
Migration: A Neglected Potential Contribution of HCl-Oxidized Au(0)


Abstract
:
1. Introduction
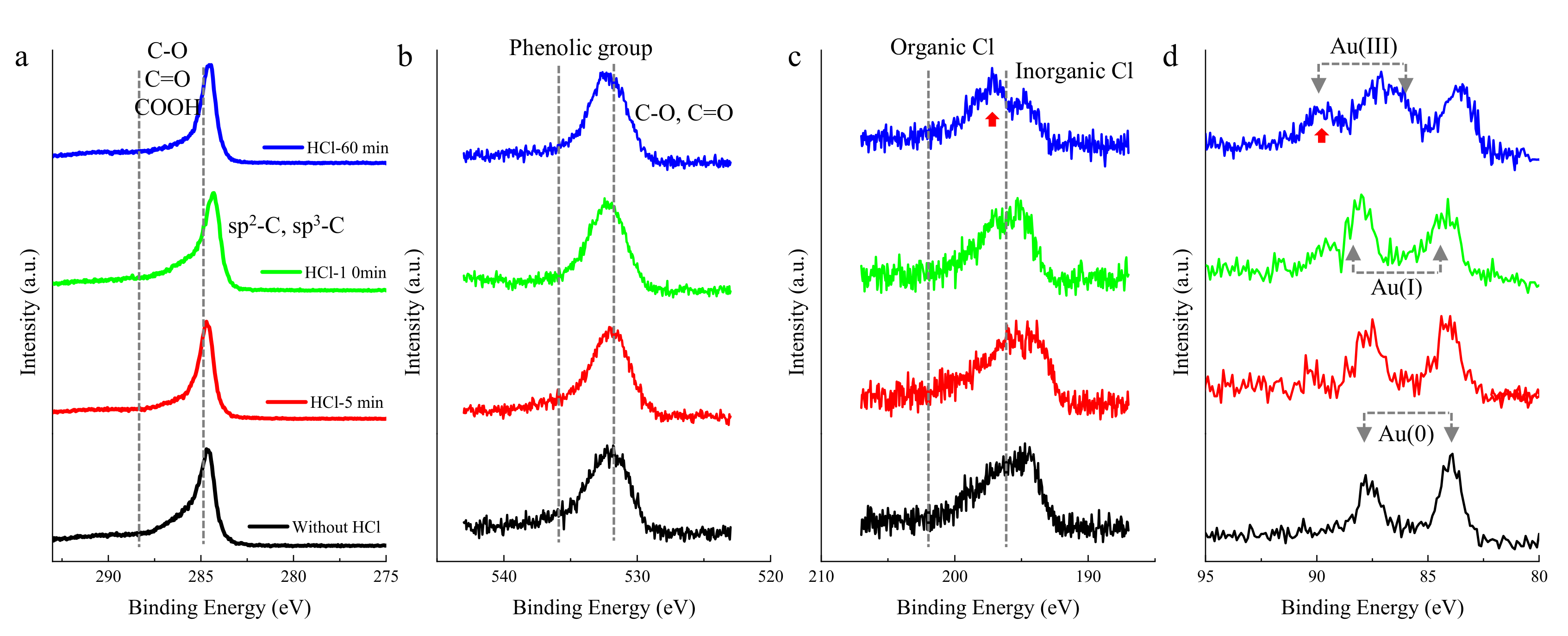
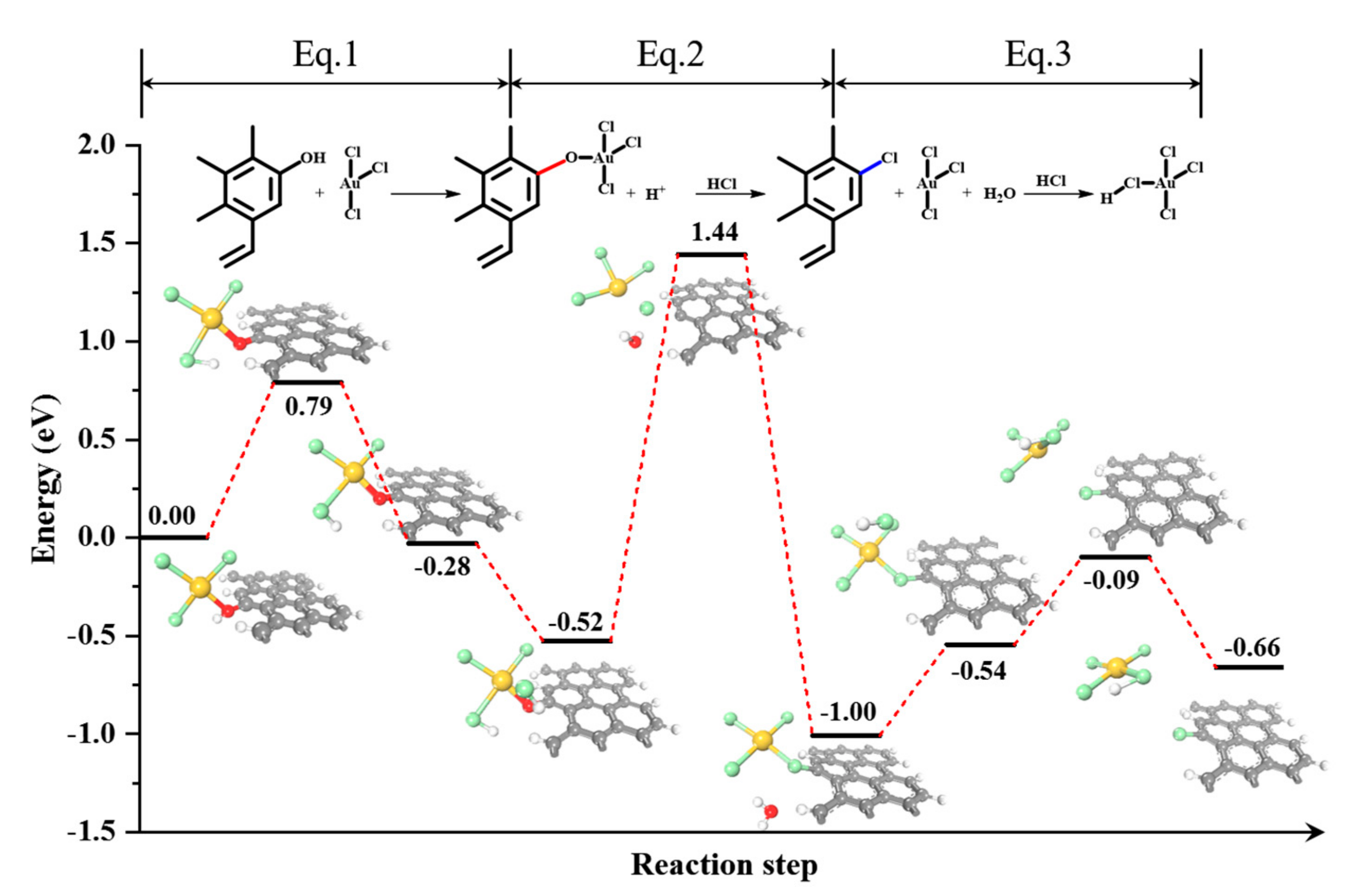
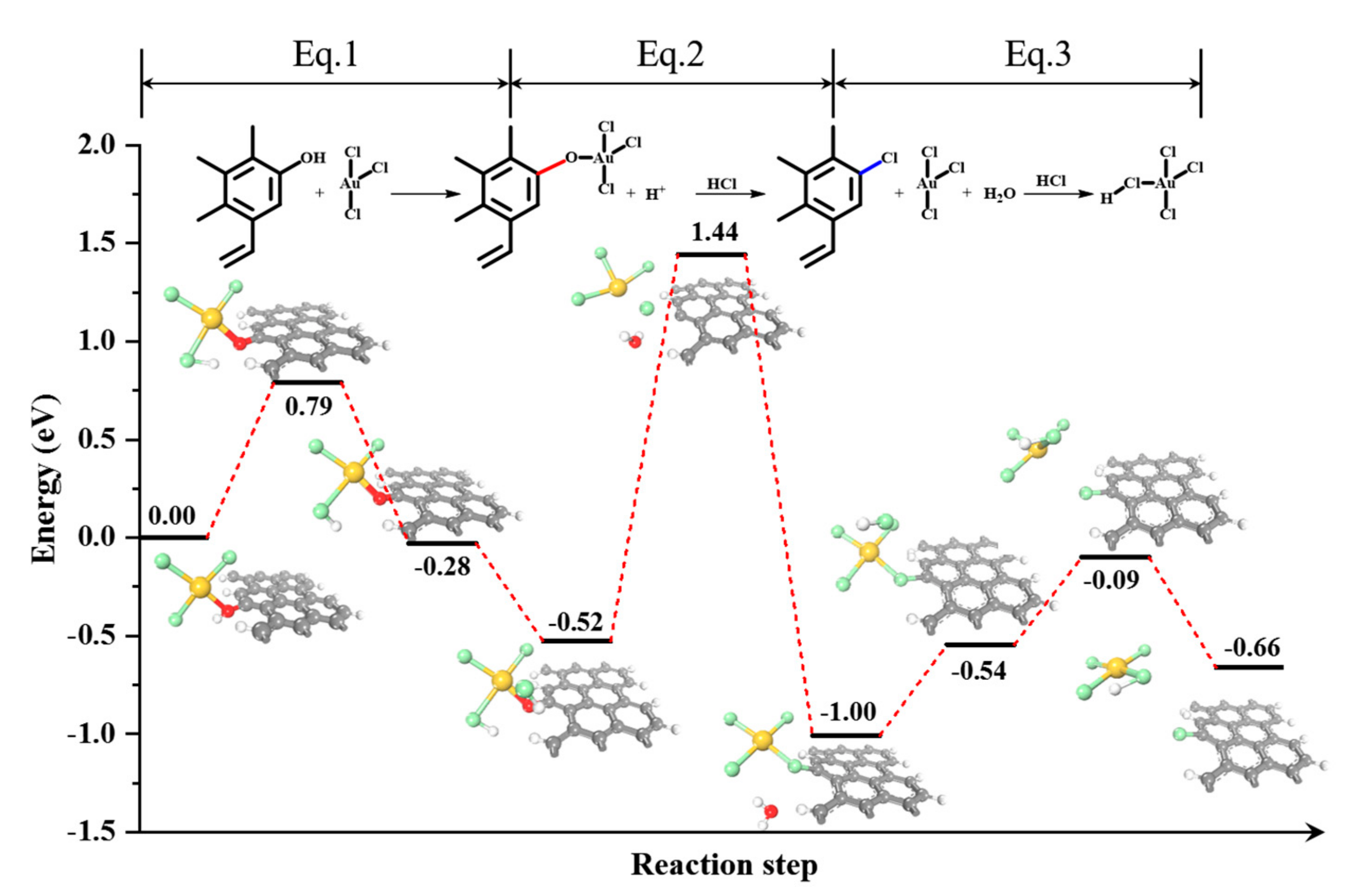
2. Results and Discussion
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bi, F.; Zhao, Z.; Yang, Y.; Gao, W.; Liu, N.; Huang, Y.; Zhang, X. Chlorine-Coordinated Pd Single Atom Enhanced the Chlorine Resistance for Volatile Organic Compound Degradation: Mechanism Study. Environ. Sci. Technol. 2022, 56, 17321–17330. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wang, X.; Qi, X.; Guo, H.; Liu, L.; Zhao, Q.; Cui, W. Effect of Ag cocatalyst on highly selective photocatalytic CO2 reduction to HCOOH over CuO/Ag/UiO-66 Z-scheme heterojunction. J. Catal. 2022, 413, 31–47. [Google Scholar] [CrossRef]
- Bi, F.; Zhao, Z.; Yang, Y.; Liu, Q.; Huang, W.; Huang, Y.; Zhang, X. Efficient degradation of toluene over ultra-low Pd supported on UiO-66 and its functional materials: Reaction mechanism, water-resistance, and influence of SO2. Environ. Funct. Mater. 2022, 1, 166–181. [Google Scholar] [CrossRef]
- Fan, J.; Chen, L.; Li, S.; Mou, J.; Zeng, L.; Jiao, Y.; Wang, J.; Chen, Y. Insights into the promotional effect of alkaline earth metals in Pt-based three-way catalysts for NO reduction. J. Catal. 2023, 418, 90–99. [Google Scholar] [CrossRef]
- Geng, P.; Wang, L.; Du, M.; Bai, Y.; Li, W.; Liu, Y.; Chen, S.; Braunstein, P.; Xu, Q.; Pang, H. MIL-96-Al for Li–S Batteries: Shape or Size? Adv. Mater. 2022, 34, 2107836. [Google Scholar] [CrossRef]
