
Theoretical Study on the Origin of Abnormal Regioselectivity in Ring-Opening Reaction of Hexafluoropropylene Oxide


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
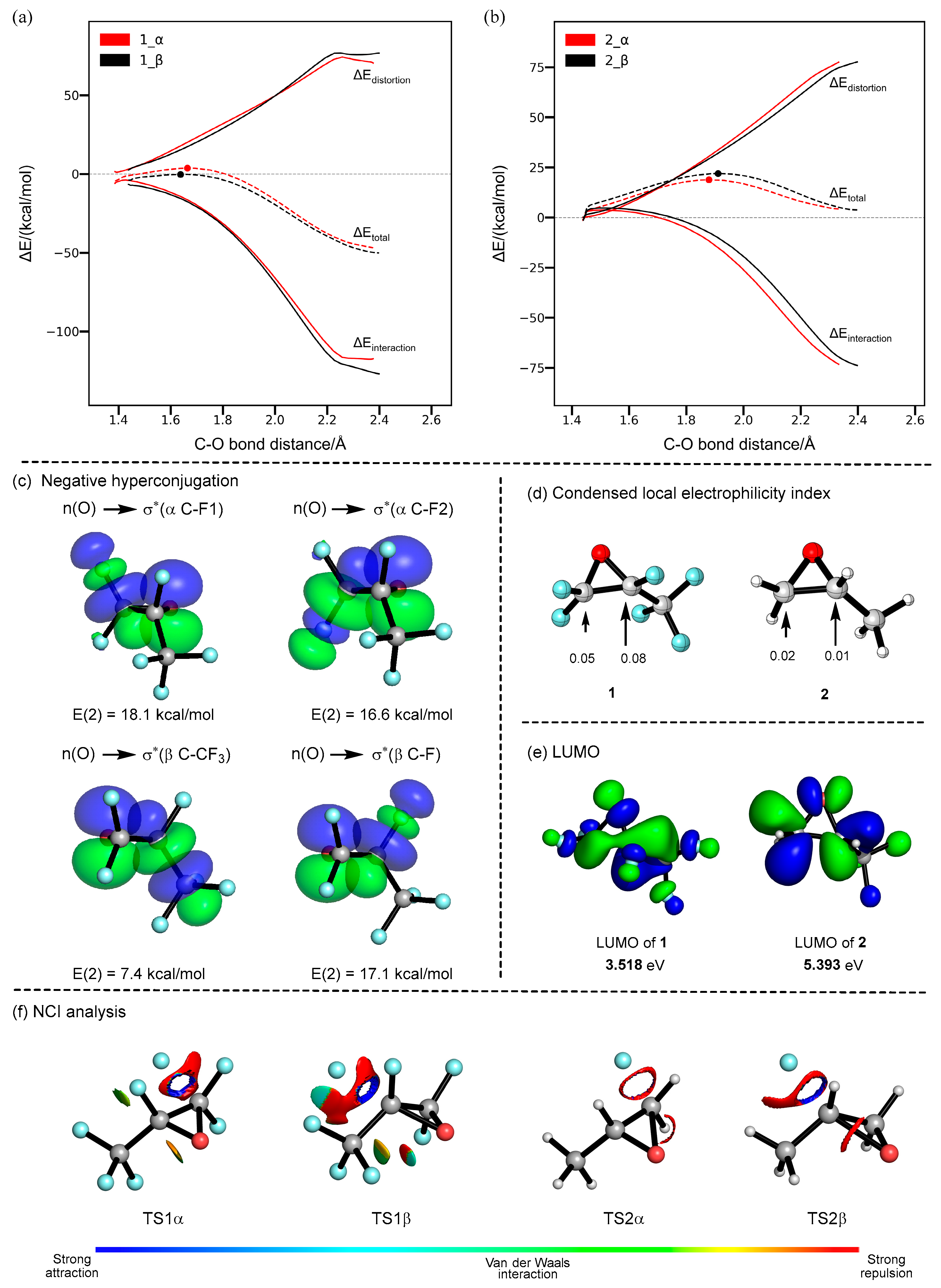
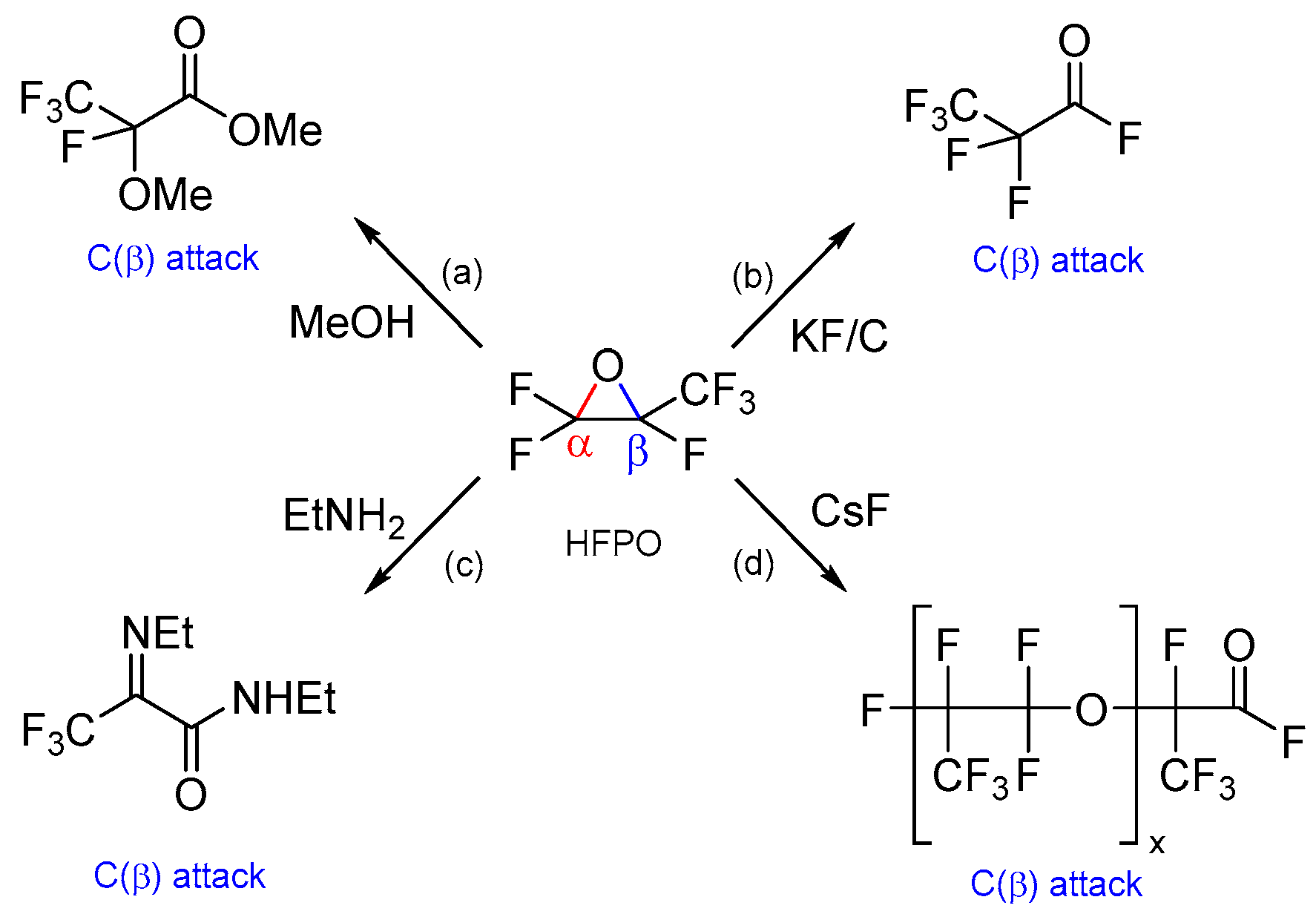
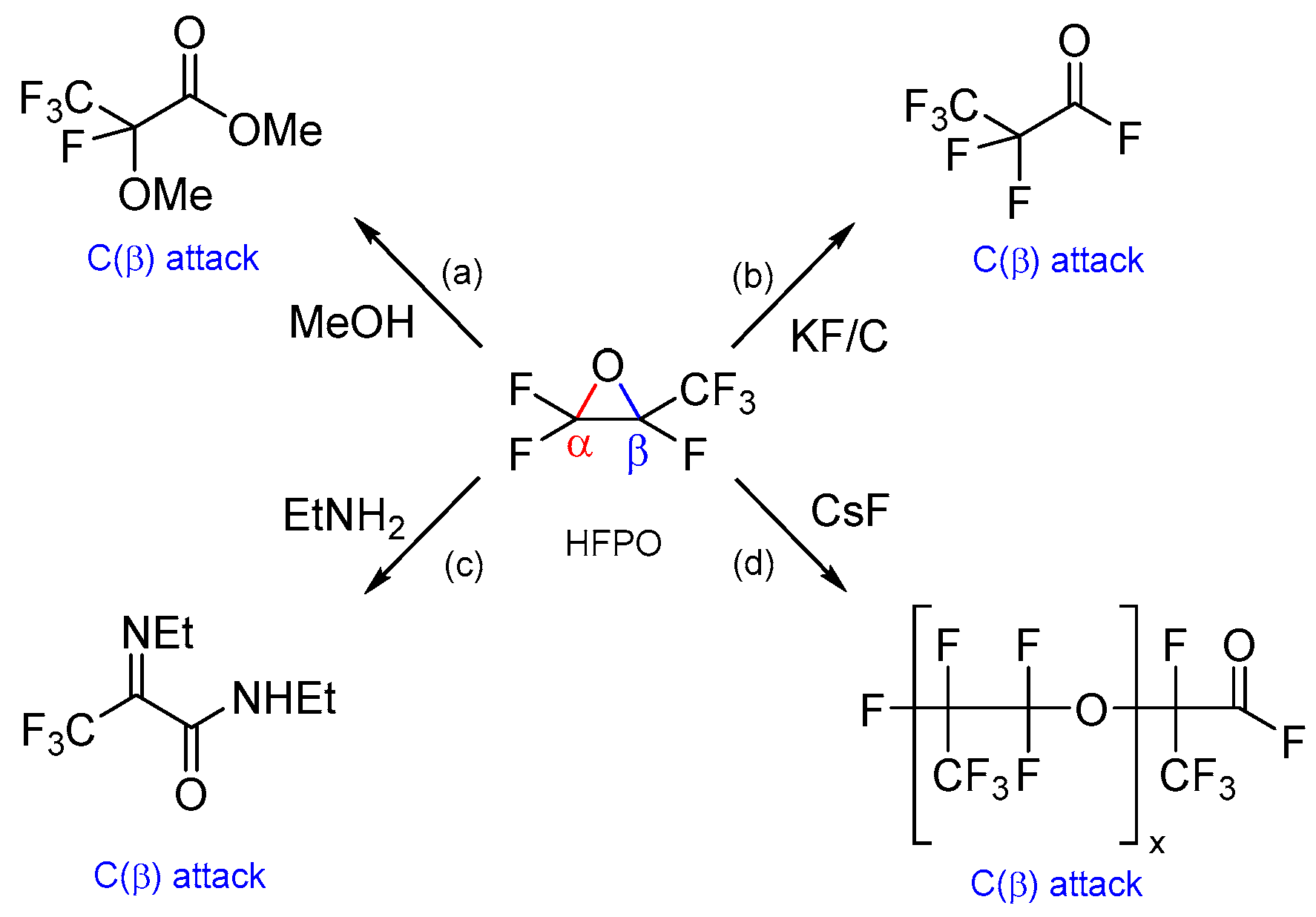
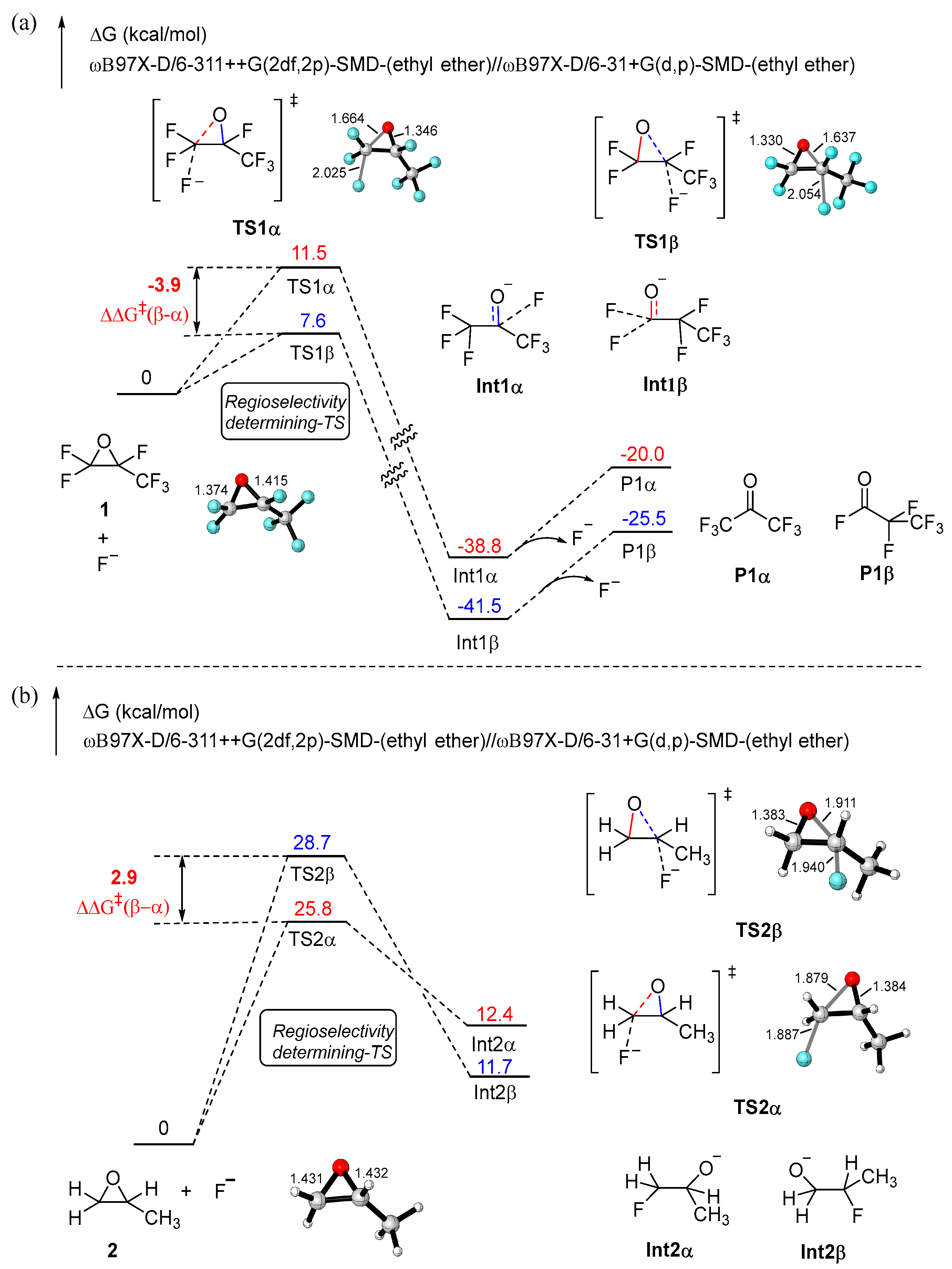
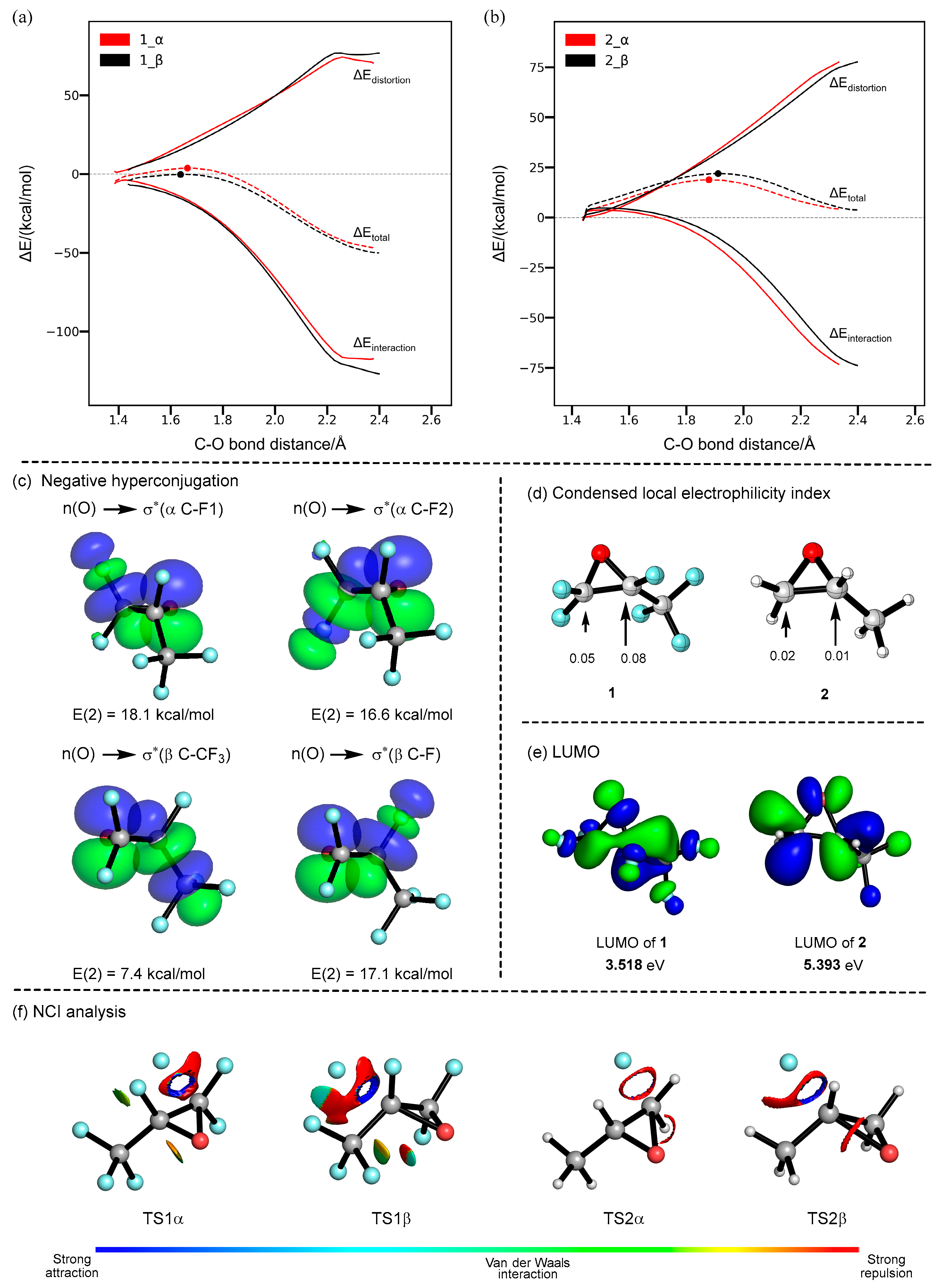
2. Results and Discussion
3. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Gomes, A.R.; Varela, C.L.; Tavares-da-Silva, E.J.; Roleira, F.M.F. Epoxide containing molecules: A good or a bad drug design approach. Eur. J. Med. Chem. 2020, 201, 112327. [Google Scholar] [PubMed]
- Sugimoto, H.; Inoue, S. Copolymerization of carbon dioxide and epoxide. J. Polym. Sci. Part A: Polym. Chem. 2004, 42, 5561–5573. [Google Scholar]
- Brocas, A.-L.; Mantzaridis, C.; Tunc, D.; Carlotti, S. Polyether synthesis: From activated or metal-free anionic ring-opening polymerization of epoxides to functionalization. Prog. Polym. Sci. 2013, 38, 845–873. [Google Scholar]
- Schneider, C. Synthesis of 1,2-Difunctionalized Fine Chemicals through Catalytic, Enantioselective Ring-Opening Reactions of Epoxides. Synthesis 2006, 23, 3919–3944. [Google Scholar] [CrossRef]
- Vilotijevic, I.; Jamison, T.F. Epoxide-opening cascades in the synthesis of polycyclic polyether natural products. Angew. Chem. Int. Ed. 2009, 48, 5250–5281. [Google Scholar]
- Vilotijevic, I.; Jamison, T.F. Synthesis of marine polycyclic polyethers via endo-selective epoxide-opening cascades. Mar. Drugs 2010, 8, 763–809. [Google Scholar] [PubMed]
- Jat, J.L.; Kumar, G. Isomerization of Epoxides. Adv. Synth. Catal. 2019, 361, 4426–4441. [Google Scholar]
- Talukdar, R. Synthetically important ring opening reactions by alkoxybenzenes and alkoxynaphthalenes. RSC. Adv. 2020, 10, 31363–31376. [Google Scholar]
