
Triphenylborane in Metal-Free Catalysis


Abstract
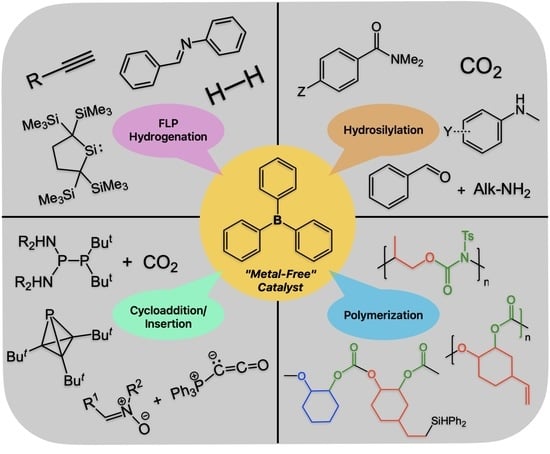
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. BPh3 in Polymerization Catalysis
3. BPh3 as an FLP Component in Hydrogenation Catalysis
4. Hydrosilylation Catalysis
5. Miscellaneous
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Krause, E.; Nitsche, R. Darstellung von Organischen Bor-Verbindungen Mit Hilfe von Borfluorid, II.: Bortriphenyl Und Phenyl-borsäure. Ber. Dtsch. Chem. Ges. A/B 1922, 55, 1261–1265. [Google Scholar] [CrossRef]
- Zettler, F.; Hausen, H.D.; Hess, H. Crystal and Molecular Structure of Triphenylborane. J. Organomet. Chem. 1974, 72, 157–162. [Google Scholar] [CrossRef]
- Guibert, C.R.; Little, J.L. Alkyl- and Arylboranes. In Ullmanns’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag: Weinheim, Germany, 2005. [Google Scholar] [CrossRef]
- Wu, Q.; Esteghamatian, M.; Hu, N.-X.; Popovic, Z.; Enright, G.; Tao, Y.; D’Iorio, M.; Wang, S. Synthesis, Structure, and Electroluminescence of BR2q (R = Et, Ph, 2-Naphthyl and q = 8-Hydroxyquinolato). Chem. Mater. 2000, 12, 79–83. [Google Scholar] [CrossRef]
- Liu, Q.-D.; Mudadu, M.S.; Thummel, R.; Tao, Y.; Wang, S. From blue to red: Syntheses, structures, electronic, and electroluminescent properties of tunable luminescent N,N chelate boron complexes. Adv. Funct. Mater. 2005, 15, 143–154. [Google Scholar] [CrossRef]
- Chen, H.-Y.; Chi, Y.; Liu, C.-S.; Yu, J.-K.; Cheng, Y.-M.; Chen, K.-S.; Chou, P.-T.; Peng, S.-M.; Lee, G.-H.; Carty, A.J.; et al. Rational color tuning and luminescent properties of functionalized boron-containing 2-pyridylpyrrolide complexes. Adv. Funct. Mater. 2005, 15, 567–574. [Google Scholar] [CrossRef]
- Liddle, B.J.; Silva, R.M.; Morin, T.J.; Macedo, F.P.; Shukla, R.; Lindeman, S.V.; Gardinier, J.R. BORAZANs: Tunable fluorophores based on 2-(pyrazolyl)aniline chelates of diphenylboron. J. Org. Chem. 2007, 72, 5637–5646. [Google Scholar] [CrossRef]
- Tokoro, Y.; Nagai, A.; Kokado, K.; Chujo, Y. Synthesis of Organoboron Quinoline-8-thiolate and Quinoline-8-selenolate Complexes and Their Incorporation into the π-Conjugated Polymer Main-Chain. Macromolecules 2009, 42, 2988–2993. [Google Scholar] [CrossRef]
- Li, D.; Wang, K.; Huang, S.; Qu, S.; Liu, X.; Zhu, Q.; Zhang, H.; Wang, Y. Brightly fluorescent red organic solids bearing boron-bridged π-conjugated skeletons. J. Mater. Chem. 2011, 21, 15298–15304. [Google Scholar] [CrossRef]
- Frath, D.; Azizi, S.; Ulrich, G.; Retailleau, P.; Ziessel, R. Facile Synthesis of Highly Fluorescent Boranil Complexes. Org. Lett. 2011, 13, 3414–3417. [Google Scholar] [CrossRef]
- Kubota, Y.; Hara, H.; Tanaka, S.; Funabiki, K.; Matsui, M. Synthesis and Fluorescence Properties of Novel Pyrazine-Boron Complexes Bearing a β-Iminoketone Ligand. Org. Lett. 2011, 13, 6544–6547. [Google Scholar] [CrossRef]
