
Recent Advances in N-O Bond Cleavage of Oximes and Hydroxylamines to Construct N-Heterocycle

{kind=link}
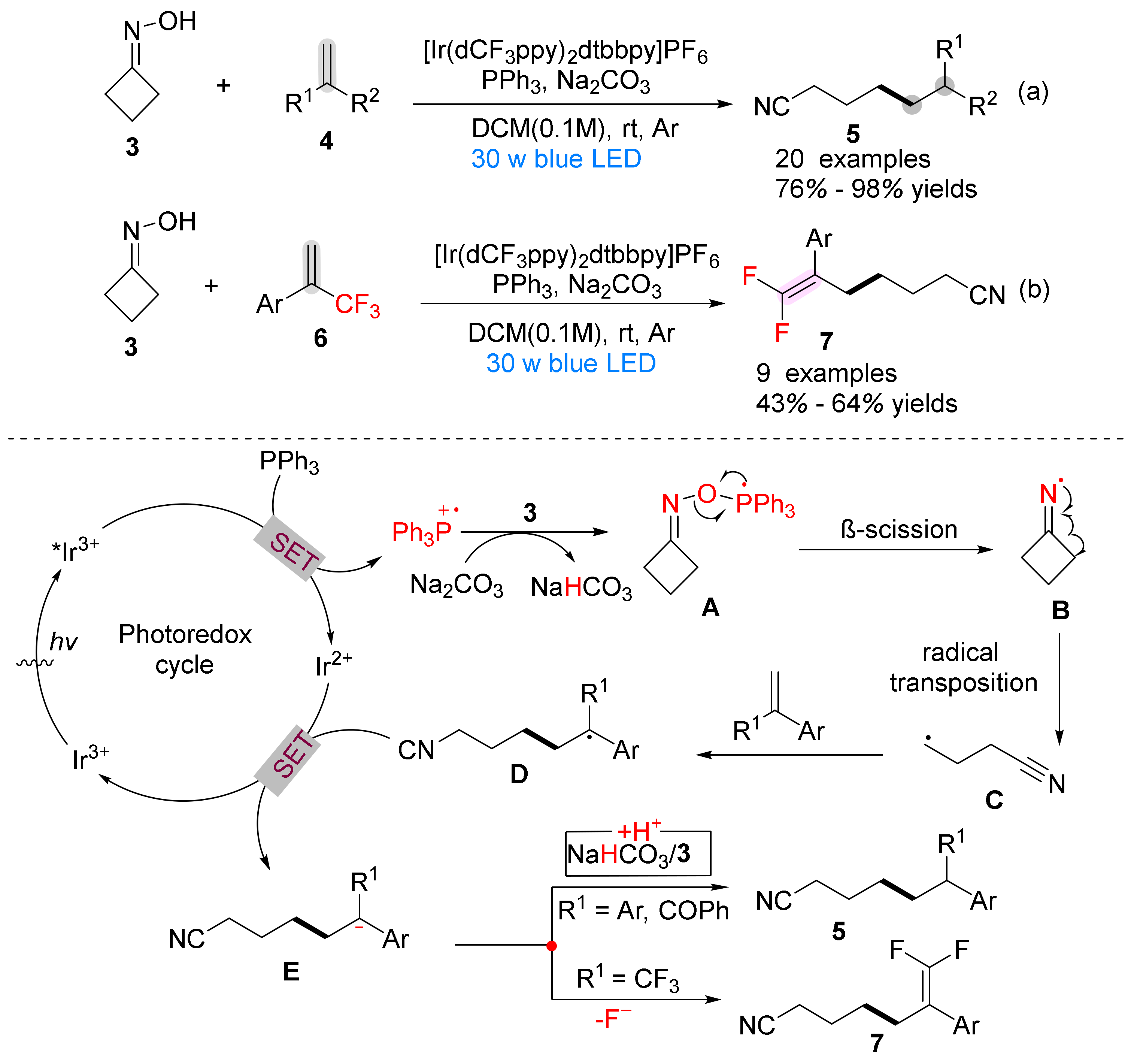{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
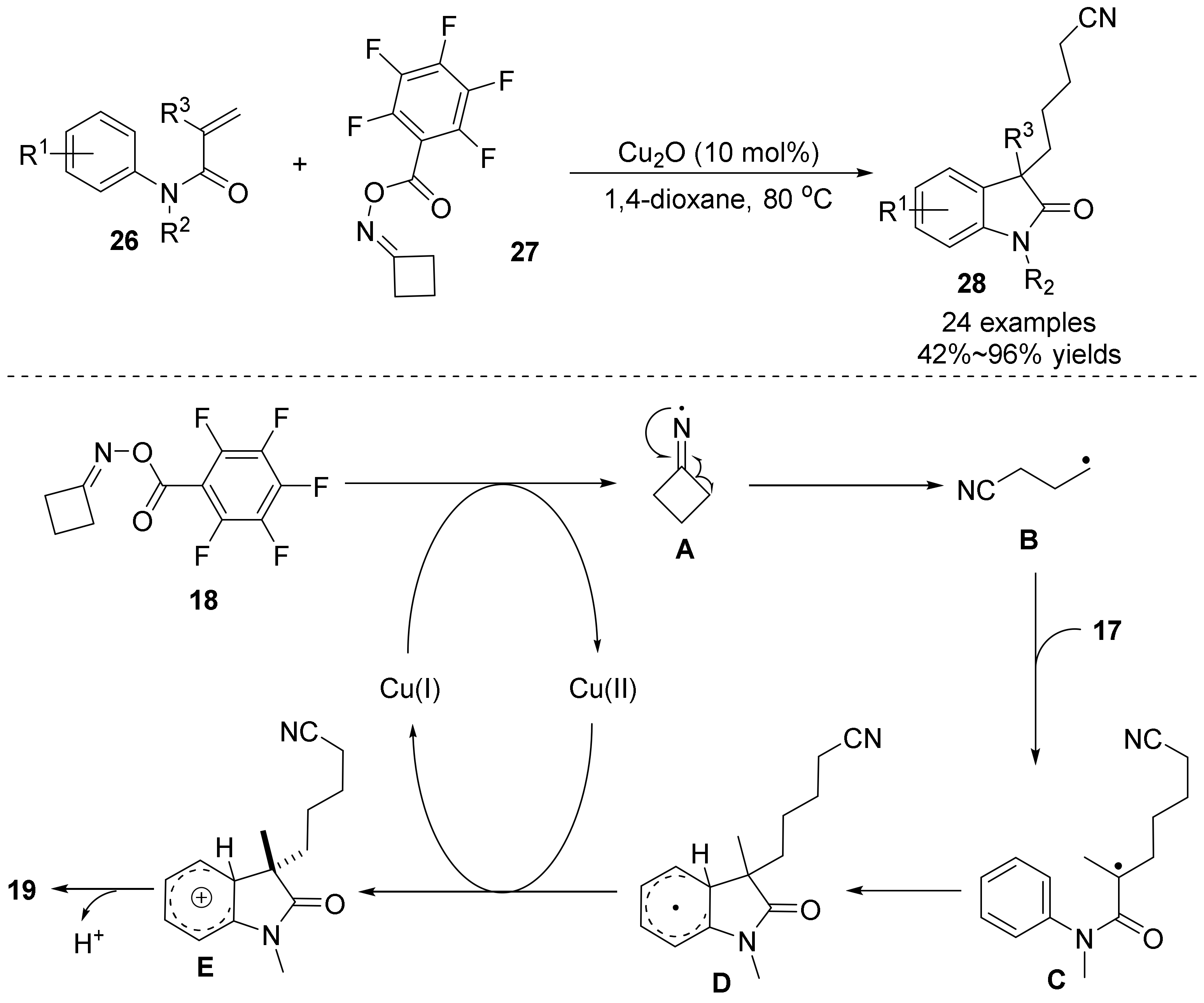{kind=link}
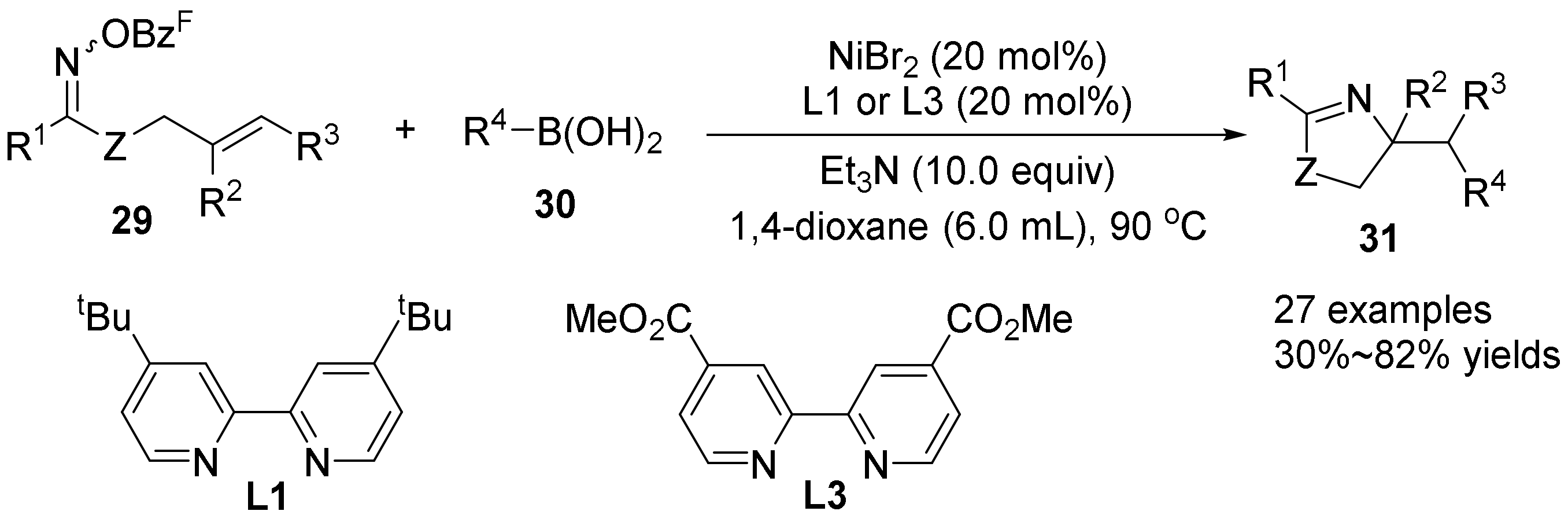{kind=link}
{kind=link}
{kind=link}
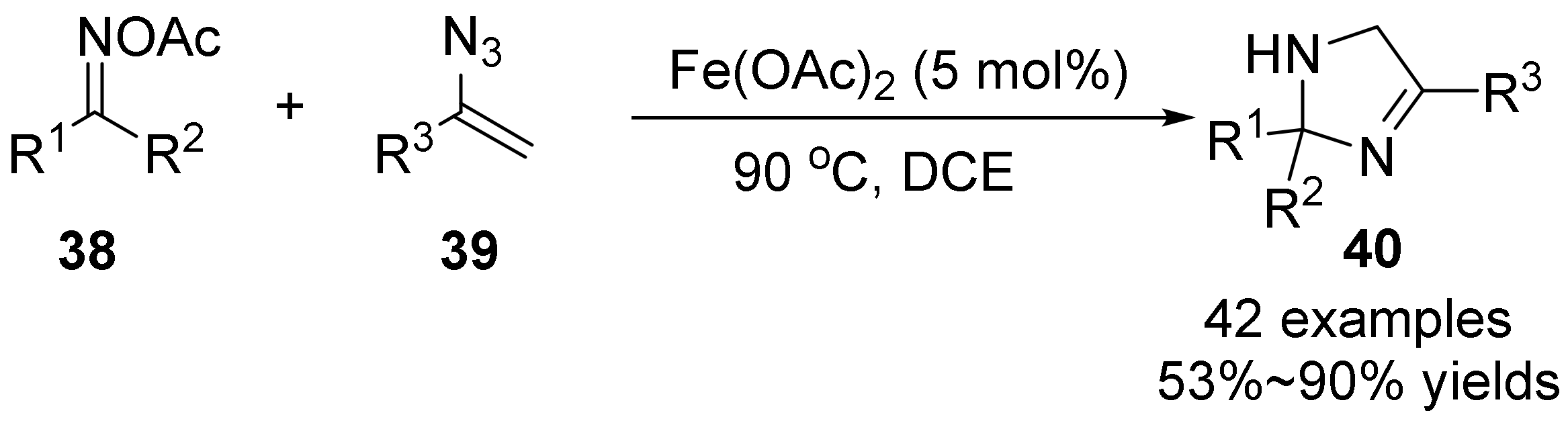{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The N-O Bond Cleavage of Oximes to Construct N-Heterocycle
2.1. Hydroxy Oximes
2.2. The N-O Bond Cleavage of the Oxime Esters to Construct 2H-Azirines
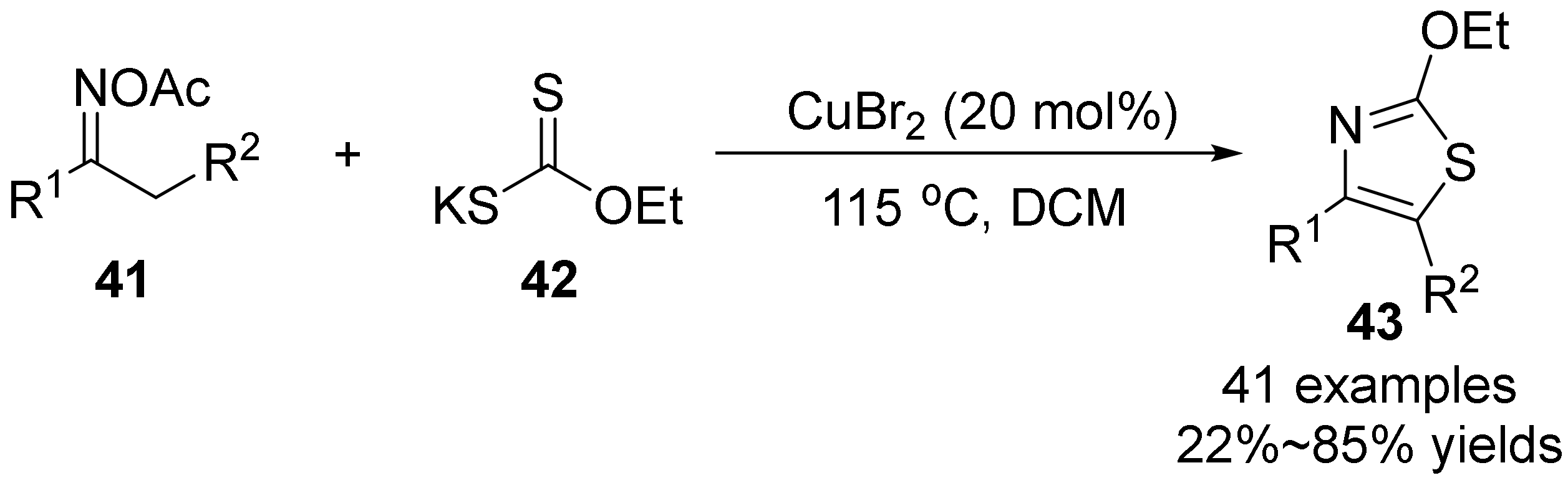
2.3. The N-O Bond Cleavage of the Oxime Esters to Construct Five-Membered N-Heterocycle
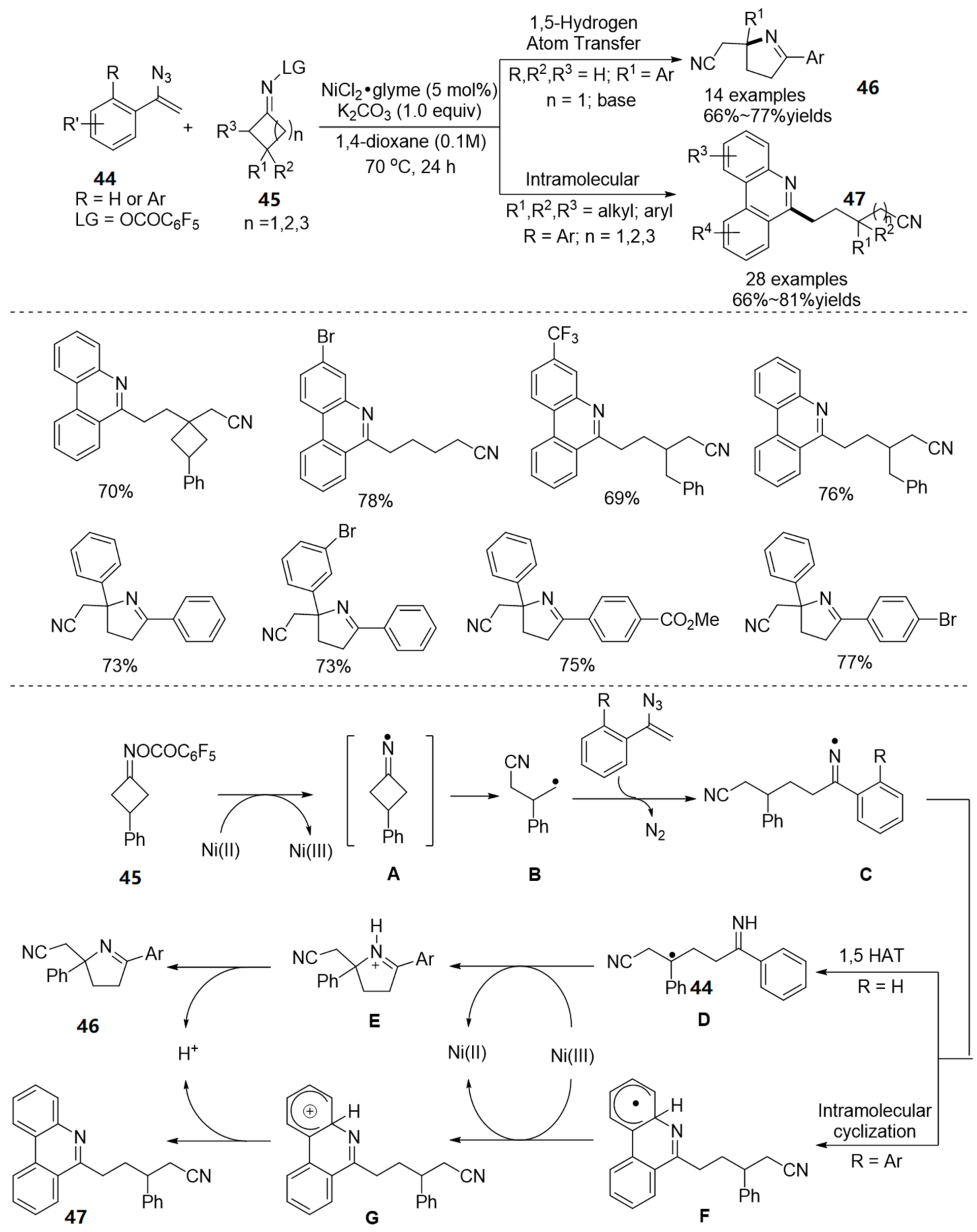
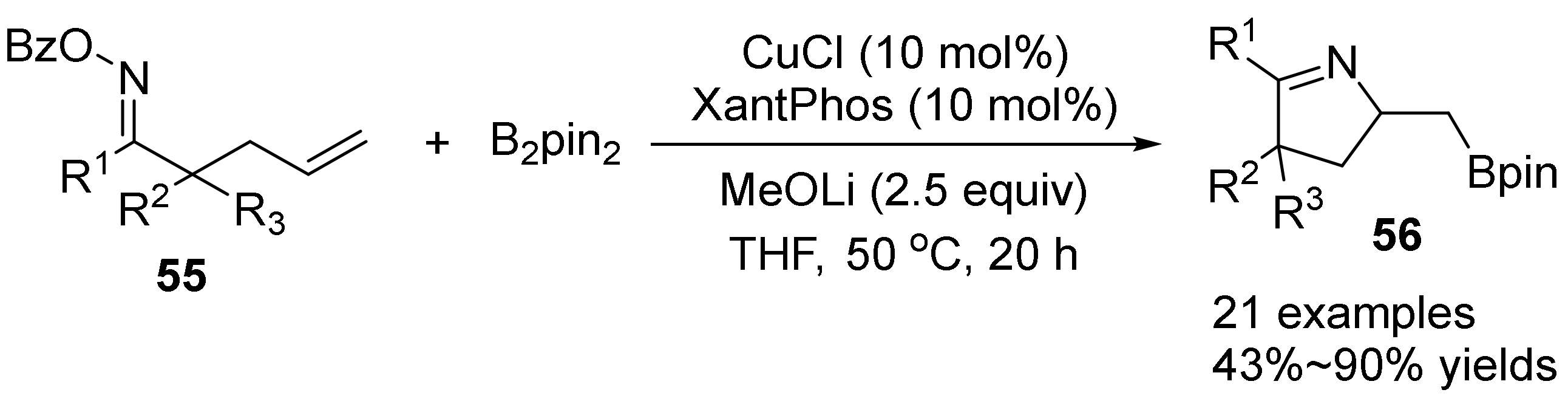
2.4. The N-O Bond Cleavage of Oxime Esters to form Spirocyclic Compounds
2.5. The N-O Bond Cleavage of Oxime Esters to form Pyridine Derivatives
3. The N-O Bond Cleavage of Hydroxylamine to Construct N-Heterocycles
4. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, J.; Hu, Y.; Zhang, D.; Liu, Q.; Dong, Y.; Liu, H. Transition Metal-Catalyzed Reactions Involving Oximes. Adv. Synth. Catal. 2017, 359, 710–771. [Google Scholar] [CrossRef]
- Murahashi, S.I.; Imada, Y. Synthesis and Transformations of Nitrones for Organic Synthesis. Chem. Rev. 2019, 119, 4684–4716. [Google Scholar] [CrossRef] [PubMed]
- Bolotin, D.S.; Bokach, N.A.; Demakova, M.Y.; Kukushkin, V.Y. Metal-Involving Synthesis and Reactions of Oximes. Chem. Rev. 2017, 117, 13039–13122. [Google Scholar] [CrossRef] [PubMed]
- Tabolin, A.A.; Ioffe, S.L. Rearrangement of N-Oxyenamines and Related Reactions. Chem. Rev. 2014, 114, 5426–5476. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.B.; March, J. March’s Advanced Organic Chemistry: Reactions, Mechanisms and Structure, 6th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2019; pp. 105–131. [Google Scholar]
- Ābele, E.; Lukevics, E. Recent Advances in the Synthesis of Heterocycles from Oximes. Heterocycles 2000, 53, 2285–2336. [Google Scholar]