- Morgan, K.M.; Ellis, J.A.; Lee, J.; Fulton, A.; Wilson, S.L.; Dupart, P.S.; Dastoori, R. Thermochemical studies of epoxides and related compounds. J. Org. Chem. 2013, 78, 4303–4311. [Google Scholar]
- Parker, R.E.; Isaacs, N.S. Mechanisms of Epoxide Reactions. Chem. Rev. 1959, 59, 737–799. [Google Scholar]
- Carey, F.A.; Sundberg, R.J. Advanced Organic Chemistry Part A: Structureand Mechanisms, 5th ed.; Springer: Boston, MA, USA, 2007; pp. 511–515. [Google Scholar]
- Millauer, H.; Schwertfeger, W.; Siegemund, G. Hexafluoropropene Oxide—A Key Compound in Organofluorine Chemistry. Angew. Chem. Int. Ed. 1985, 24, 161–179. [Google Scholar] [CrossRef]
- Eleuterio, H.S. Polymerization of Perfluoro Epoxides. J. Macromol. Sci. Part A Pure Appl. Chem. 1972, 6, 1027–1052. [Google Scholar]
- Sianesi, D.; Pasetti, A.; Tarli, F. The Chemistry of Hexafluoropropene Epoxide. J. Org. Chem. 1966, 31, 2312–2316. [Google Scholar] [CrossRef]
- Ishikawa, N.; Sasaki, S. Preparation of Perfluoroalkylated Benzoheterocyclic Compounds Using Hexafluoro-1,2-epoxypropane. Bull. Chem. Soc. Jpn. 1977, 50, 2164–2167. [Google Scholar]
- Coe, P.L.; Löhr, M.; Rochin, C. Reactions involving hexafluoropropylene oxide: Novel ring opening reactions and resolution of a racemic mixture of a bromofluoro ester, ultrasound mediated Reformatsky reactions and stereoselectivity. J. Chem. Soc. Perkin Trans. 1998, 17, 2803–2812. [Google Scholar] [CrossRef]
- Hill, J.T. Polymers from Hexafluoropropylene Oxide (HFPO). J. Macromol. Sci. Part A: Pure Appl. Chem. 1974, 8, 499–520. [Google Scholar] [CrossRef]
