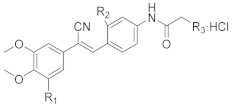
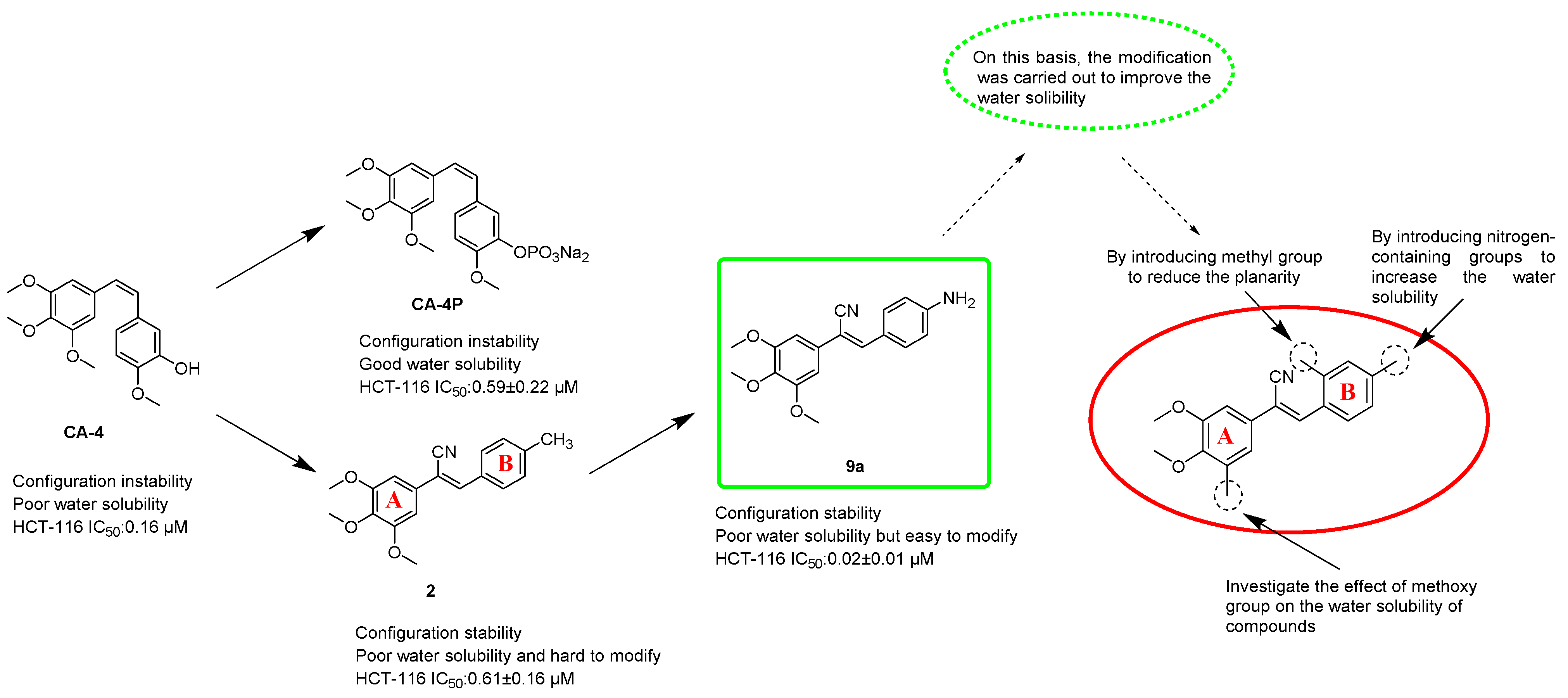
2.2.1. Evaluation of the Cytotoxicity and Aqueous Solubility In Vitro
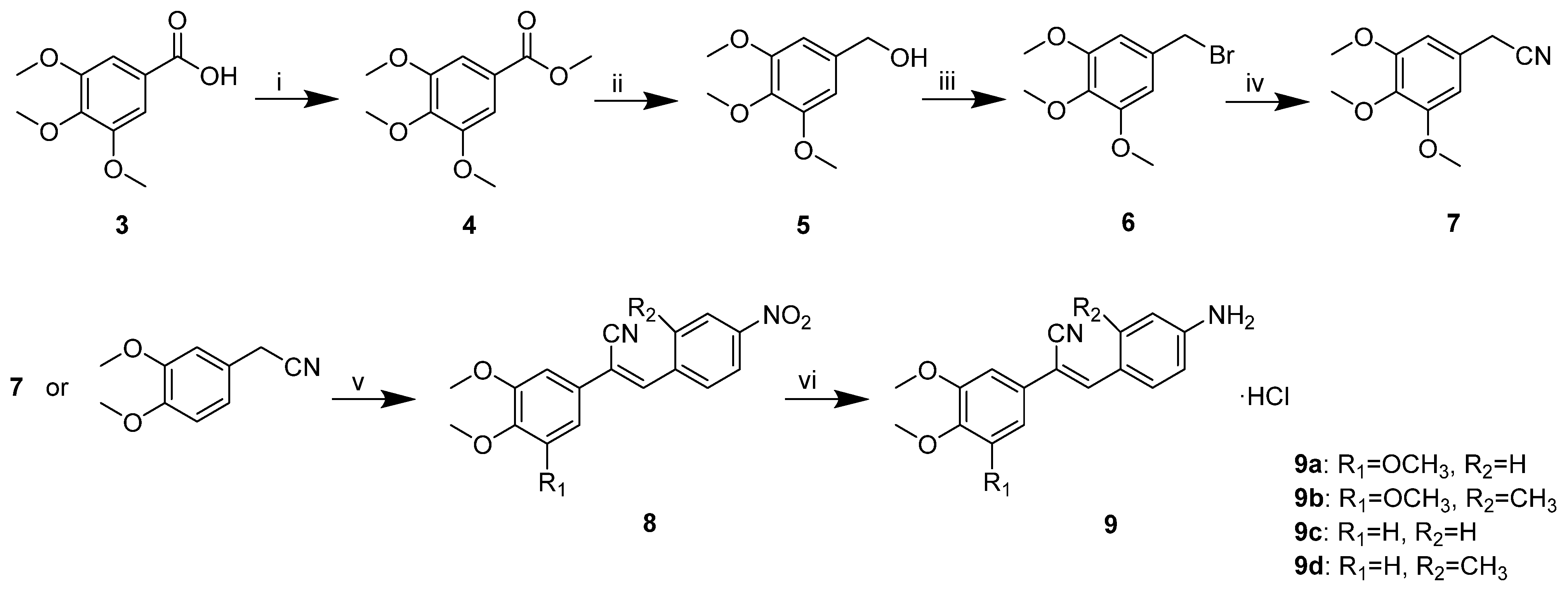
Initially, we designed and synthesised seven compounds—
9a and
12a1–
12a6—and evaluated the anticancer activity and aqueous solubility. We evaluated the cytotoxic effects of these stilbene nitrile analogues against a panel of four human cancer cell lines (HCT-116, BEL-7402, MCF-7 and AGS) and two human non-cancerous normal cell lines (L-02 and MCF-10A), using CA-4 as a control. The compounds were screened for aqueous solubility in 0.01 M PBS using a high throughput UV method [
28,
29,
30]. The results are shown in
Table 3. Compared with
9a,
12a1–
12a6 showed reduced activity against cancer cells. The activity of the six different nitrogenous compounds
12a1–
12a6 against cancer cells was compared, and it was found that the changes in these compounds had little effect on the rate of growth inhibition of the cancer cells. It is possible that the size of the nitrogen heterocycle had little effect on the activity of these compounds. The compounds containing a morpholine ring were generally slightly less active against cancer cells than the other nitrogen-containing compounds. The aqueous solubility of all the compounds
12a1–
12a6 was higher than that of
9a, particularly that of
12a1 and
12a2 containing piperazine rings. The water solubility of
12a1 and
12a2 was at least 1687 and 2494 times higher than that of
9a. Of the six compounds with improved aqueous solubility,
12a1 had the best activity. These results indicated that the introduction of a piperazine ring gave the best results for increasing the solubility of the stilbene nitrile parent structure compared with the other amine compounds.
Another strategy to improve the aqueous solubility of compounds is to reduce the planarity [
27]. Therefore, we introduced a methyl group into the
meta position of the benzene B ring to give
9b and the derivatives
12b1–2, which contained a piperazine ring (
Table 2; notably, similar to the
12a series, a series of compounds containing the other four nitrogen-containing groups (morpholine, pyrrolidine, diethylamine and 4-piperidinemethanol) were also synthesised. Still, these compounds had low activity and poor aqueous solubility). By synthesizing
9c and
9d and the respective derivatives,
12c1–2 and
12d1–2 (
Table 2), we could also explore the effect of the methoxy group on the solubility of the stilbene nitrile parent. The anticancer activity and aqueous solubility of all the synthesised compounds were determined, as before, to explore the structure–solubility relationship. The results are shown in
Table 4.