- Li, D.; Zhang, H.; Wang, C.; Huang, S.; Guo, J.; Wang, Y. Construction of full-color-tunable and strongly emissive materials by functionalizing a boron-chelate four-ring-fused π-conjugated core. J. Mater. Chem. 2012, 22, 4319–4328. [Google Scholar] [CrossRef]
- Shimizu, S.; Murayama, A.; Haruyama, T.; Iino, T.; Mori, S.; Furuta, H.; Kobayashi, N. Benzo[c,d]indole-Containing Aza-BODIPY Dyes: Asymmetrization-Induced Solid-State Emission and Aggregation-Induced Emission Enhancement as New Properties of a Well-Known Chromophore. Chem. Eur. J. 2015, 21, 12996–13003. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wu, Y.; Liu, Q.; Li, Z.; Yan, H.; Ji, C.; Duan, J.; Liu, Z. Aggregation-induced emission (AIE) of pyridyl-enamido-based organoboron luminophores. Chem. Commun. 2015, 51, 784–787. [Google Scholar] [CrossRef] [PubMed]
- Dou, C.; Ding, Z.; Zhang, Z.; Xie, Z.; Liu, J.; Wang, L. Developing Conjugated Polymers with High Electron Affinity by Replacing a C-C Unit with a B-N Unit. Angew. Chem., Int. Ed. 2015, 54, 3648–3652. [Google Scholar] [CrossRef]
- Liu, F.; Ding, Z.; Liu, J.; Wang, L. An organoboron compound with a wide absorption spectrum for solar cell applications. Chem. Commun. 2017, 53, 12213–12216. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Nakamuro, T.; Yamashita, K.; Yanagisawa, H.; Nureki, O.; Kikkawa, M.; Gao, H.; Tian, J.; Shang, R.; Nakamura, E. B/N-Doped p-Arylenevinylene Chromophores: Synthesis, Properties, and Microcrystal Electron Crystallographic Study. J. Am. Chem. Soc. 2020, 142, 18990–18996. [Google Scholar] [CrossRef]
- Krause, E. Valence problem of boron: A series of strikingly stable secondary valence compounds of boron triphenyl. Ber. Dtsch. Chem. Ges. 1924, 57B, 813–818. [Google Scholar] [CrossRef]
- Nakamura, T.; Umeno, M. Triarylborane-Amine Compounds, and Their (co)Polymers, and Antifouling Agents. Japan Patent JP2000143673 A, 26 May 2000. [Google Scholar]
- Shimada, A.; Kohara, M.; Shibuya, Y. Triphenylboron-Containing Polymers and Their Use as Marine Antifouling Agents. WO9833829 A1, 6 August 1998. [Google Scholar]
- Saeki, Y.; Kumagaya, H. Synergistic Marine Antifouling Agents Containing Triphenylboranes and Higher Aliphatic Polyamines. Japan Patent JP10182322 A, 7 July 1998. [Google Scholar]
- Lawson, K.R.; Mound, W.R.; Whittingham, W.G. Preparation of Pyrazolylborane Derivatives as Fungicides in Plants. World Intellectual Property Organization. WO9711952 A1, 3 April 1997. [Google Scholar]
- Lawson, K.R.; Mound, W.R.; Whittingham, W.G. Preparation of Pyrimidinylborane Derivatives as Fungicides. World Intellectual Property Organization. WO9711951 A1, 3 April 1997. [Google Scholar]
- Tsang, T.H.; Strutzel, J.L. Preparation of Bis(amineborane)alkanes as Agrochemical Fungicides. U.S. Patent US5075292 A, 24 December 1991. [Google Scholar]
