/hexane = 30/70 (0.01% DEA), flow = 0.7 mL/min, λ = 285 nm.
- 6,7-dimethoxy-1-(naphthalen-1-yl)-1,2,3,4-tetrahydroisoquinoline (V): 1° isomer: 14.7 min (min); 2° isomer: 16.4 min (maj); column: Chiralcel OD-H, eluent: 2-propanol/hexane = 30/70 (0.01% DEA), flow = 0.7 mL/min, λ = 285 nm.
- 6,7-dimethoxy-1-(2-nitrophenyl)-1,2,3,4-tetrahydroisoquinoline (VI): 1° isomer: 15.2 min (min); 2° isomer: 17.3 min (maj) column: Chiralcel OD-H, eluent: 2-propanol/hexane = 30/70 (0.01% DEA), flow = 0.7 mL/min, λ = 285 nm.
- 1-(3,5-bis(trifluoromethyl)phenyl)-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline (VII): 1° isomer: 6.9 min (maj); 2° isomer: 9.4 min (min); column: Lux Amilose-2, eluent: 2-propanol/hexane = 10/90 (0.01% DEA), flow = 0.8 mL/min, λ = 220 nm.
- 1-phenyl-1,2,3,4-tetrahydroisoquinoline (VIII): S-isomer: 13.1 (min); R-isomer: 15.4 (maj); column: Chiralpak AD, eluent: 2-propanol/hexane = 4/96, flow = 0.8 mL/min, λ = 240 nm.
- 1-(4-fluorophenyl)-1,2,3,4-tetrahydroisoquinoline (IX): S-isomer: 7.0 (min); R-isomer: 7.5 (maj); column: AD-H, eluent: 2-propanol/hexane = 10/90, flow = 1.0 mL/min, λ = 220 nm.
- 1-(4-nitrophenyl)-1,2,3,4-tetrahydroisoquinoline (X): S-isomer: 17.9 (min); R-isomer: 19.9 (maj); column: Lux Amylose-2, eluent: 2-propanol/hexane = 10/90 (0.01% DEA), flow = 0.8 mL/min, λ = 254 nm.
- 1-(3,5-bis(trifluoromethyl)phenyl)-1,2,3,4-tetrahydroisoquinoline (XI): 1° isomer: 4.4 (min); 2° isomer: 5.0 (maj); column: Lux Cellulose-2, eluent: 2-propanol/hexane = 5/110, flow = 0.6 mL/min, λ = 220 nm.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Patil, M.D.; Grogan, G.; Bommarius, A.; Yun, H. Oxidoreductase-Catalyzed Synthesis of Chiral Amines. ACS Catal. 2018, 8, 10985–11015. [Google Scholar] [CrossRef]
- Liu, A.; Han, J.; Nakano, A.; Konno, H.; Moriwaki, H.; Abe, H.; Izawa, K.; Soloshonok, V.A. New pharmaceuticals approved by FDA in 2020: Small-molecule drugs derived from amino acids and related compounds. Chirality 2022, 34, 86–103. [Google Scholar] [CrossRef] [PubMed]
- Nugent, T.C. Chiral Amine Synthesis: Methods, Developments and Applications; Wiley-VCH: Weinheim, Germany, 2010. [Google Scholar]
- Tian, Y.; Hu, L.A.; Wang, Y.-Z.; Zhang, X.; Yin, Q. Recent advances on transition-metal-catalysed asymmetric reductive amination. Org. Chem. Front. 2021, 8, 2328–2342. [Google Scholar] [CrossRef]