- Haruta, M.; Kobayashi, T.; Sano, H.; Yamada, N. Novel gold catalysts for the oxidation of carbon monoxide at a temperature far below 0 °C. Chem. Lett. 1987, 16, 405–408. [Google Scholar] [CrossRef]
- Hutchings, G.J. Vapor phase hydrochlorination of acetylene: Correlation of catalytic activity of supported metal chloride catalysts. J. Catal. 1985, 96, 292–295. [Google Scholar] [CrossRef]
- Peter, J.; Nicholas, C. Discovery, Development, and Commercialization of Gold Catalysts for Acetylene Hydrochlorination. J. Am. Chem. Soc. 2015, 137, 14548–14557. [Google Scholar]
- Conte, M.; Carley, A.F.; Heirene, C.; Willock, D.J.; Johnston, P.; Herzing, A.A.; Kiely, C.J.; Hutchings, G.J. Hydrochlorination of acetylene using a supported gold catalyst: A study of the reaction mechanism. J. Catal. 2007, 250, 231–239. [Google Scholar] [CrossRef]
- Wang, B.; Jiang, Z.; Wang, T.; Tang, Q.; Yu, M.; Feng, T.; Tian, M.; Chang, R.; Yue, Y.; Pan, Z. Controllable synthesis of vacancy-defect Cu site and its catalysis for the manufacture of vinyl chloride monomer. ACS Catal. 2021, 11, 11016–11028. [Google Scholar] [CrossRef]
- Chen, Z.; Chen, Y.; Chao, S.; Dong, X.; Chen, W.; Luo, J.; Liu, C.; Wang, D.; Chen, C.; Li, W. Single-atom AuI–N3 site for acetylene hydrochlorination reaction. ACS Catal. 2020, 10, 1865–1870. [Google Scholar] [CrossRef]
- Wang, B.; Yue, Y.; Jin, C.; Lu, J.; Wang, S.; Yu, L.; Guo, L.; Li, R.; Hu, Z.-T.; Pan, Z. Hydrochlorination of acetylene on single-atom Pd/N-doped carbon catalysts: Importance of pyridinic-N synergism. Appl. Catal. 2020, 272, 118944. [Google Scholar] [CrossRef]