- Massey, A.G.; Park, A.J.; Stone, F.G.A. Tris(pentafluorophenyl)boron. Proc. Chem. Soc. 1963, 2, 212. [Google Scholar]
- Mayer, R.J.; Hampel, N.; Ofial, A.R. Lewis Acidic Boranes, Lewis Bases, and Equilibrium Constants: A Reliable Scaffold for a Quantitative Lewis Acidity/Basicity Scale. Chem. Eur. J. 2021, 27, 4070–4080. [Google Scholar] [CrossRef]
- Gao, H.; Battley, A.; Leitao, E.M. The Ultimate Lewis Acid Catalyst: Using Tris(Pentafluorophenyl) Borane to Create Bespoke Siloxane Architectures. Chem. Commun. 2022, 58, 7451–7465. [Google Scholar] [CrossRef] [PubMed]
- Nori, V.; Pesciaioli, F.; Sinibaldi, A.; Giorgianni, G.; Carlone, A. Boron-Based Lewis Acid Catalysis: Challenges and Perspectives. Catalysts 2022, 12, 5. [Google Scholar] [CrossRef]
- Kumar, G.; Roy, S.; Chatterjee, I. Tris(Pentafluorophenyl)Borane Catalyzed C–C and C–Heteroatom Bond Formation. Org. Biomol. Chem. 2021, 19, 1230–1267. [Google Scholar] [CrossRef] [PubMed]
- Hackel, T.; McGrath, N.A. Tris(Pentafluorophenyl)Borane-Catalyzed Reactions Using Silanes. Molecules 2019, 24, 432. [Google Scholar] [CrossRef]
- Park, S. B(C6F5)3-Catalyzed Sp3 C—Si Bond Forming Consecutive Reactions. Chin. J. Chem. 2019, 37, 1057–1071. [Google Scholar] [CrossRef]
- Lawson, J.R.; Melen, R.L. Tris(Pentafluorophenyl)Borane and Beyond: Modern Advances in Borylation Chemistry. Inorg. Chem. 2017, 56, 8627–8643. [Google Scholar] [CrossRef]
- Oestreich, M.; Hermeke, J.; Mohr, J. A Unified Survey of Si–H and H–H Bond Activation Catalysed by Electron-Deficient Boranes. Chem. Soc. Rev. 2015, 44, 2202–2220. [Google Scholar] [CrossRef]
- Brook, M.A.; Grande, J.B.; Ganachaud, F. New Synthetic Strategies for Structured Silicones Using B(C6F5)3. In Silicon Polymers; Muzafarov, A.M., Ed.; Advances in Polymer Science; Springer: Berlin/Heidelberg, Germany, 2010; Volume 235, pp. 161–183. ISBN 978-3-642-16047-9. [Google Scholar]
- Erker, G. Tris(Pentafluorophenyl)Borane: A Special Boron Lewis Acid for Special Reactions. Dalton Trans. 2005, 36, 1883–1890. [Google Scholar] [CrossRef]
- Piers, W.E.; Chivers, T. Pentafluorophenylboranes: From Obscurity to Applications. Chem. Soc. Rev. 1997, 26, 345. [Google Scholar] [CrossRef]
- Welch, G.C.; Juan, R.R.S.; Masuda, J.D.; Stephan, D.W. Reversible, Metal-Free Hydrogen Activation. Science 2006, 314, 1124–1126. [Google Scholar] [CrossRef]
- Welch, G.C.; Stephan, D.W. Facile Heterolytic Cleavage of Dihydrogen by Phosphines and Boranes. J. Am. Chem. Soc. 2007, 129, 1880–1881. [Google Scholar] [CrossRef]
- Spies, P.; Erker, G.; Kehr, G.; Bergander, K.; Fröhlich, R.; Grimme, S.; Stephan, D.W. Rapid Intramolecular Heterolytic Dihydrogen Activation by a Four-Membered Heterocyclic Phosphane–Borane Adduct. Chem. Commun. 2007, 5072–5074. [Google Scholar] [CrossRef]
- Mummadi, S.; Brar, A.; Wang, G.; Kenefake, D.; Diaz, R.; Unruh, D.K.; Li, S.; Krempner, C. “Inverse” Frustrated Lewis Pairs: An Inverse FLP Approach to the Catalytic Metal Free Hydrogenation of Ketones. Chem. Eur. J. 2018, 24, 16526–16531. [Google Scholar] [CrossRef]