- Latrache, M.; Hoffmann, N. Photochemical radical cyclization reactions with imines, hydrazones, oximes and related compounds Recent Advances in the Synthesis of Heterocycles from Oximes. Chem. Soc. Rev. 2021, 50, 7418–7435. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Ji, X.; Wu, W.; Jiang, H. Transition metal-catalyzed C–H functionalization of N-oxyenamine internal oxidants. Chem. Soc. Rev. 2015, 44, 1155–1171. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Wu, J. Recent advances for the photoinduced C-C bond cleavage of cycloketone oximes. Chin. Chem. Lett. 2020, 31, 3083–3094. [Google Scholar]
- Xiao, F.; Guo, Y.; Zeng, Y.-F. Recent Developments in Radical Cross-Coupling of Redox-Active Cycloketone Oximes. Adv. Synth. Catal. 2021, 363, 120–143. [Google Scholar] [CrossRef]
- Zhao, P.; Wang, F.; Han, K.; Li, X. Rhodium(III)-Catalyzed Cyclization–Olefination of N-Acetoxyl Ketoimine-Alkynes. Org. Lett. 2012, 14, 3400–3403. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Huang, L.; Qi, C.; Wu, W.; Jiang, H. An efficient synthesis of polysubstituted pyrroles via copper-catalyzed coupling of oxime acetates with dialkyl acetylenedicarboxylates under aerobic conditions. Chem. Commun. 2013, 49, 9597–9599. [Google Scholar] [CrossRef] [PubMed]
- Krylov, I.B.; Segida, O.O.; Budnikov, A.S.; Terent’eva, A.O. Oxime-Derived Iminyl Radicals in Selective Processes of Hydrogen Atom Transfer and Addition to Carbon-Carbon π-Bonds. Adv. Synth. Catal. 2021, 363, 2502–2528. [Google Scholar] [CrossRef]
- Liao, J.-Y.; Wu, Q.-Y.; Lu, X.; Zou, N.; Pan, C.-X.; Liang, C.; Su, G.-F.; Mo, D.-L. A copper-catalyzed diastereoselective O-transfer reaction of N-vinyl-α,β-unsaturated nitrones with ketenes into γ-lactones through [5 + 2] cycloaddition and N–O bond cleavage. Green Chem. 2019, 21, 6567–6573. [Google Scholar] [CrossRef]
- Song, L.; Zhang, X.; Tang, X.; Van Meervelt, L.; Van der Eycken, J.; Harvey, J.N.; Van der Eycken, E.V. Ruthenium-catalyzed cascade C-H activation/annulation of N-alkoxybenzamides: Reaction development and mechanistic insight. Chem. Sci. 2020, 11, 11562–11569. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.-P.; Li, K.; Wu, S.-Y.; Liang, C.; Su, G.-F.; Mo, D.-L. Construction of 2,3-quaternary fused indolines from alkynyl tethered oximes and diaryliodonium salts through a cascade strategy of N-arylation/cycloaddition/[3,3]-rearrangement. Green Chem. 2017, 19, 5761–5766. [Google Scholar] [CrossRef]