- Murugesan, K.; Wei, Z.; Chandrashekhar, V.G.; Neumann, H.; Spannenberg, A.; Jiao, H.; Beller, M.; Jagadeesh, R.V. Homogeneous cobalt-catalyzed reductive amination for synthesis of functionalized primary amines. Nat. Commun. 2019, 10, 5443. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Wang, W.; Guo, H.; Gao, G.; Huang, H.; Chang, M. Iridium-catalyzed direct asymmetric reductive amination utilizing primary alkyl amines as the N-sources. Nat. Commun. 2022, 13, 3344. [Google Scholar] [CrossRef] [PubMed]
- Stivala, C.E.; Zbieg, J.R.; Liu, P.; Krische, M.J. Chiral Amines via Enantioselective π-Allyliridium-C,O-Benzoate-Catalyzed Allylic Alkylation: Student Training via Industrial–Academic Collaboration. Acc. Chem. Res. 2022, 55, 2138–2147. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Shen, M.-L.; Fan, L.-F.; Zhou, X.-L.; Wang, P.-S.; Gong, L.-Z. Palladium-Catalyzed Branch- and Z-Selective Allylic C−H Amination with Aromatic Amines. Angew. Chem. Int. Ed. 2003, 62, e202211631. [Google Scholar] [CrossRef]
- Steck, V.; Carminati, D.M.; Johnson, N.R.; Fasan, R. Enantioselective Synthesis of Chiral Amines via Biocatalytic Carbene N–H Insertion. ACS Catal. 2020, 10, 10967–10977. [Google Scholar] [CrossRef]
- Zhu, S.-F.; Zhou, Q.-L. Transition-Metal-Catalyzed Enantioselective Heteroatom–Hydrogen Bond Insertion Reactions. Acc. Chem. Res. 2012, 45, 1365–1377. [Google Scholar] [CrossRef]
- Cosgrove, S.C.; Thompson, M.P.; Ahmed, S.T.; Parmeggiani, F.; Turner, N.J. One-Pot Synthesis of Chiral N-Arylamines by Combining Biocatalytic Aminations with Buchwald–Hartwig N-Arylation. Angew. Chem. Int. Ed. 2020, 59, 18156–18160. [Google Scholar] [CrossRef]
- Mathew, S.; Sagadevan, A.; Renn, D.; Rueping, M. One-Pot Chemoenzymatic Conversion of Alkynes to Chiral Amines. ACS Catal. 2021, 11, 12565–12569. [Google Scholar] [CrossRef]
- Gandolfi, R.; Coffetti, G.; Facchetti, G.; Rimoldi, I. Double approaches for obtaining an asymmetric one-pot addition/reduction reaction. Mol. Catal. 2022, 532, 112716. [Google Scholar] [CrossRef]
- Tang, P.; Wang, H.; Zhang, W.; Chen, F.-E. Asymmetric catalytic hydrogenation of imines and enamines in natural product synthesis. Green Synth. Catal. 2020, 1, 26–41. [Google Scholar] [CrossRef]
- Facchetti, G.; Christodoulou, M.S.; Binda, E.; Fusè, M.; Rimoldi, I. Asymmetric Hydrogenation of 1-aryl substituted-3,4-Dihydroisoquinolines with Iridium Catalysts Bearing Different Phosphorus-Based Ligands. Catalysts 2020, 10, 914. [Google Scholar] [CrossRef]
- Li, B.; Liu, R.; Yang, J.; Luo, J.; Yao, L.; Li, M.; Zheng, X.; Jiang, R.; Nie, H.; Zhang, S. Iridium-Catalyzed Asymmetric Hydrogenation of Sterically Hindered Cyclic Imines for Enantioselective Synthesis of Tetrahydroisoquinolines. Org. Lett. 2021, 23, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Nie, H.; Zhu, Y.; Hu, X.; Wei, Z.; Yao, L.; Zhou, G.; Wang, P.; Jiang, R.; Zhang, S. Josiphos-Type Binaphane Ligands for Iridium-Catalyzed Enantioselective Hydrogenation of 1-Aryl-Substituted Dihydroisoquinolines. Org. Lett. 2019, 21, 8641–8645. [Google Scholar] [CrossRef]