- Paradies, J. Metal-Free Hydrogenation of Unsaturated Hydrocarbons Employing Molecular Hydrogen. Angew. Chem. Int. Ed. 2014, 53, 3552–3557. [Google Scholar] [CrossRef] [PubMed]
- Farrell, J.M.; Posaratnanathan, R.T.; Stephan, D.W. A Family of N-Heterocyclic Carbene-Stabilized Borenium Ions for Metal-Free Imine Hydrogenation Catalysis. Chem. Sci. 2015, 6, 2010–2015. [Google Scholar] [CrossRef]
- Erős, G.; Mehdi, H.; Pápai, I.; Rokob, T.A.; Király, P.; Tárkányi, G.; Soós, T. Expanding the Scope of Metal-Free Catalytic Hydrogenation through Frustrated Lewis Pair Design. Angew. Chem. Int. Ed. 2010, 49, 6559–6563. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.-H.; Kehr, G.; Fröhlich, R.; Wibbeling, B.; Schirmer, B.; Grimme, S.; Erker, G. Reaction of Frustrated Lewis Pairs with Conjugated Ynones-Selective Hydrogenation of the Carbon-Carbon Triple Bond. Angew. Chem. Int. Ed. 2011, 50, 7183–7186. [Google Scholar] [CrossRef]
- Chernichenko, K.; Madarász, Á.; Pápai, I.; Nieger, M.; Leskelä, M.; Repo, T. A Frustrated-Lewis-Pair Approach to Catalytic Reduction of Alkynes to Cis-Alkenes. Nature Chem. 2013, 5, 718–723. [Google Scholar] [CrossRef]
- Kobayashi, M.; Sanda, F.; Endo, T. Application of (Triphenylphosphinemethyl-Ene)Boranes to Thermally Latent Catalysts for Polyaddition of Bisphenol A Diglycidyl Ether with Bisphenol A: Model System of Epoxy−Novolac Resin. Macromolecules 2001, 34, 1134–1136. [Google Scholar] [CrossRef]
- Kobayashi, M.; Sanda, F.; Endo, T. Substituent Effect of (Triphenylphosphinemethylene)Boranes on Latent Catalytic Activity for Polyaddition of Bisphenol A Diglycidyl Ether with Bisphenol A: Model System of Epoxy−Novolac Resin. Macromolecules 2002, 35, 346–348. [Google Scholar] [CrossRef]
- Wang, L.; Kodamaa, K.; Hirose, T. DBU/benzyl bromide: An efficient catalytic system for the chemical fixation of CO2 into cyclic carbonates under metal- and solvent-free conditions. Catal. Sci. Technol. 2016, 6, 3872–3877. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, G.; Kodamaa, K.; Hirose, T. An efficient metal- and solvent-free organocatalytic system for chemical fixation of CO2 into cyclic carbonates under mild conditions. Green Chem. 2016, 18, 1229–1233. [Google Scholar] [CrossRef]
- Zhang, D.; Boopathi, S.K.; Hadjichristidis, N.; Gnanou, Y.; Feng, X. Metal-Free Alternating Copolymerization of CO2 with Epoxides: Fulfilling “Green” Synthesis and Activity. J. Am. Chem. Soc. 2016, 138, 11117–11120. [Google Scholar] [CrossRef]
- Yang, J.-L.; Wu, H.-L.; Li, Y.; Zhang, X.-H.; Darensbourg, D.J. Perfectly Alternating and Regioselective Copolymerization of Carbonyl Sulfide and Epoxides by Metal-Free Lewis Pairs. Angew. Chem. Int. Ed. 2017, 56, 5774–5779. [Google Scholar] [CrossRef] [PubMed]