- Ye, L.; Duan, X.; Wu, S.; Wu, T.; Zhao, Y.; Robertson, A.; Chou, H.; Zheng, J.; Ayvalı, T.; Day, S. Self-regeneration of Au/CeO2 based catalysts with enhanced activity and ultra-stability for acetylene hydrochlorination. Nat. Commun. 2019, 10, 914. [Google Scholar] [CrossRef]
- Wang, B.; Jin, C.; Shao, S.; Yue, Y.; Zhang, Y.; Wang, S.; Chang, R.; Zhang, H.; Zhao, J.; Li, X. Electron-deficient Cu site catalyzed acetylene hydrochlorination. Green Energy Environ. 2022. [Google Scholar] [CrossRef]
- Conte, M.; Carley, A.F.; Hutchings, G.J. Reactivation of a carbon-supported gold catalyst for the hydrochlorination of acetylene. Catal. Lett. 2008, 124, 165–167. [Google Scholar] [CrossRef]
- Malta, G.; Kondrat, S.A.; Freakley, S.J.; Morgan, D.J.; Gibson, E.K.; Wells, P.P.; Aramini, M.; Gianolio, D.; Thompson, P.B.J.; Johnston, P. In situ K-edge X-ray absorption spectroscopy of the ligand environment of single-site Au/C catalysts during acetylene hydrochlorination. Chem. Sci. 2020, 11, 7040–7052. [Google Scholar] [CrossRef]
- Wang, B.; Yue, Y.; Pang, X.; Yu, M.; Wang, T.; Chang, R.; Pan, Z.; Zhao, J.; Li, X. Nature of HCl oxidation Au anomalies and activation of non-carbon-material-supported Au catalyst. J. Catal. 2021, 404, 198–203. [Google Scholar] [CrossRef]
- Malta, G.; Kondrat, S.A.; Freakley, S.J.; Davies, C.J.; Dawson, S.; Liu, X.; Lu, L.; Dymkowski, K.; Fernandez-Alonso, F.; Mukhopadhyay, S. Deactivation of a single-site gold-on-carbon acetylene hydrochlorination catalyst: An X-ray absorption and inelastic neutron scattering study. ACS Catal. 2018, 8, 8493–8505. [Google Scholar] [CrossRef]