- Takeda, N.; Futaki, E.; Kobori, Y.; Ueda, M.; Miyata, O. Nucleophilic Arylation of N,O-Ketene Acetals with Triaryl Aluminum Reagents: Access to alpha-Aryl Amides through an Umpolung Process. Angew. Chem. Int. Ed. 2017, 56, 16342–16346. [Google Scholar] [CrossRef]
- Budnikov, A.S.; Krylov, I.B.; Lastovko, A.V.; Yu, B.; Terent’ev, A.O. N-Alkoxyphtalimides as Versatile Alkoxy Radical Precursors in Modern Organic Synthesis. Asian J. Org. Chem. 2022, 11, e202200262. [Google Scholar] [CrossRef]
- Usuki, A.; Kojima, Y.; Okada, A.; Kawasumi, M.; Fukushima, Y.; Kurauchi, T.; Kamigaito, O. Synthesis of nylon 6-clay hybrid. J. Mater. Res. 1993, 8, 1179–1184. [Google Scholar] [CrossRef]
- Si, Y.F.; Lv, Q.Y.; Yu, B. Radical Cascade Reactions of β,γ-Unsaturated Hydrazones/Oximes. Adv. Synth. Catal. 2021, 363, 4640–4666. [Google Scholar] [CrossRef]
- Wang, T.; Wang, Y.N.; Wang, R.; Zhang, B.C.; Yang, C.; Li, Y.L.; Wang, X.S. Enantioselective cyanation via radical-mediated C-C single bond cleavage for synthesis of chiral dinitriles. Nat. Commun. 2019, 10, 5373. [Google Scholar] [CrossRef]
- Yang, S.; Wang, L.; Wang, L.; Li, H. Visible-Light Photoredox-Catalyzed Regioselective Sulfonylation of Alkenes Assisted by Oximes via [1,5]-H Migration. J. Org. Chem. 2020, 85, 564–573. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Jiang, K.; Du, F.; Yang, J.; Shuai, L.; Ouyang, Q.; Chen, Y.-C.; Wei, Y. Iron-Catalyzed, Iminyl Radical-Triggered Cascade 1,5-Hydrogen Atom Transfer/(5+2) or (5+1) Annulation: Oxime as a Five-Atom Assembling Unit. Angew. Chem. Int. Ed. 2020, 59, 19222–19228. [Google Scholar] [CrossRef] [PubMed]
- Baars, H.; Engel, J.; Mertens, L.; Meister, D.; Bolm, C. The Reactivity of Difluorocarbene with Hydroxylamines: Synthesis of Carbamoyl Fluorides. Adv. Synth. Catal. 2016, 358, 2293–2299. [Google Scholar] [CrossRef]
- Chen, F.; Zhu, C.; Jiang, H. [3 + 1 + 1] Annulation Reaction of Benzo-1,2-Quinones, Aldehydes and Hydroxylamine Hydrochloride: Access to Benzoxazoles with Inorganic Nitrogen Source. Adv. Synth. Catal. 2021, 363, 2124–2132. [Google Scholar] [CrossRef]
- Yang, Z.; Jiang, K.; Chen, Y.C.; Wei, Y. Copper-Catalyzed Dihydroquinolinone Synthesis from Isocyanides and O-Benzoyl Hydroxylamines. J. Org. Chem. 2019, 84, 3725–3734. [Google Scholar] [CrossRef]
- Chen, J.; Xu, Y.; Shao, W.; Ji, J.; Wang, B.; Yang, M.; Mao, G.; Xiao, F.; Deng, G.J. Pd-Catalyzed C-O Bond Formation Enabling the Synthesis of Congested N,N,O-Trisubstituted Hydroxylamines. Org. Lett. 2022, 24, 8271–8276. [Google Scholar] [CrossRef]