- Koyama, M.; Akiyama, M.; Kashiwagi, K.; Nozaki, K.; Okazoe, T. Synthesis of Crystalline CF3-Rich Perfluoropolyethers from Hexafluoropropylene Oxide and (Trifluoromethyl)Trimethylsilane. Macromol. Rapid. Commun. 2022, 43, 2200038. [Google Scholar]
- Zapevalov, A.; Filyakova, T.; Kolenko, I. Abstracts of papers. In Proceedings of the Fifth Regular Meeting of Soviet-Japanese Fluorine Chemists, Tokyo, Japan, 25–26 January 1988; pp. 55–74. [Google Scholar]
- Huang, W. Organofluorine Chemistry in China; Shanghai Scientific and Technical Publisher: Shanghai, China, 1996; pp. 20–23. ISBN 7-5323-3742-1. [Google Scholar]
- Furuyama, S.; Golden, D.M.; Benson, S.W. Kinetic Study of the Reaction CHI3 + HI ⇄ CH2I2 + I2. A Summary of Thermochemical Properties of Halomethanes and Halomethyl Radicals. J. Am. Chem. Soc. 1969, 91, 7564–7569. [Google Scholar]
- Egger, K.W.; Cocks, A.T. Homopolar and Heteropolar Bond Dissociation Energies and Heats of Formation of Radicals and Ions in the Gas Phase II. The relationship between structure and bond dissociation in organic molecules. Helv. Chim. Acta 1973, 56, 1537–1552. [Google Scholar] [CrossRef]
- Lemal, D.M.; Ramanathan, S. Fluorinated Three-Membered Ring Heterocycles. In Fluorinated Heterocyclic Compounds; Petrov, V.A., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2009; pp. 1–64. [Google Scholar]
- Nilsson Lill, S.O.; Rauhut, G.; Anders, E. Chemical Reactivity Controlled by Negative Hyperconjugation: A Theoretical Study. Chem. Eur. J. 2003, 9, 3143–3153. [Google Scholar]
- Wang, C.Q.; Zhang, Y.; Feng, C. Fluorine Effects on Group Migration via a Rhodium(V) Nitrenoid Intermediate. Angew. Chem. Int. Ed. 2017, 56, 14918–14922. [Google Scholar]
- Suenaga, M.; Nakata, K.; Abboud, J.-L.M.; Mishima, M. A natural bond orbital analysis of aryl-substituted polyfluorinated carbanions: Negative hyperconjugation. J. Phys. Org. Chem. 2018, 31, e3721. [Google Scholar]