- Facchetti, G.; Cesarotti, E.; Pellizzoni, M.; Zerla, D.; Rimoldi, I. “In situ” Activation of Racemic RuII Complexes: Separation of trans and cis Species and Their Application in Asymmetric Reduction. Eur. J. Inorg. Chem. 2012, 2012, 4365–4370. [Google Scholar] [CrossRef]
- Facchetti, G.; Fusè, M.; Pecoraro, T.; Nava, D.; Rimoldi, I. New sp3 diphosphine-based rhodium catalysts for the asymmetric conjugate addition of aryl boronic acids to 3-azaarylpropenones. New J. Chem. 2021, 45, 18769–18775. [Google Scholar] [CrossRef]
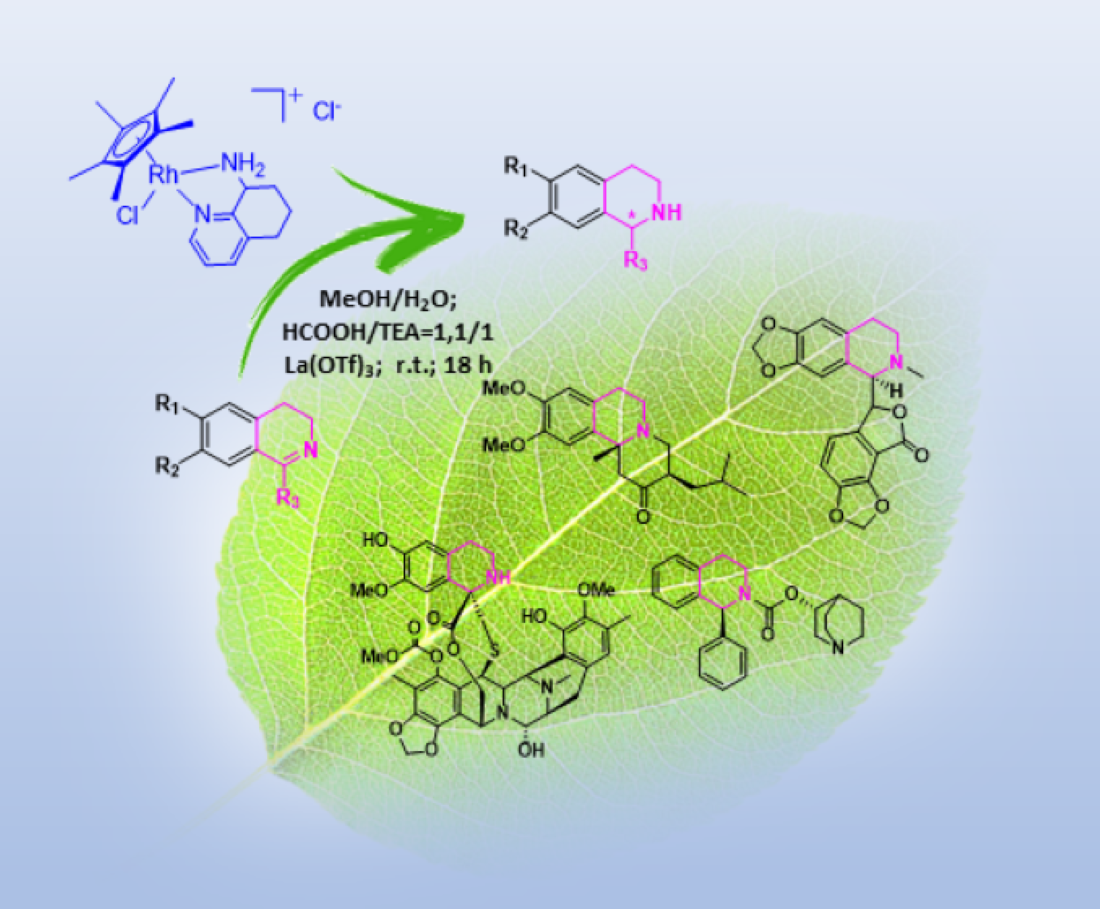
- Shang, X.-F.; Yang, C.-J.; Morris-Natschke, S.L.; Li, J.-C.; Yin, X.-D.; Liu, Y.-Q.; Guo, X.; Peng, J.-W.; Goto, M.; Zhang, J.-Y.; et al. Biologically active isoquinoline alkaloids covering 2014–2018. Med. Res. Rev. 2020, 40, 2212–2289. [Google Scholar] [CrossRef]
- Plazas, E.; Avila, M.M.C.; Muñoz, D.R.; Cuca, S.L.E. Natural isoquinoline alkaloids: Pharmacological features and multi-target potential for complex diseases. Pharmacol. Res. 2022, 177, 106126. [Google Scholar] [CrossRef]
- Wang, Y.; Zheng, T.; Huo, Y.; Du, W. Exploration of Isoquinoline Alkaloids as Potential Inhibitors against Human Islet Amyloid Polypeptide. ACS Chem. Neurosci. 2022, 13, 2164–2175. [Google Scholar] [CrossRef] [PubMed]
- Facchetti, G.; Christodoulou, M.S.; Mendoza, L.B.; Cusinato, F.; Dalla Via, L.; Rimoldi, I. Biological Properties of New Chiral 2-Methyl-5,6,7,8-tetrahydroquinolin-8-amine-based Compounds. Molecules 2020, 25, 5561. [Google Scholar] [CrossRef] [PubMed]
- Pyne, M.E.; Martin, V.J.J. Microbial synthesis of natural, semisynthetic, and new-to-nature tetrahydroisoquinoline alkaloids. Curr. Opin. Green Sustain. Chem. 2022, 33, 100561. [Google Scholar] [CrossRef]
- Ehrenworth, A.M.; Peralta-Yahya, P. Accelerating the semisynthesis of alkaloid-based drugs through metabolic engineering. Nat. Chem. Biol. 2017, 13, 249–258. [Google Scholar] [CrossRef]
- Facchetti, G.; Ferri, N.; Lupo, M.G.; Giorgio, L.; Rimoldi, I. Monofunctional PtII Complexes Based on 8-Aminoquinoline: Synthesis and Pharmacological Characterization. Eur. J. Inorg. Chem. 2019, 2019, 3389–3395. [Google Scholar] [CrossRef]
- Facchetti, G.; Bucci, R.; Fusè, M.; Rimoldi, I. Asymmetric Hydrogenation vs Transfer Hydrogenation in the Reduction of Cyclic Imines. ChemistrySelect 2018, 3, 8797–8800. [Google Scholar] [CrossRef]