- Fan, L.; Hao, J.; Yu, J.; Ma, X.; Liu, J.; Luan, X. Hydroxylamines as Bifunctional Single-Nitrogen Sources for the Rapid Assembly of Diverse Tricyclic Indole Scaffolds. J. Am. Chem. Soc. 2020, 142, 6698–6707. [Google Scholar] [CrossRef]
- Wang, S.; Guo, Y.Q.; Ren, Z.H.; Wang, Y.Y.; Guan, Z.H. K2CO3-Mediated Cyclization and Rearrangement of gamma,delta-Alkynyl Oximes To Form Pyridols. Org. Lett. 2017, 19, 1574–1577. [Google Scholar] [CrossRef]
- Xia, P.-J.; Ye, Z.-P.; Hu, Y.-Z.; Song, D.; Xiang, H.-Y.; Chen, X.-Q.; Yang, H. Photocatalytic, Phosphoranyl Radical-Mediated N–O Cleavage of Strained Cycloketone Oximes. Org. Lett. 2019, 21, 2658–2662. [Google Scholar] [CrossRef]
- Qi, Z.; Wang, S. Chemodivergent Synthesis of Oxazoles and Oxime Ethers Initiated by Selective C-N/C-O Formation of Oximes and Diazo Esters. Org. Lett. 2021, 23, 8549–8553. [Google Scholar] [CrossRef]
- Qi, Z.; Wang, S. Copper-Catalyzed beta-Lactam Formation Initiated by 1,3-Azaprotio Transfer of Oximes and Methyl Propiolate. Org. Lett. 2021, 23, 5777–5781. [Google Scholar] [CrossRef]
- Zhao, J.-Q.; Yue, D.-F.; Zhang, X.-M.; Xu, X.-Y.; Yuan, W.-C. The organocatalytic asymmetric Neber reaction for the enantioselective synthesis of spirooxindole 2H-azirines. Org. Biomol. Chem. 2016, 14, 10946–10952. [Google Scholar] [CrossRef] [PubMed]
- Ning, Y.; Otani, Y.; Ohwada, T. Contrasting C- and O-Atom Reactivities of Neutral Ketone and Enolate Forms of 3-Sulfonyloxyimino-2-methyl-1-phenyl-1-butanones. J. Org. Chem. 2018, 83, 203–219. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.-N.; Zhang, W.; Wang, X.-C.; Zhang, Y.; Yang, D.-S.; Guan, Z.-H. Modular 2,3-diaryl-2H-azirine synthesis from ketoxime acetates via Cs2CO3-mediated cyclization. Org. Biomol. Chem. 2018, 16, 4333–4337. [Google Scholar] [CrossRef]
- Huang, Y.-J.; Qiao, B.; Zhang, F.-G.; Ma, J.-A. Facile construction of trifluoromethyl-azirines via one-pot metal-free Neber reaction. Tetrahedron 2018, 74, 3791–3796. [Google Scholar] [CrossRef]