- Alabugin, I.V.; Kuhn, L.; Medvedev, M.G.; Krivoshchapov, N.V.; Vil, V.A.; Yaremenko, I.A.; Mehaffy, P.; Yarie, M.; Terent’ev, A.O.; Zolfigol, M.A. Stereoelectronic power of oxygen in control of chemical reactivity: The anomeric effect is not alone. Chem. Soc. Rev. 2021, 50, 10253–10345. [Google Scholar] [PubMed]
- Farnham, W.B.; Smart, B.E.; Middleton, W.J.; Calabrese, J.C.; Dixon, D.A. Crystal and molecular structure of tris(dimethylamino)sulfonium trifluoromethoxide. Evidence for negative fluorine hyperconjugation. J. Am. Chem. Soc. 1985, 107, 4565–4567. [Google Scholar] [CrossRef]
- Dixon, D.A.; Fukunaga, T.; Smart, B.E. Structures and stabilities of fluorinated carbanions. Evidence for anionic hyperconjugation. J. Am. Chem. Soc. 1986, 108, 4027–4031. [Google Scholar]
- Levandowski, B.J.; Raines, R.T.; Houk, K.N. Hyperconjugative pi --> sigma*(CF) Interactions Stabilize the Enol Form of Perfluorinated Cyclic Keto-Enol Systems. J. Org. Chem. 2019, 84, 6432–6436. [Google Scholar]
- Zhou, B.; Yan, T.; Xue, X.S.; Cheng, J.P. Mechanism of Silver-Mediated Geminal Difluorination of Styrenes with a Fluoroiodane Reagent: Insights into Lewis-Acid-Activation Model. Org. Lett. 2016, 18, 6128–6131. [Google Scholar] [CrossRef]
- Yan, T.; Zhou, B.; Xue, X.S.; Cheng, J.P. Mechanism and Origin of the Unexpected Chemoselectivity in Fluorocyclization of o-Styryl Benzamides with a Hypervalent Fluoroiodane Reagent. J. Org. Chem. 2016, 81, 9006–9011. [Google Scholar]
- Li, M.; Guo, J.; Xue, X.S.; Cheng, J.P. Quantitative Scale for the Trifluoromethylthio Cation-Donating Ability of Electrophilic Trifluoromethylthiolating Reagents. Org. Lett. 2016, 18, 264–267. [Google Scholar]