- Andrea, K.A.; Kerton, F.M. Triarylborane-Catalyzed Formation of Cyclic Organic Carbonates and Polycarbonates. ACS Catal. 2019, 9, 1799–1809. [Google Scholar] [CrossRef]
- Andrea, K.A.; Kerton, F.M. Functionalized Polycarbonates via Triphenylborane Catalyzed Polymerization-Hydrosilylation. RSC Adv. 2019, 9, 26542–26546. [Google Scholar] [CrossRef]
- Andrea, K.A.; Wheeler, M.D.; Kerton, F.M. Borane Catalyzed Polymerization and Depolymerization Reactions Controlled by Lewis Acidic Strength. Chem. Commun. 2021, 57, 7320–7322. [Google Scholar] [CrossRef] [PubMed]
- Jia, M.; Hadjichristidis, N.; Gnanou, Y.; Feng, X. Polyurethanes from Direct Organocatalytic Copolymerization of p -Tosyl Isocyanate with Epoxides. Angew. Chem. Int. Ed. 2021, 60, 1593–1598. [Google Scholar] [CrossRef]
- Stephan, D.W. Catalysis without Precious Metals; Bullock, R.M., Ed.; Wiley-VCH: Weinheim, Germany, 2010; pp. 261–275. ISBN 978-3-527-32354-32358. [Google Scholar]
- Stephan, D.W.; Greenberg, S.; Graham, T.W.; Chase, P.; Hastie, J.J.; Geier, S.J.; Farrell, J.M.; Brown, C.C.; Heiden, Z.M.; Welch, G.C.; et al. Metal-Free Catalytic Hydrogenation of Polar Substrates by Frustrated Lewis Pairs. Inorg. Chem. 2011, 50, 12338–12348. [Google Scholar] [CrossRef] [PubMed]
- Stephan, D.W. “Frustrated Lewis Pair” Hydrogenations. Org. Biomol. Chem. 2012, 10, 5740. [Google Scholar] [CrossRef]
- Erker, G.; Stephan, D.W. Frustrated Lewis Pairs I: Uncovering and Understanding; Topics in current chemistry; Springer: Heidelberg, Germany; New York, NY, USA, 2013; ISBN 978-3-642-36697-0. [Google Scholar]
- Hounjet, L.J.; Stephan, D.W. Hydrogenation by Frustrated Lewis Pairs: Main Group Alternatives to Transition Metal Catalysts? Org. Process Res. Dev. 2014, 18, 385–391. [Google Scholar] [CrossRef]
- Rokob, T.A.; Hamza, A.; Pápai, I. Rationalizing the Reactivity of Frustrated Lewis Pairs: Thermodynamics of H 2 Activation and the Role of Acid−Base Properties. J. Am. Chem. Soc. 2009, 131, 10701–10710. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Aquino, A.J.A.; Cordes, D.B.; Hung-Low, F.; Hase, W.L.; Krempner, C. A Zwitterionic Carbanion Frustrated by Boranes—Dihydrogen Cleavage with Weak Lewis Acids via an “Inverse” Frustrated Lewis Pair Approach. J. Am. Chem. Soc. 2013, 135, 16066–16069. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.J.M.; Labinger, J.A.; Bercaw, J.E. Homogeneous CO Hydrogenation: Dihydrogen Activation Involves a Frustrated Lewis Pair Instead of a Platinum Complex. J. Am. Chem. Soc. 2010, 132, 3301–3303. [Google Scholar] [CrossRef]
- Mummadi, S.; Unruh, D.K.; Zhao, J.; Li, S.; Krempner, C. “Inverse” Frustrated Lewis Pairs—Activation of Dihydrogen with Organosuperbases and Moderate to Weak Lewis Acids. J. Am. Chem. Soc. 2016, 138, 3286–3289. [Google Scholar] [CrossRef] [PubMed]