- Huang, H.; Cai, J.; Ji, X.; Xiao, F.; Chen, Y.; Deng, G.J. Internal Oxidant-Triggered Aerobic Oxygenation and Cyclization of Indoles under Copper Catalysis. Angew. Chem. Int. Ed. 2016, 55, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhang, J.Y.; Gao, P.; Xu, S.L.; Guo, L.N. Copper-Catalyzed Redox-Neutral Cyanoalkylarylation of Activated Alkenes with Cyclobutanone Oxime Esters. J. Org. Chem. 2018, 83, 1046–1055. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.J.; Duan, X.H.; Wu, Y.; Yang, J.C.; Guo, L.N. Transition-metal free C-C bond cleavage/borylation of cycloketone oxime esters. Chem. Sci. 2019, 10, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.-B.; Pathipati, S.R.; Selander, N. Nickel-Catalyzed 1,2-Aminoarylation of Oxime Ester-Tethered Alkenes with Boronic Acids. ACS Catal. 2017, 7, 8441–8445. [Google Scholar] [CrossRef]
- Zhu, C.; Zeng, H.; Chen, F.; Liu, C.; Zhu, R.; Wu, W.; Jiang, H. Copper-catalyzed coupling of oxime acetates and aryldiazonium salts: An azide-free strategy toward N-2-aryl-1,2,3-triazoles. Org. Chem. Front. 2018, 5, 571–576. [Google Scholar] [CrossRef]
- Gao, S.; Gao, L.; Meng, H.; Luo, M.; Zeng, X. Iron-catalyzed synthesis of benzoxazoles by oxidative coupling/cyclization of phenol derivatives with benzoyl aldehyde oximes. Chem. Commun. 2017, 53, 9886–9889. [Google Scholar] [CrossRef]
- Zhu, Z.; Tang, X.; Li, J.; Li, X.; Wu, W.; Deng, G.; Jiang, H. Iron-Catalyzed Synthesis of 2H-Imidazoles from Oxime Acetates and Vinyl Azides under Redox-Neutral Conditions. Org. Lett. 2017, 19, 1370–1373. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Tang, X.; Cen, J.; Li, J.; Wu, W.; Jiang, H. Copper-catalyzed synthesis of thiazol-2-yl ethers from oxime acetates and xanthates under redox-neutral conditions. Chem. Commun. 2018, 54, 3767–3770. [Google Scholar] [CrossRef]
- Tang, Y.Q.; Yang, J.C.; Wang, L.; Fan, M.; Guo, L.N. Ni-Catalyzed Redox-Neutral Ring-Opening/Radical Addition/Ring-Closing Cascade of Cycloketone Oxime Esters and Vinyl Azides. Org. Lett. 2019, 21, 5178–5182. [Google Scholar] [CrossRef]
- Wang, Y.; Ding, J.; Zhao, J.; Sun, W.; Lian, C.; Chen, C.; Zhu, B. Iminyl radical-promoted imino sulfonylation, imino cyanogenation and imino thiocyanation of γ,δ-unsaturated oxime esters: Synthesis of versatile functionalized pyrrolines. Org. Chem. Front. 2019, 6, 2240–2244. [Google Scholar] [CrossRef]