- Li, M.; Xue, X.S.; Guo, J.; Wang, Y.; Cheng, J.P. An Energetic Guide for Estimating Trifluoromethyl Cation Donor Abilities of Electrophilic Trifluoromethylating Reagents: Computations of X-CF3 Bond Heterolytic Dissociation Enthalpies. J. Org. Chem. 2016, 81, 3119–3126. [Google Scholar] [CrossRef]
- Li, M.; Wang, Y.; Xue, X.S.; Cheng, J.P. A Systematic Assessment of Trifluoromethyl Radical Donor Abilities of Electrophilic Trifluoromethylating Reagents. Asian J. Org. Chem. 2017, 6, 235–240. [Google Scholar]
- Li, M.; Xue, X.-S.; Cheng, J.-P. Mechanism and Origins of Stereoinduction in Natural Cinchona Alkaloid Catalyzed Asymmetric Electrophilic Trifluoromethylthiolation of β-Keto Esters with N-Trifluoromethylthiophthalimide as Electrophilic SCF3 Source. ACS Catal. 2017, 7, 7977–7986. [Google Scholar] [CrossRef]
- Li, M.; Zhou, B.; Xue, X.S.; Cheng, J.P. Establishing the Trifluoromethylthio Radical Donating Abilities of Electrophilic SCF(3)-Transfer Reagents. J. Org. Chem. 2017, 82, 8697–8702. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.D.; Wang, Y.; Xue, X.S.; Cheng, J.P. A Systematic Evaluation of the N-F Bond Strength of Electrophilic N-F Reagents: Hints for Atomic Fluorine Donating Ability. J. Org. Chem. 2017, 82, 4129–4135. [Google Scholar]
- Zhou, B.; Xue, X.S.; Cheng, J.P. Theoretical study of Lewis acid activation models for hypervalent fluoroiodane reagent: The generality of "F-coordination” activation model. Tetrahedron Lett. 2017, 58, 1287–1291. [Google Scholar]
- Li, M.; Kang, H.; Xue, X.S.; Cheng, J.P. Computational Study of the Trifluoromethyl Radical Donor Abilities of CF3 Sources. Acta Chim. Sin. 2018, 76, 988–996. [Google Scholar]
- Zhou, B.; Haj, M.K.; Jacobsen, E.N.; Houk, K.N.; Xue, X.S. Mechanism and Origins of Chemo- and Stereoselectivities of Aryl Iodide-Catalyzed Asymmetric Difluorinations of beta-Substituted Styrenes. J. Am. Chem. Soc. 2018, 140, 15206–15218. [Google Scholar] [CrossRef]