- Malta, G.; Kondrat, S.A.; Freakley, S.J.; Davies, C.J.; Lu, L.; Dawson, S.; Thetford, A.; Gibson, E.K.; Morgan, D.J.; Jones, W. Identification of single-site gold catalysis in acetylene hydrochlorination. Science 2017, 355, 1399–1403. [Google Scholar] [CrossRef]
- Wang, B.; Zhao, J.; Yue, Y.; Sheng, G.; Lai, H.; Rui, J.; He, H.; Hu, Z.t.; Feng, F.; Zhang, Q. Carbon with Surface-Enriched Nitrogen and Sulfur Supported Au Catalysts for Acetylene Hydrochlorination. ChemCatChem 2019, 11, 1002–1009. [Google Scholar] [CrossRef]
- Du, M.; Geng, P.; Pei, C.; Jiang, X.; Shan, Y.; Hu, W.; Ni, L.; Pang, H.J. High-Entropy Prussian Blue Analogues and Their Oxide Family as Sulfur Hosts for Lithium-Sulfur Batteries. Angew. Chem. 2022, 134, e202209350. [Google Scholar] [CrossRef]
- Li, W.; Guo, X.; Geng, P.; Du, M.; Jing, Q.; Chen, X.; Zhang, G.; Li, H.; Xu, Q.; Braunstein, P. Rational design and general synthesis of multimetallic metal–organic framework nano-octahedra for enhanced Li–S battery. Adv. Mater. 2021, 33, 2105163. [Google Scholar] [CrossRef] [PubMed]
- Malta, G.; Freakley, S.J.; Kondrat, S.A.; Hutchings, G.J. Acetylene hydrochlorination using Au/carbon: A journey towards single site catalysis. Chem. Commun. 2017, 53, 11733–11746. [Google Scholar] [CrossRef]
- Buono, C.; Davies, P.R.; Davies, R.J.; Jones, T.; Kulhavý, J.; Lewis, R.; Morgan, D.J.; Robinson, N.; Willock, D.J. Spectroscopic and atomic force studies of the functionalisation of carbon surfaces: New insights into the role of the surface topography and specific chemical states. Faraday Discuss. 