The results showed that the aqueous solubility of 9b, 9d, 12b1–2 and 12d1–2 decreased when a methyl group was introduced into the meta position of the benzene B ring. This was inconsistent with our previous expectations. Compared with 9a, the aqueous solubility of the methyl-containing 9b was decreased by nine times, and this effect was more marked in compounds 12b1–2 compared with 12a1–2. In addition, compounds 9b and 12b1–2 showed a slight decrease in the activity against cancer cells, compared with 9a and 12a1–2. The toxicity of 9b and 12b1 toward two types of normal cells was increased, but the toxicity of 12b2 toward normal L-02 cells was reduced, compared with 9a and 12a1–2. A similar trend for the anticancer activity and toxicity was observed between compounds 9c and 9d and the piperazine ring-containing derivatives 12c1–2 and 12d1–2. In short, the results in several cells we screened indicated that compounds without a methyl group on the benzene B ring had increased anticancer activity, increased aqueous solubility and lower toxicity compared with compounds with a methyl group.
The effect of the methoxy group of the benzene A ring on the activity and solubility was investigated by comparing
9a and
9c with the respective piperazine derivatives
12a1–2 and
12c1–2. The activities of
12c1–2 against several cancer cells screened were lower than those of
12a1–2, respectively. It was evident that the three methoxy groups on the benzene A ring are an important part of the pharmacophore, which was in agreement with previous studies [
3,
4,
30]. The solubility was also decreased when one of the methoxy groups of the benzene A ring was replaced with hydrogen (
Table 3 and
Table 4). This result indicated that when the number of methoxy groups on the benzene A ring was decreased, the lipophilicity of the parent stilbene nitrile was enhanced. This enhancement can offset the effect of the piperazine ring on the aqueous solubility of the parent structure. In summary, the trimethoxyphenyl structure of the parent stilbene nitrile is important for the anticancer activity and water solubility of the compound.
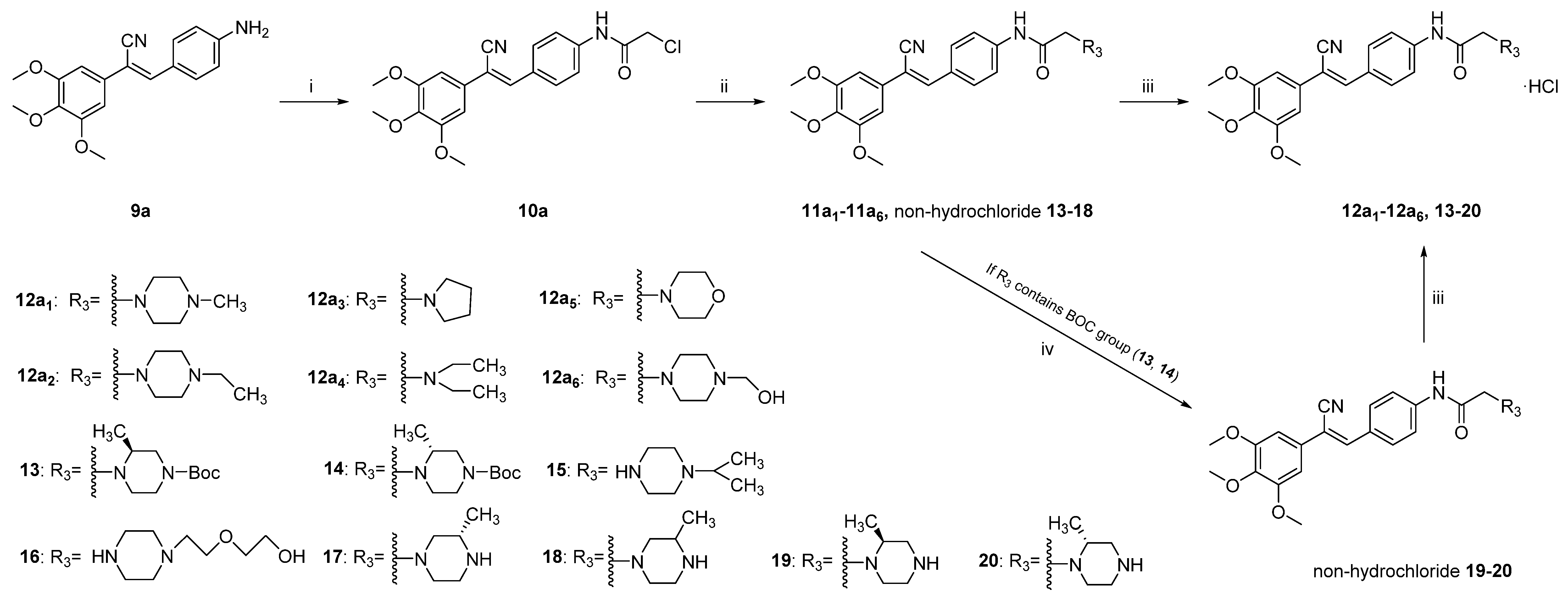
Comprehensive analysis of
9 and
12a1–6 showed that
12a1 was the most promising compound. Although the activity was slightly reduced, the water solubility was greatly improved. To further investigate the solubility of compounds containing a piperazine group, six piperazine derivatives (
Table 2,
15–
20) with different substituents or configurations were selected for further study. The compounds were synthesised as described in
Scheme 2, and the anticancer activity in vitro and solubility were characterised (
Table 5). We found that the aqueous solubility of
15 and
16 was significantly improved compared with
9a; the solubility was increased by more than 231 times compared with
9a.