- Li, M.; Sang, Y.; Xue, X.S.; Cheng, J.P. Origin of Stereocontrol in Photoredox Organocatalysis of Asymmetric alpha-Functionalizations of Aldehydes. J. Org. Chem. 2018, 83, 3333–3338. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, J.D.; Zheng, H.; Xue, X.S.; Mayr, H.; Cheng, J.P. Exploration of the Synthetic Potential of Electrophilic Trifluoromethylthiolating and Difluoromethylthiolating Reagents. Angew. Chem. Int. Ed. 2018, 57, 12690–12695. [Google Scholar] [CrossRef]
- Li, M.; Zheng, H.; Xue, X.S.; Cheng, J.P. Ordering the relative power of electrophilic fluorinating, trifluoromethylating, and trifluoromethylthiolating reagents: A summary of recent efforts. Tetrahedron Lett. 2018, 59, 1278–1285. [Google Scholar]
- Kang, H.; Zhou, B.; Li, M.; Xue, X.S.; Cheng, J.P. Quantification of the Activation Capabilities of Lewis/Brønsted Acid for Electrophilic Trifluoromethylthiolating Reagents. Chin. J. Chem. 2019, 38, 130–134. [Google Scholar] [CrossRef]
- Li, M.; Xue, X.S.; Cheng, J.P. Establishing Cation and Radical Donor Ability Scales of Electrophilic F, CF(3), and SCF(3) Transfer Reagents. Acc. Chem. Res. 2020, 53, 182–197. [Google Scholar] [PubMed]
- Zheng, H.; Li, Z.; Jing, J.; Xue, X.S.; Cheng, J.P. The Acidities of Nucleophilic Monofluoromethylation Reagents: An Anomalous α-Fluorine Effect. Angew. Chem. Int. Ed. 2021, 60, 9401–9406. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.M.; Tan, H.D.; Sang, Y.; Xue, X.S.; Cheng, J.P. Computational insights into the effects of reagent structure and bases on nucleophilic monofluoromethylation of aldehydes. Chin. Chem. Lett. 2022, 33, 2387–2390. [Google Scholar]