- Chen, C.; Bao, Y.; Zhao, J.; Zhu, B. Silver-promoted cascade radical cyclization of gamma,delta-unsaturated oxime esters with P(O)H compounds: Synthesis of phosphorylated pyrrolines. Chem. Commun. 2019, 55, 14697–14700. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wu, X.-F. Copper-catalyzed borylative cyclization of γ,δ-unsaturated aromatic oxime esters to (borylmethyl)pyrrolidines. Org. Chem. Front. 2020, 7, 3382–3386. [Google Scholar] [CrossRef]
- Zhang, Y.-C.; Wu, X.-F. Iron-catalyzed carbonylative cyclization of γ,δ-unsaturated aromatic oxime esters with amines. Chem. Commun. 2020, 56, 14605–14608. [Google Scholar] [CrossRef]
- Miao, C.B.; Zheng, A.Q.; Zhou, L.J.; Lyu, X.; Yang, H.T. Copper-Catalyzed Annulation of Oxime Acetates with alpha-Amino Acid Ester Derivatives: Synthesis of 3-Sulfonamido/Imino 4-Pyrrolin-2-ones. Org. Lett. 2020, 22, 3381–3385. [Google Scholar] [CrossRef]
- Yu, J.X.; Teng, F.; Xiang, J.N.; Deng, W.; Li, J.H. One-Carbon Incorporation Using Cyclobutanone Oxime Ester Enabled [2 + 2 + 1] Carboannulation of 1,7-Enynes by C-C/N-O Bond Cleavage and C-H Functionalization. Org. Lett. 2019, 21, 9434–9437. [Google Scholar] [CrossRef]
- Jia, X.G.; Yao, Q.W.; Shu, X.-Z. Enantioselective Reductive N-Cyclization-Alkylation Reaction of Alkene-Tethered Oxime Esters and Alkyl Iodides by Nickel Catalysis. J. Am. Chem. Soc. 2022, 144, 13461–13467. [Google Scholar] [CrossRef]
- Liu, L.; Jian, Y.; Hu, W.; Zhao, S.; Shi, Z.-J.; Selander, N.; Zhou, T. Ni and Fe catalyzed cascade radical reactions of oxime esters with diselenides. Org. Chem. Front. 2022, 9, 3480–3485. [Google Scholar] [CrossRef]
- Zhao, B.; Liang, H.-W.; Yang, J.; Yang, Z.; Wei, Y. Copper-Catalyzed Intermolecular Cyclization between Oximes and Alkenes: A Facile Access to Spiropyrrolines. ACS Catal. 2017, 7, 5612–5617. [Google Scholar] [CrossRef]
- Sun, L.; Liu, B.; Zhao, Y.; Chang, J.; Kong, L.; Wang, F.; Deng, W.-Q.; Li, X. Rhodium(iii)-catalyzed asymmetric [4+1] spiroannulations of O-pivaloyl oximes with α-diazo compounds. Chem. Commun. 2021, 57, 8268–8271. [Google Scholar] [CrossRef]
- Huang, H.; Cai, J.; Tang, L.; Wang, Z.; Li, F.; Deng, G.J. Metal-Free Assembly of Polysubstituted Pyridines from Oximes and Acroleins. J. Org. Chem. 2016, 81, 1499–1505. [Google Scholar] [CrossRef]
- Fu, Y.; Wang, P.; Guo, X.; Wu, P.; Meng, X.; Chen, B. Synthesis of Polyfunctional Pyridines via Copper-Catalyzed Oxidative Coupling Reactions. J. Org. Chem. 2016, 81, 11671–11677. [Google Scholar] [CrossRef]