2014, 173, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Wojnicki, M.; Luty-Błocho, M.; Socha, R.P.; Mech, K.; Pedzich, Z.; Fitzner, K.; Rudnik, E. Kinetic studies of sorption and reduction of gold (III) chloride complex ions on activated carbon Norit ROX 0.8. J. Ind. Eng. Chem. 2015, 29, 289–297. [Google Scholar] [CrossRef]
- Xu, J.; Zhao, J.; Xu, J.; Zhang, T.; Li, X.; Di, X.; Ni, J.; Wang, J.; Cen, J. Influence of surface chemistry of activated carbon on the activity of gold/activated carbon catalyst in acetylene hydrochlorination. Ind. Eng. Chem. Res. 2014, 53, 14272–14281. [Google Scholar] [CrossRef]
- Lan, G.; Qiu, Y.; Fan, J.; Wang, X.; Tang, H.; Han, W.; Liu, H.; Liu, H.; Song, S.; Li, Y. Defective graphene@diamond hybrid nanocarbon material as an effective and stable metal-free catalyst for acetylene hydrochlorination. Chem. Commun. 2019, 55, 1430–1433. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, B.; Yue, Y.; Di, S.; Zhai, Y.; He, H.; Sheng, G.; Lai, H.; Zhu, Y.; Guo, L. Towards a greener approach for the preparation of highly active gold/carbon catalyst for the hydrochlorination of ethyne. J. Catal. 2018, 365, 153–162. [Google Scholar] [CrossRef]
- Brown, M.A.; Fujimori, Y.; Ringleb, F.; Shao, X.; Stavale, F.; Nilius, N.; Sterrer, M.; Freund, H. Oxidation of Au by surface OH: Nucleation and electronic structure of gold on hydroxylated MgO (001). J. Am. Chem. Soc. 2011, 133, 10668–10676. [Google Scholar] [CrossRef]