- Bickelhaupt, F.M. Understanding reactivity with Kohn-Sham molecular orbital theory: E2-SN2 mechanistic spectrum and other concepts. J. Comput. Chem. 1999, 20, 114–128. [Google Scholar]
- Bickelhaupt, F.M.; Houk, K.N. Analyzing Reaction Rates with the Distortion/Interaction-Activation Strain Model. Angew. Chem. Int. Ed. 2017, 56, 10070–10086. [Google Scholar]
- Vermeeren, P.; van der Lubbe, S.C.C.; Fonseca Guerra, C.; Bickelhaupt, F.M.; Hamlin, T.A. Understanding chemical reactivity using the activation strain model. Nat. Protoc. 2020, 15, 649–667. [Google Scholar]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. Natural bond orbital methods. Wires. Comput. Mol. Sci. 2011, 2, 1–42. [Google Scholar]
- Glendenning, E.; Reed, A.; Carpenter, J.; Weinhold, F. NBO Version 3.1, Madison, WI, USA. 2001. Available online: https://www.cup.uni-muenchen.de/ch/compchem/pop/nbo2.html (accessed on 2 October 2022).
- Beagley, B.; Pritchard, R.G.; Banks, R.E. Gas-phase electron diffraction determination of the molecular structure of perfluoro(methyloxirane). J. Fluorine Chem. 1981, 18, 159–171. [Google Scholar] [CrossRef]
- Parr, R.G.; Szentpály, L.; Liu, S. Electrophilicity Index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Shu-Bin, L.I.U. Conceptual Density Functional Theory and Some Recent Developments. Acta. Phys. Chim. Sin. 2009, 25, 590–600. [Google Scholar]
- Hansen, T.; Vermeeren, P.; Haim, A.; van Dorp, M.J.H.; Codée, J.D.C.; Bickelhaupt, F.M.; Hamlin, T.A. Regioselectivity of Epoxide Ring-Openings via SN2 Reactions Under Basic and Acidic Conditions. Eur. J. Org. Chem. 2020, 25, 3822–3828. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. A.03, Wallingford, CT, USA. 2016. Available online: https://gaussian.com/citation_a03/ (accessed on 2 October 2022).