- Facchetti, G.; Rimoldi, I. 8-Amino-5,6,7,8-tetrahydroquinoline in iridium(iii) biotinylated Cp* complex as artificial imine reductase. New J. Chem. 2018, 42, 18773–18776. [Google Scholar] [CrossRef]
- Gandolfi, R.; Facchetti, G.; Christodoulou, M.S.; Fusè, M.; Meneghetti, F.; Rimoldi, I. Cascade Reaction by Chemo- and Biocatalytic Approaches to Obtain Chiral Hydroxy Ketones and anti 1,3-Diols. ChemistryOpen 2018, 7, 393–400. [Google Scholar] [CrossRef]
- Zerla, D.; Facchetti, G.; Fuse, M.; Pellizzoni, M.; Castellano, C.; Cesarotti, E.; Gandolfi, R.; Rimoldi, I. 8-Amino-5,6,7,8-tetrahydroquinolines as ligands in iridium(III) catalysts for the reduction of aryl ketones by asymmetric transfer hydrogenation (ATH). Tetrahedron Asymmetry 2014, 25, 1031–1037. [Google Scholar] [CrossRef]
- Rimoldi, I.; Facchetti, G.; Cesarotti, E.; Pellizzoni, M.; Fuse, M.; Zerla, D. Enantioselective transfer hydrogenation of aryl ketones: Synthesis and 2D-NMR characterization of new 8-amino-5,6,7,8-tetrahydroquinoline Ru(II)-complexes. Curr. Org. Chem. 2012, 16, 2982–2988. [Google Scholar] [CrossRef]
- Du, Q.; Liu, L.; Zhou, T. General and Efficient Copper-Catalyzed Oxazaborolidine Complex in Transfer Hydrogenation of Isoquinolines under Mild Conditions. ACS Omega 2020, 5, 21219–21225. [Google Scholar] [CrossRef] [PubMed]
- Letko, C.S.; Heiden, Z.M.; Rauchfuss, T.B. Activation and Deactivation of Cp*Ir(TsDPEN) Hydrogenation Catalysts in Water. Eur. J. Inorg. Chem. 2009, 2009, 4927–4930. [Google Scholar] [CrossRef]
- Toubiana, J.; Medina, L.; Sasson, Y. The Nature of the True Catalyst in Transfer Hydrogenation with Alcohol Donors Using (arene)2Ru2Cl4(II)/TsDPEN Precursor. Mod. Res. Catal. 2014, 3, 68–88. [Google Scholar] [CrossRef]
- Li, J.; Yu, B.; Lu, Z. Chiral Imidazoline Ligands and Their Applications in Metal-Catalyzed Asymmetric Synthesis†. Chin. J. Chem. 2021, 39, 488–514. [Google Scholar] [CrossRef]
- Piyasaengthong, A.; Williams, L.J.; Yufit, D.S.; Walton, J.W. Novel ruthenium complexes bearing bipyridine-based and N-heterocyclic carbene-supported pyridine (NCN) ligands: The influence of ligands on catalytic transfer hydrogenation of ketones. Dalton Trans. 2022, 51, 340–351. [Google Scholar] [CrossRef]
- Singh, P. Recent Developments in the Synthesis and Applications of Pyridines; Elsevier: Amsterdam, The Netherlands, 2022; Volume 15. [Google Scholar]
- Samec, J.S.M.; Bäckvall, J.-E.; Andersson, P.G.; Brandt, P. Mechanistic aspects of transition metal-catalyzed hydrogen transfer reactions. Chem. Soc. Rev. 2006, 35, 237–248. [Google Scholar] [CrossRef]