Interestingly,
17 and
18 had similar anticancer activity, compared with
19 and
20, respectively, but had increased aqueous solubility. Importantly, racemic
18 was significantly more soluble than
17 with an (
S)-configuration. In contrast, the solubility of
20 was not changed appreciably compared with that of
19. We attributed this result to the different crystal forms of
17 and
18 in water, which affected their solubility in water [
33].
The toxicity of anti-tumour drugs toward normal tissues is a very important issue in chemotherapy, so we investigated the cytotoxic effects of these derivatives on two kinds of normal human cells (
Table 3,
Table 4 and
Table 5). The results indicated that the reference compound CA-4 was very toxic to L-02 and MCF-10A normal cells, with IC
50 values of 1.10 and 3.23 µM, respectively, but most of the synthesised compounds showed only slight cytotoxicity toward the two kinds of normal cells (IC
50 > 15 µM), the IC
50 value of some of the compounds, such as
12c1, was >100 µM. In addition, the most active compound
9a was 1000 times more selective for HCT-116 cancer cells over L-02 normal cells.
After analysing the biological activity and aqueous solubility of these compounds, we further investigated the drug-like properties of 9a and 12a1, which were the most promising lead compounds.
2.2.2. Determination of the Drug-Plasma Protein Binding Rate
Most drugs are delivered to disease targets through the blood after entering the human body. However, when drugs are mixed with blood, the drugs may combine with many components in the blood, including cells and proteins. Plasma proteins can absorb a large proportion of drug molecules. The binding of a drug with plasma protein will retain the drug in the plasma, limit the distribution of the drug to the diseased tissue, and reduce the metabolism of the drug. A higher dose of the drug will be required to achieve the therapeutic concentration, which increases the potential for toxicity and side effects.
We selected compound
12a1, which had excellent water solubility and activity, and determined the plasma protein binding rate with rat plasma; the results are summarised in
Table 6. Compared with the 72.3% binding rate demonstrated for CA-4 in rat plasma [
34], a relatively low protein binding rate (60.7%) was observed for
12a1 in rat plasma, which may be related to the low lipophilicity of
12a1. However, it should be recognised that this may also be caused by objective differences in experimental conditions, but in general,
12a1 maintains at least the same level of plasma protein binding rate as CA-4.
2.2.3. Plasma Stability
Blood contains a large number of hydrolases, and hydrolysis in plasma is a major cause of compound clearance, which can result in pharmacologically efficacious concentrations being unable to be achieved in vivo. Therefore, in vitro determination of the plasma stability of compounds can not only improve the screening of compounds to enable the better selection of lead compounds but can also guide the subsequent modification of selected compounds. The plasma stability of 12a1 was investigated because it contains an amide group susceptible to plasma degradation. Compound 9a is the lead compound of this series of compounds, and it had the best activity against cancer cells, so we tested its stability in plasma as a control.