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.n.W.; Schleyer, P.V.R. Efficient diffuse function-augmented basis sets for anion calculations. III. The 3-21+G basis set for first-row elements, Li-F. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar]
- Hariharan, P.C.; Pople, J.A. Accuracy of AHn equilibrium geometries by single determinant molecular orbital theory. Mol. Phys. 2006, 27, 209–214. [Google Scholar] [CrossRef]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B. 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Galabov, B.; Koleva, G.; Schaefer, H.F., 3rd; Allen, W.D. Nucleophilic Influences and Origin of the S(N)2 Allylic Effect. Chem. Eur. J. 2018, 24, 11637–11648. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar]
- Gonzales, J.M.; Cox, R.S.; Brown, S.T.; Allen, W.D.; Schaefer, H.F. Assessment of Density Functional Theory for Model SN2 Reactions: CH3X + F− (X = F, Cl, CN, OH, SH, NH2, PH2). J. Phys. Chem. A 2001, 105, 11327–11346. [Google Scholar]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Contreras-Garcia, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.P.; Beratan, D.N.; Yang, W. NCIPLOT: A program for plotting non-covalent interaction regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, Q. Realization of Conceptual Density Functional Theory and Information-Theoretic Approach in Multiwfn Program. In Conceptual Density Functional Theory; Liu, S., Ed.; WILEY-VCH GmbH: Weinheim, Germany, 2022; pp. 631–647. [Google Scholar]
- Svatunek, D.; Houk, K.N. autoDIAS: A python tool for an automated distortion/interaction activation strain analysis. J. Comput. Chem. 2019, 40, 2509–2515. [Google Scholar]
- Schrödinger, LLC. The PyMOL Molecular Graphics System, Version 2.0.4. Available online: http://www.pymol.org/pymol (accessed on 1 October 2022).
- Legault, C.Y. Université de Sherbrooke, CYLview20. 2020. Available online: https://www.cylview.org (accessed on 10 October 2022).
- Ni, C.; Hu, J. The unique fluorine effects in organic reactions: Recent facts and insights into fluoroalkylations. Chem. Soc. Rev. 2016, 45, 5441–5454. [Google Scholar]
- Liu, H.; Tian, L.; Wang, H.; Li, Z.-Q.; Zhang, C.; Xue, F.; Feng, C. A novel type of donor–acceptor cyclopropane with fluorine as the donor: (3 + 2)-cycloadditions with carbonyls. Chem. Sci. 2022, 13, 2686–2691. [Google Scholar]
- Wu, H.; Li, X.; Tang, X.; Feng, C.; Huang, G. Mechanisms of Rhodium (III)-Catalyzed C–H Functionalizations of Benzamides with α,α-Difluoromethylene Alkynes. J. Org. Chem. 2018, 83, 9220–9230. [Google Scholar]
- Kang, L.; Lin, Y.; Gao, Z.; Zhang, J.; Yang, H.; Qian, J.; Peng, Q.; Jiang, G. Fluorine Effects for Tunable C–C and C–S Bond Cleavage in Fluoro-Julia–Kocienski Intermediates. CCS Chem. 2020, 3, 1678–1689. [Google Scholar] [CrossRef]
- Peng, S.Q.; Zhang, X.W.; Zhang, L.; Hu, X.G. Esterification of Carboxylic Acids with Difluoromethyl Diazomethane and Interrupted Esterification with Trifluoromethyl Diazomethane: A Fluorine Effect. Org. Lett. 2017, 19, 5689–5692. [Google Scholar] [CrossRef]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual density functional theory. Chem. Rev. 2003, 103, 1793–1873. [Google Scholar]
- Rong, F.U.; Tian, L.U.; Fei-Wu, C. Comparing Methods for Predicting the Reactive Site of Electrophilic Substitution. Acta Phys. Chim. Sin. 2014, 30, 628–639. [Google Scholar]
- Domingo, L.R.; Rios-Gutierrez, M.; Perez, P. Applications of the Conceptual Density Functional Theory Indices to Organic Chemistry Reactivity. Molecules 2016, 21, 748. [Google Scholar] [PubMed]
- Parr, R.G.; Yang, W. Density functional approach to the frontier-electron theory of chemical reactivity. J. Am. Chem. Soc. 1984, 106, 4049–4050. [Google Scholar]
- Yang, W.; Mortier, W.J. The use of global and local molecular parameters for the analysis of the gas-phase basicity of amines. J. Am. Chem. Soc. 1986, 108, 5708–5711. [Google Scholar] [PubMed]
- Morell, C.; Grand, A.; Toro-Labbe, A. New dual descriptor for chemical reactivity. J. Phys. Chem. A 2005, 109, 205–212. [Google Scholar] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, C.; Sang, Y.; Li, Y.; Xue, X. Theoretical Study on the Origin of Abnormal Regioselectivity in Ring-Opening Reaction of Hexafluoropropylene Oxide. Molecules 2023, 28, 1669. https://doi.org/10.3390/molecules28041669
Yu C, Sang Y, Li Y, Xue X. Theoretical Study on the Origin of Abnormal Regioselectivity in Ring-Opening Reaction of Hexafluoropropylene Oxide. Molecules. 2023; 28(4):1669. https://doi.org/10.3390/molecules28041669
Chicago/Turabian StyleYu, Cui, Yueqian Sang, Yao Li, and Xiaosong Xue. 2023. "Theoretical Study on the Origin of Abnormal Regioselectivity in Ring-Opening Reaction of Hexafluoropropylene Oxide" Molecules 28, no. 4: 1669. https://doi.org/10.3390/molecules28041669
APA StyleYu, C., Sang, Y., Li, Y., & Xue, X. (2023). Theoretical Study on the Origin of Abnormal Regioselectivity in Ring-Opening Reaction of Hexafluoropropylene Oxide. Molecules, 28(4), 1669. https://doi.org/10.3390/molecules28041669