- Mwansa, J.M.; Stirling, M.J.; Page, M.I. Changing the kinetic order of enantiomer formation and distinguishing between iminium ion and imine as the reactive species in the asymmetric transfer hydrogenation of substituted imines using a cyclopentadienyl iridium (III) complex. Pure Appl. Chem. 2020, 92, 107–121. [Google Scholar] [CrossRef]
- Martins, J.E.D.; Clarkson, G.J.; Wills, M. Ru(II) Complexes of N-Alkylated TsDPEN Ligands in Asymmetric Transfer Hydrogenation of Ketones and Imines. Org. Lett. 2009, 11, 847–850. [Google Scholar] [CrossRef]
- Itoh, T.; Nagata, K.; Kurihara, A.; Miyazaki, M.; Ohsawa, A. Reductive amination of aldehydes and ketones by a Hantzsch dihydropyridine using scandium triflate as a catalyst. Tetrahedron Lett. 2002, 43, 3105–3108. [Google Scholar] [CrossRef]
- Evanno, L.; Ormala, J.; Pihko, P.M. A Highly Enantioselective Access to Tetrahydroisoquinoline and β-Carboline Alkaloids with Simple Noyori-Type Catalysts in Aqueous Media. Chem. Eur. J. 2009, 15, 12963–12967. [Google Scholar] [CrossRef]
- Wu, X.; Li, X.; King, F.; Xiao, J. Insight into and Practical Application of pH—Controlled Asymmetric Transfer Hydrogenation of Aromatic Ketones in Water. Angew. Chem. Int. Ed. 2005, 117, 3473–3477. [Google Scholar] [CrossRef]
- Kuzma, M.; Václavík, J.; Novák, P.; Přech, J.; Januščák, J.; Červený, J.; Pecháček, J.; Šot, P.; Vilhanová, B.; Matoušek, V.; et al. New insight into the role of a base in the mechanism of imine transfer hydrogenation on a Ru(ii) half-sandwich complex. Dalton Trans. 2013, 42, 5174–5182. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhu, J.; Sun, C.; Deng, Z.; Qu, X. Biosynthesis of plant tetrahydroisoquinoline alkaloids through an imine reductase route. Chem. Sci. 2020, 11, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Shende, V.S.; Deshpande, S.H.; Shingote, S.K.; Joseph, A.; Kelkar, A.A. Asymmetric Transfer Hydrogenation of Imines in Water by Varying the Ratio of Formic Acid to Triethylamine. Org. Lett. 2015, 17, 2878–2881. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.M.R.; Berry, D.B.G.; Crossley, J.N.; Codina, A.; Clegg, I.; Lowe, J.P.; Buchard, A.; Hintermair, U. Does the Configuration at the Metal Matter in Noyori–Ikariya Type Asymmetric Transfer Hydrogenation Catalysts? ACS Catal. 2021, 11, 13649–13659. [Google Scholar] [CrossRef]
- Perez, M.; Wu, Z.; Scalone, M.; Ayad, T.; Ratovelomanana-Vidal, V. Enantioselective Synthesis of 1-Aryl-Substituted Tetrahydroisoquinolines Through Ru-Catalyzed Asymmetric Transfer Hydrogenation. Eur. J. Org. Chem. 2015, 2015, 6503–6514. [Google Scholar] [CrossRef]