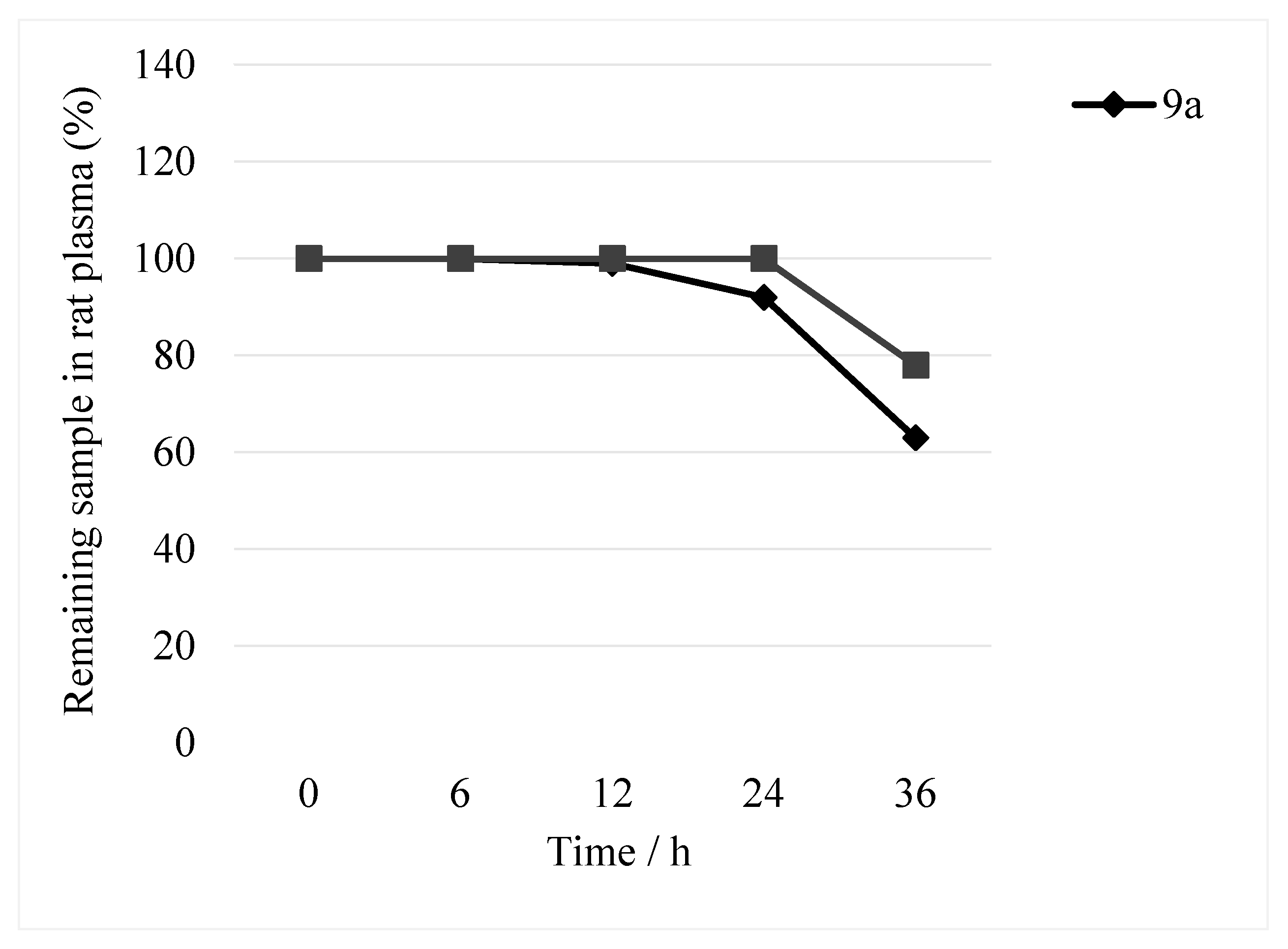
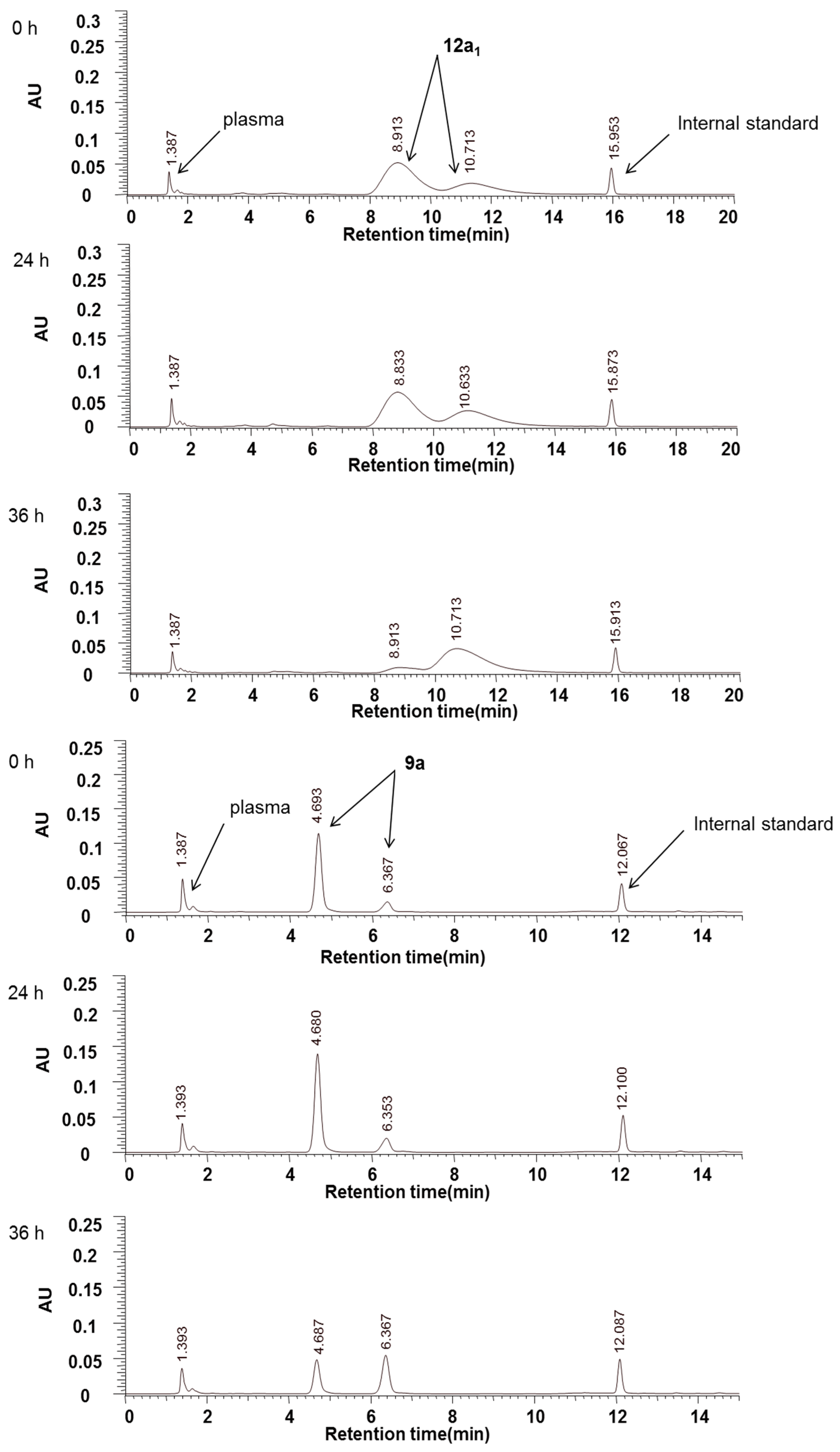
Compounds
9a and
12a1 were incubated in rat plasma and analysed via HPLC with UV detection at 294 nm (
Figure 2). Within 36 h, the peak areas of
9a and
12a1 decreased by approximately 33% and 22%, respectively. However, no additional peaks because of the hydrolysis of these two compounds were detected (
Figure 3). We believe that this result may be because (1) the degradation products lacked UV absorption at 294 nm or were not retained on the column, or (2) compounds
9a and
12a1 are, in fact, stable in blood plasma and the decrease in the peak areas was because of the adsorption of protein in the injection bottle or the plasma. In addition, the decrease in the peak area mainly occurred after 24 h, and almost no change occurred before then. Therefore, we tend to believe that the decrease was because of hydrolysis rather than adsorption; that is, owing to a lack of UV absorption and retention, the hydrolysates of
9a and
12a1 were not detected. In either case, the peak areas of the two compounds were almost unchanged within 24 h (
Figure 2). Thus, we concluded that these two compounds were stable in the plasma for 24 h.