- Geng, J.; Wu, Y.; Hu, X. Controlling the Lewis Acidity and Polymerizing Effectively Prevent Frustrated Lewis Pairs from Deactivation in the Hydrogenation of Terminal Alkynes. Org. Lett. 2021, 23, 3685–3690. [Google Scholar] [CrossRef]
- Tsushima, D.; Igarashi, M.; Sato, K.; Shimada, S. Ir-Catalyzed Hydrogenolysis Reaction of Silyl Triflates and Halides with H2. Chem. Lett. 2017, 46, 1532–1534. [Google Scholar] [CrossRef]
- Beppu, T.; Sakamoto, K.; Nakajima, Y.; Matsumoto, K.; Sato, K.; Shimada, S. Hydrosilane Synthesis via Catalytic Hydrogenolysis of Halosilanes Using a Metal-Ligand Bifunctional Iridium Catalyst. J. Organomet. Chem. 2018, 869, 75–80. [Google Scholar] [CrossRef]
- Glüer, A.; Schweizer, J.I.; Karaca, U.S.; Würtele, C.; Diefenbach, M.; Holthausen, M.C.; Schneider, S. Hydrosilane Synthesis by Catalytic Hydrogenolysis of Chlorosilanes and Silyl Triflates. Inorg. Chem. 2018, 57, 13822–13828. [Google Scholar] [CrossRef]
- Durin, G.; Berthet, J.-C.; Nicolas, E.; Thuéry, P.; Cantat, T. The Role of (tBuPOCOP)Ir(I) and Iridium(III) Pincer Complexes in the Catalytic Hydrogenolysis of Silyl Triflates into Hydrosilanes. Organometallics 2022, 41, 1786–1796. [Google Scholar] [CrossRef]
- Durin, G.; Berthet, J.-C.; Nicolas, E.; Cantat, T. Unlocking the Catalytic Hydrogenolysis of Chlorosilanes into Hydrosilanes with Superbases. ACS Catal. 2021, 11, 10855–10861. [Google Scholar] [CrossRef]
- Durin, G.; Fontaine, A.; Berthet, J.; Nicolas, E.; Thuéry, P.; Cantat, T. Metal-Free Catalytic Hydrogenolysis of Silyl Triflates and Halides into Hydrosilanes. Angew. Chem. Int. Ed. 2022, 61, e202200911. [Google Scholar] [CrossRef]
- Kira, M.; Müller, T. Dihydrogen Splitting Using Dialkylsilylene-Based Frustrated Lewis Pairs. Chem. Asian J. 2017, 12, 1204–1207. [Google Scholar] [CrossRef]
- Li, Y.; Molina de La Torre, J.A.; Grabow, K.; Bentrup, U.; Junge, K.; Zhou, S.; Brückner, A.; Beller, M. Selective Reduction of Amides to Amines by Boronic Acid Catalyzed Hydrosilylation. Angew. Chem. Int. Ed. 2013, 52, 11577–11580. [Google Scholar] [CrossRef]
- Chadwick, R.C.; Kardelis, V.; Lim, P.; Adronov, A. Metal-Free Reduction of Secondary and Tertiary N -Phenyl Amides by Tris(Pentafluorophenyl)Boron-Catalyzed Hydrosilylation. J. Org. Chem. 2014, 79, 7728–7733. [Google Scholar] [CrossRef]
- Blondiaux, E.; Cantat, T. Efficient Metal-Free Hydrosilylation of Tertiary, Secondary and Primary Amides to Amines. Chem. Commun. 2014, 50, 9349–9352. [Google Scholar] [CrossRef]
- Mukherjee, D.; Shirase, S.; Mashima, K.; Okuda, J. Chemoselective Reduction of Tertiary Amides to Amines Catalyzed by Triphenylborane. Angew. Chem. Int. Ed. 2016, 55, 13326–13329. [Google Scholar] [CrossRef]
- Mukherjee, D.; Sauer, D.F.; Zanardi, A.; Okuda, J. Selective Metal-Free Hydrosilylation of CO2 Catalyzed by Triphenylborane in Highly Polar, Aprotic Solvents. Chem. Eur. J. 2016, 22, 7730–7733. [Google Scholar] [CrossRef] [PubMed]