- Bulushev, D.A.; Yuranov, I.; Suvorova, E.; Buffat, P.A.; Kiwi-Minsker, L. Highly dispersed gold on activated carbon fibers for low-temperature CO oxidation. J. Catal. 2004, 224, 8–17. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Catalysts | Composition | |||||||
|---|---|---|---|---|---|---|---|---|
| Au/C (wt.%) | Au(III) (μmol g(Cat.)−1) | Au(III)/Au(0) (mol.%) | ||||||
| Nominal | ICP | XPS a | XRF | XPS a | TPR b | XPS a | TPR b | |
| Au/C | 1.00 | 0.98 | 0.01 | 1.07 | 0 | 9.65 | 0 | 19.01 |
| Au/C HCl-5 min | 1.00 | 1.01 | 0.04 | 0.95 | 0.005 | 9.12 | 23 | 17.97 |
| Au/C HCl-10 min | 1.00 | 0.98 | 0.08 | 1.02 | 0.020 | 10.08 | 51 | 19.86 |
| Au/C HCl-60 min | 1.00 | 0.97 | 0.16 | 0.98 | 0.045 | 9.44 | 56 | 18.60 |
| Au(0)/C H2-180 min, HCl-60 min | 1.00 | 1.03 | 0.01 | 1.11 | 0 | 0 | 0 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Zhang, H.; Wang, B.; Yue, Y.; Zhao, J. Migration: A Neglected Potential Contribution of HCl-Oxidized Au(0). Molecules 2023, 28, 1600. https://doi.org/10.3390/molecules28041600
Zhang Z, Zhang H, Wang B, Yue Y, Zhao J. Migration: A Neglected Potential Contribution of HCl-Oxidized Au(0). Molecules. 2023; 28(4):1600. https://doi.org/10.3390/molecules28041600
Chicago/Turabian StyleZhang, Zilong, Haifeng Zhang, Bolin Wang, Yuxue Yue, and Jia Zhao. 2023. "Migration: A Neglected Potential Contribution of HCl-Oxidized Au(0)" Molecules 28, no. 4: 1600. https://doi.org/10.3390/molecules28041600
APA StyleZhang, Z., Zhang, H., Wang, B., Yue, Y., & Zhao, J. (2023). Migration: A Neglected Potential Contribution of HCl-Oxidized Au(0). Molecules, 28(4), 1600. https://doi.org/10.3390/molecules28041600