- Tan, W.W.; Ong, Y.J.; Yoshikai, N. Synthesis of Highly Substituted Pyridines through Copper-Catalyzed Condensation of Oximes and alpha,beta-Unsaturated Imines. Angew. Chem. Int. Ed. 2017, 56, 8240–8244. [Google Scholar] [CrossRef] [PubMed]
- Bai, D.; Wang, X.; Zheng, G.; Li, X. Redox-Divergent Synthesis of Fluoroalkylated Pyridines and 2-Pyridones through Cu-Catalyzed N-O Cleavage of Oxime Acetates. Angew. Chem. Int. Ed. 2018, 57, 6633–6637. [Google Scholar] [CrossRef] [PubMed]
- Zhan, J.-L.; Wu, M.-W.; Wei, D.; Wei, B.-Y.; Jiang, Y.; Yu, W.; Han, B. 4-HO-TEMPO-Catalyzed Redox Annulation of Cyclopropanols with Oxime Acetates toward Pyridine Derivatives. ACS. Catal. 2019, 9, 4179–4188. [Google Scholar] [CrossRef]
- Yang, X.; Liu, S.; Yu, S.; Kong, L.; Lan, Y.; Li, X. Redox-Neutral Access to Isoquinolinones via Rhodium(III)-Catalyzed Annulations of O-Pivaloyl Oximes with Ketenes. Org. Lett. 2018, 20, 2698–2701. [Google Scholar] [CrossRef] [PubMed]
- Sabir, S.; Pandey, C.B.; Yadav, A.K.; Tiwari, B.; Jat, J.L. Direct N-H/N-Me Aziridination of Unactivated Olefins Using O-(Sulfonyl)hydroxylamines as Aminating Agents. J. Org. Chem. 2018, 83, 12255–12260. [Google Scholar] [CrossRef]
- Farndon, J.J.; Young, T.A.; Bower, J.F. Stereospecific Alkene Aziridination Using a Bifunctional Amino-Reagent: An Aza-Prilezhaev Reaction. J. Am. Chem. Soc. 2018, 140, 17846–17850. [Google Scholar] [CrossRef]
- Kirby, G.; Grimaud, L.; Vitale, M.R.; Prestat, G.; Berhal, F. Iron(ii)-catalyzed intermolecular aziridination of alkenes employing hydroxylamine derivatives as clean nitrene sources. Green Chem. 2021, 23, 9428–9432. [Google Scholar] [CrossRef]
- Xiong, Z.; Wang, J.; Wang, Y.; Luo, S.; Zhu, Q. Palladium-catalyzed C(sp2)–H aminoimidoylation of isocyano-containing arenes: Synthesis of amino substituted N-heterocycles. Org. Chem. Front. 2017, 4, 1768–1771. [Google Scholar] [CrossRef]
- Ueda, M. Radical Addition-Initiated Domino Reactions of Conjugated Oxime Ethers. Chem. Pharm. Bull. 2014, 62, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Chen, P.; Liu, G. Copper-Catalyzed Radical Relay for Asymmetric Radical Transformations. Acc. Chem. Res. 2018, 51, 2036–2046. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Liang, Y.-J.; Wang, P.-Z.; Li, G.-Q.; Zhang, B.; Qian, H.; Huan, X.-D.; Guan, W.; Xiao, W.-J.; Chen, J.-R. Photoinduced Copper-Catalyzed Asymmetric C–O Cross-Coupling. J. Am. Chem. Soc. 2021, 14, 13382–13392. [Google Scholar] [CrossRef] [PubMed]