- Uenishi, J.; Hamada, M. Synthesis of Enantiomerically Pure 8-Substituted 5,6,7,8-Tetrahydroquinolines. Synthesis 2002, 2002, 0625–0630. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
| Entry | Complex | Additive | Hydrogen Donor | Media | Conversion % | ee % |
| 1 | RhCp*TsDPEN | none | HCOOH/TEA 1.1:1 | H2O/MeOH 1:1 | 84 | 7 |
| 2 | C1 | none | HCOOH/TEA 1.1:1 | MOPS buffer 1.2 M pH 7.8 | 94 | 27 |
| 3 | C1 | La(OTf)3 | HCOOH | H2O/MeOH 1:1 | 99 | 45 |
| 4 | C2 | none | HCOOH/TEA 1.1:1 | K2HPO4/NaH2PO4 0.1 M pH 8 | 38 | 22 |
| 5 | C2 | La(OTf)3 | HCOOH | MOPS buffer 1.2 M pH 7.8 | 95 | 18 |
| 6 | C3 | none | HCOOH/TEA 1.1:1 | H2O/MeOH 1:1 | 49 | 69 |
| 7 | C3 | La(OTf)3 | HCOOH/TEA 1.1:1 | H2O/MeOH 1:1 | 95 | 69 |
| 8 | C4 | none | HCOOH/TEA 1.1:1 | H2O/MeOH 1:1 | 60 | 57 |
| 9 | C4 | La(OTf)3 | HCOOH/TEA 1.1:1 | H2O/MeOH 1:1 | 95 | 60 |
| 10 | C5 | none | n.r. | n.r. | ||
| 11 | C5 | La(OTf)3 | HCOOH/TEA 1.1:1 | H2O/MeOH 1:1 | >10 | 5 |
| 12 | C6 | none | n.r. | n.r. | ||
| 13 | C6 | La(OTf)3 | n.r. | n.r. | ||
| 14 | C7 | none | HCOOH/TEA 1.1:1 | H2O/MeOH 1:1 | 13 | rac |
| 15 | C8 | none | HCOOH/TEA 1.1:1 | H2O/MeOH 1:1 | 94 | rac |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Facchetti, G.; Neva, F.; Coffetti, G.; Rimoldi, I. Chiral 8-Amino-5,6,7,8-tetrahydroquinoline Derivatives in Metal Catalysts for the Asymmetric Transfer Hydrogenation of 1-Aryl Substituted-3,4-dihydroisoquinolines as Alkaloids Precursors. Molecules 2023, 28, 1907. https://doi.org/10.3390/molecules28041907
Facchetti G, Neva F, Coffetti G, Rimoldi I. Chiral 8-Amino-5,6,7,8-tetrahydroquinoline Derivatives in Metal Catalysts for the Asymmetric Transfer Hydrogenation of 1-Aryl Substituted-3,4-dihydroisoquinolines as Alkaloids Precursors. Molecules. 2023; 28(4):1907. https://doi.org/10.3390/molecules28041907
Chicago/Turabian StyleFacchetti, Giorgio, Francesca Neva, Giulia Coffetti, and Isabella Rimoldi. 2023. "Chiral 8-Amino-5,6,7,8-tetrahydroquinoline Derivatives in Metal Catalysts for the Asymmetric Transfer Hydrogenation of 1-Aryl Substituted-3,4-dihydroisoquinolines as Alkaloids Precursors" Molecules 28, no. 4: 1907. https://doi.org/10.3390/molecules28041907
APA StyleFacchetti, G., Neva, F., Coffetti, G., & Rimoldi, I. (2023). Chiral 8-Amino-5,6,7,8-tetrahydroquinoline Derivatives in Metal Catalysts for the Asymmetric Transfer Hydrogenation of 1-Aryl Substituted-3,4-dihydroisoquinolines as Alkaloids Precursors. Molecules, 28(4), 1907. https://doi.org/10.3390/molecules28041907