2.2.4. Molecular Modelling
To further understand the SAR observed in the cell assays,
9a,
12a1 and
12c1 were selected for molecular docking studies using
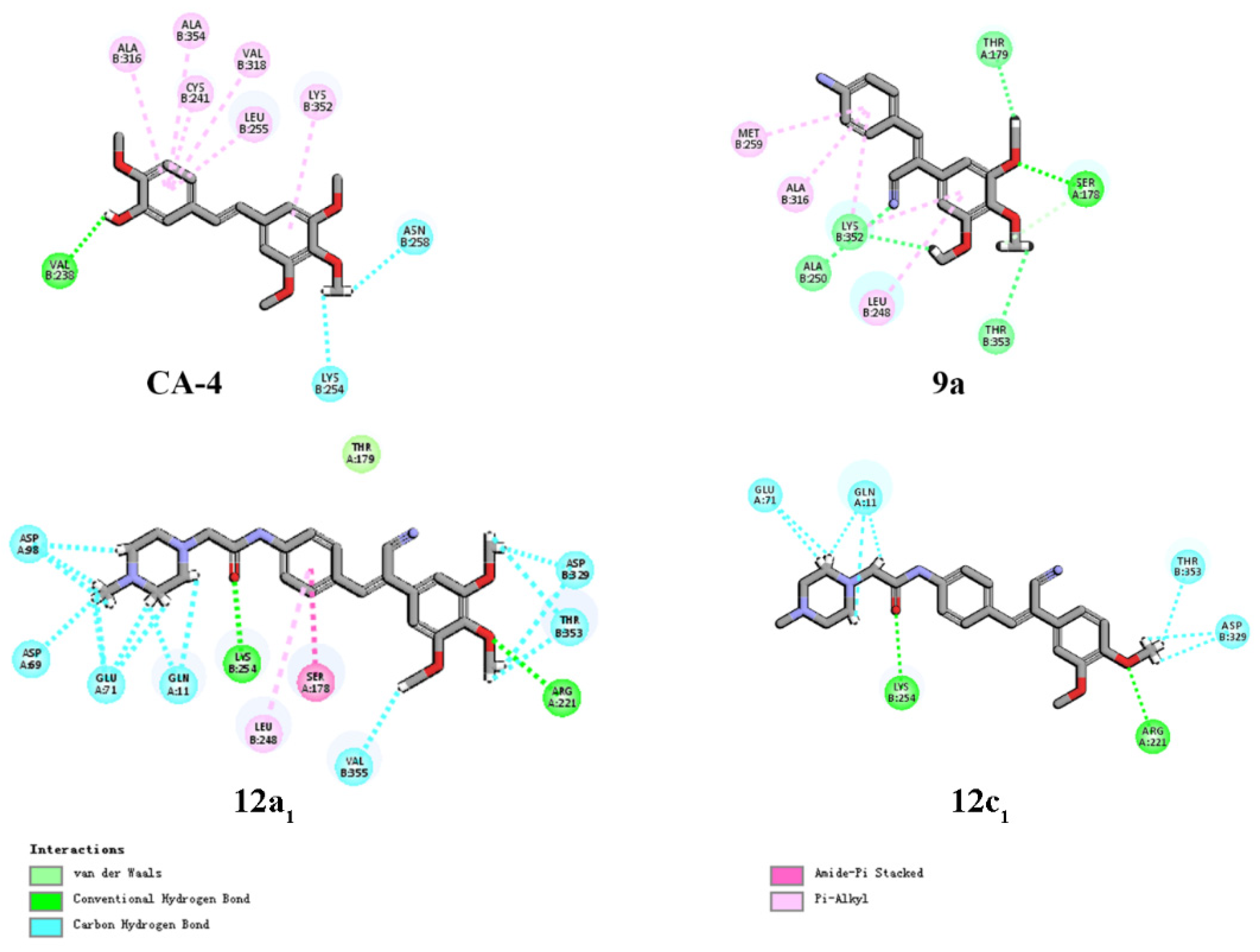
Discovery Studio 3.2 (PDB: 1SA0). In the two-dimensional model shown in
Figure 4,
9a was able to establish hydrogen bonds with more amino acids than CA-4 could, including hydrogen bonds with tubulin. The H and O atoms of the 3,4,5-trimethoxyphenyl group of
9a were also able to form interactions with Ser-178, Lys-352, Thr-179 and Thr-353. In addition, the cyano group of
9a formed hydrogen bonds with Lys-352 and Ala-250 (
Table 7). Notably, Lys-352, Thr-179 and Ala-250 are vital in the composition of the colchicine binding site, as well as in the formation of hydrogen bonds [
35]. In contrast to CA-4, which only acts on β-tubulin,
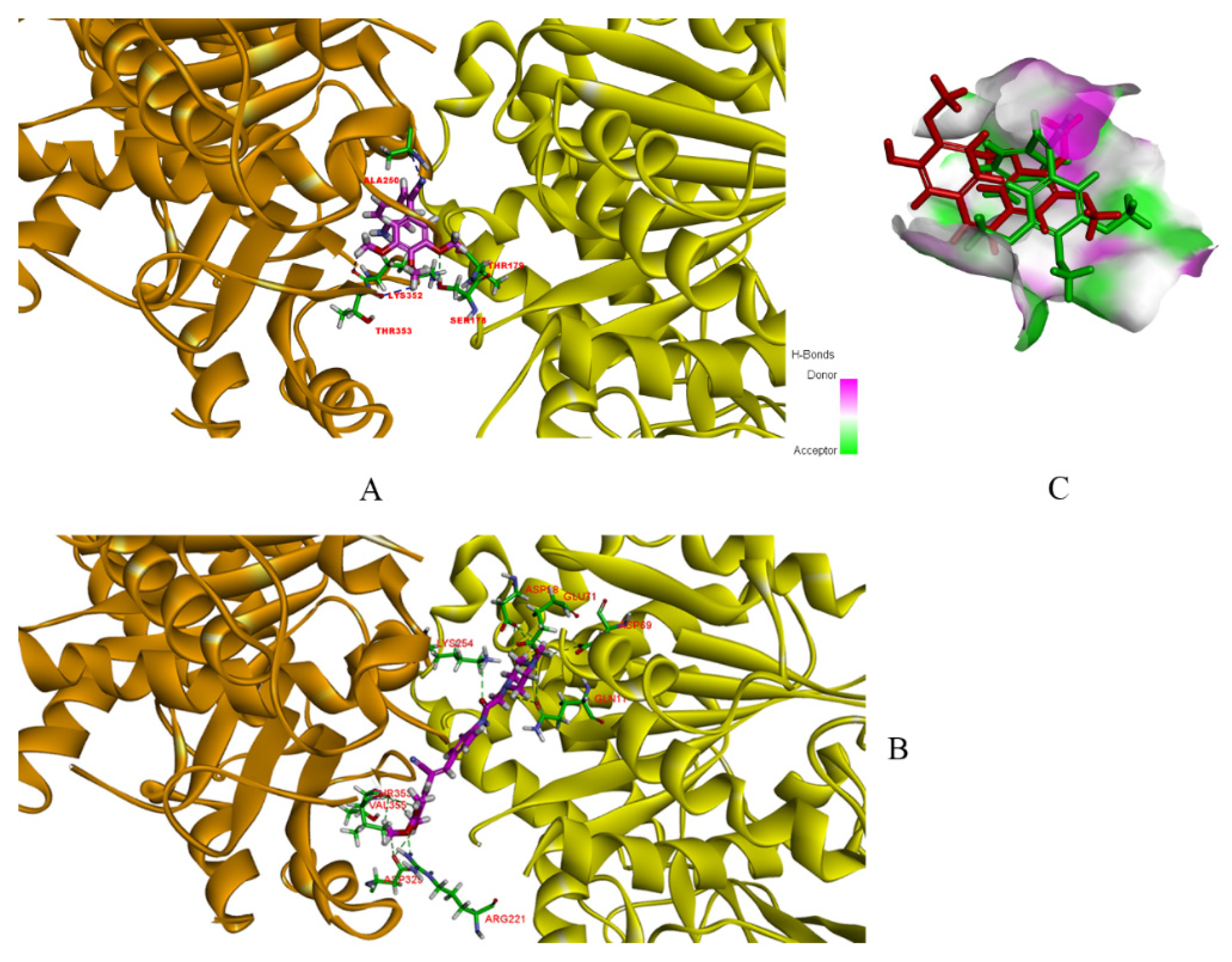
9a can interact with both α- and β-tubulin in a more compact way (
Figure 5). The docking results for compound
12a1 showed that the compound formed more hydrogen bonding interactions, compared with
9a (
Table 7), but lacked interactions with important amino acids, such as Lys-352 and Ala-250, which may be the reason for its reduced anticancer activity. However,
12a1, similar to
9a, could interact with both α- and β-tubulin, which increased its selectivity index compared with CA-4 [
17]. Compared with
12a1,
12c1 with two methoxy groups on the benzene A ring also lacked the hydrogen bond interactions with important amino acids (
Figure 4), and the number of hydrogen bonds was appreciably less than for
12a1 (
Table 7), which may lead to the observed decrease in the anticancer activity. In general, molecular docking studies could qualitatively explain the SAR observed in the biological assays.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}