- Tsymbal, A.V.; Bizzini, L.D.; MacMillan, D.W.C. Nickel Catalysis via SH2 Homolytic Substitution: The Double Decarboxylative Cross-Coupling of Aliphatic Acids. J. Am. Chem. Soc. 2022, 144, 21278–21286. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.Y.; Perry, I.B.; Bissonnette, N.B.; Buksh, B.F.; Edwards, G.A.; Frye, L.I.; Garry, O.L.; Lavagnino, M.N.; Li, B.X.; Liang, Y.; et al. Metallaphotoredox: The Merger of Photoredox and Transition Metal Catalysis. Chem. Rev. 2022, 122, 1485–1542. [Google Scholar] [CrossRef]
- Yu, X.-Y.; Zhao, Q.-Q.; Chen, J.; Xiao, W.-J.; Chen, J.-R. When Light Meets Nitrogen-Centered Radicals: From Reagents to Catalysts. Acc. Chem. Res. 2020, 53, 1066–1083. [Google Scholar] [CrossRef]
- Chen, J.-R.; Hu, X.-Q.; Lu, L.-Q.; Xiao, W.-J. Visible Light Photoredox-Controlled Reactions of N-Radicals and Radical Ions. Chem. Soc. Rev. 2016, 45, 2044–2056. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xia, W. Recent Advances in Radical-Based C-N Bond Formation via Photo-/Electrochemistry. Chem. Soc. Rev. 2018, 47, 2591–2608. [Google Scholar] [CrossRef] [PubMed]





































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, H.-M.; Zhao, Y.-L.; Sun, Q.; Ouyang, X.-H.; Li, J.-H. Recent Advances in N-O Bond Cleavage of Oximes and Hydroxylamines to Construct N-Heterocycle. Molecules 2023, 28, 1775. https://doi.org/10.3390/molecules28041775
Jiang H-M, Zhao Y-L, Sun Q, Ouyang X-H, Li J-H. Recent Advances in N-O Bond Cleavage of Oximes and Hydroxylamines to Construct N-Heterocycle. Molecules. 2023; 28(4):1775. https://doi.org/10.3390/molecules28041775
Chicago/Turabian StyleJiang, Hui-Min, Yi-Lin Zhao, Qing Sun, Xuan-Hui Ouyang, and Jin-Heng Li. 2023. "Recent Advances in N-O Bond Cleavage of Oximes and Hydroxylamines to Construct N-Heterocycle" Molecules 28, no. 4: 1775. https://doi.org/10.3390/molecules28041775
APA StyleJiang, H.-M., Zhao, Y.-L., Sun, Q., Ouyang, X.-H., & Li, J.-H. (2023). Recent Advances in N-O Bond Cleavage of Oximes and Hydroxylamines to Construct N-Heterocycle. Molecules, 28(4), 1775. https://doi.org/10.3390/molecules28041775