- Berkefeld, A.; Piers, W.E.; Parvez, M. Tandem Frustrated Lewis Pair/Tris(Pentafluorophenyl)Borane-Catalyzed Deoxygenative Hydrosilylation of Carbon Dioxide. J. Am. Chem. Soc. 2010, 132, 10660–10661. [Google Scholar] [CrossRef] [PubMed]
- Murata, T.; Hiyoshi, M.; Maekawa, S.; Saiki, Y.; Ratanasak, M.; Hasegawa, J.; Ema, T. Deoxygenative CO2 Conversions with Triphenylborane and Phenylsilane in the Presence of Secondary Amines or Nitrogen-Containing Aromatics. Green Chem. 2022, 24, 2385–2390. [Google Scholar] [CrossRef]
- Ratanasak, M.; Murata, T.; Adachi, T.; Hasegawa, J.; Ema, T. Mechanism of BPh3-Catalyzed N-Methylation of Amines with CO2 and Phenylsilane: Cooperative Activation of Hydrosilane. Chem. A Eur. J. 2022, 28, e202202210. [Google Scholar] [CrossRef]
- Scott, D.J.; Simmons, T.R.; Lawrence, E.J.; Wildgoose, G.G.; Fuchter, M.J.; Ashley, A.E. Facile Protocol for Water-Tolerant “Frustrated Lewis Pair”-Catalyzed Hydrogenation. ACS Catal. 2015, 5, 5540–5544. [Google Scholar] [CrossRef]
- Fasano, V.; Ingleson, M.J. Expanding Water/Base Tolerant Frustrated Lewis Pair Chemistry to Alkylamines Enables Broad Scope Reductive Aminations. Chem. Eur. J. 2017, 23, 2217–2224. [Google Scholar] [CrossRef] [PubMed]
- Fasano, V.; Radcliffe, J.E.; Ingleson, M.J. B(C6F5)3-Catalyzed Reductive Amination Using Hydrosilanes. ACS Catal. 2016, 6, 1793–1798. [Google Scholar] [CrossRef]
- Brar, A.; Unruh, D.K.; Ling, N.; Krempner, C. BPh3-Catalyzed [2+3] Cycloaddition of Ph3PCCO with Aldonitrones: Access to 5-Isoxazolidinones with Exocyclic Phosphonium Ylide Moieties. Org. Lett. 2019, 21, 6305–6309. [Google Scholar] [CrossRef]
- Brar, A.; Unruh, D.K.; Aquino, A.J.; Krempner, C. Lewis Acid Base Chemistry of Bestmann’s Ylide, Ph3PCCO, and Its Bulkier Analogue, (Cyclohexyl)3PCCO. Chem. Commun. 2019, 55, 3513–3516. [Google Scholar] [CrossRef]
- Riu, M.-L.Y.; Eckhardt, A.K.; Cummins, C.C. Dimerization and Cycloaddition Reactions of Transient Tri- Tert -Butylphosphacyclobutadiene Generated by Lewis Acid Induced Isomerization of Tri- Tert -Butylphosphatetrahedrane. J. Am. Chem. Soc. 2021, 143, 13005–13009. [Google Scholar] [CrossRef] [PubMed]
- Szynkiewicz, N.; Ponikiewski, Ł.; Grubba, R. Diphosphination of CO2 and CS2 Mediated by Frustrated Lewis Pairs—Catalytic Route to Phosphanyl Derivatives of Formic and Dithioformic Acid. Chem. Commun. 2019, 55, 2928–2931. [Google Scholar] [CrossRef] [PubMed]



















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mummadi, S.; Krempner, C. Triphenylborane in Metal-Free Catalysis. Molecules 2023, 28, 1340. https://doi.org/10.3390/molecules28031340
Mummadi S, Krempner C. Triphenylborane in Metal-Free Catalysis. Molecules. 2023; 28(3):1340. https://doi.org/10.3390/molecules28031340
Chicago/Turabian StyleMummadi, Suresh, and Clemens Krempner. 2023. "Triphenylborane in Metal-Free Catalysis" Molecules 28, no. 3: 1340. https://doi.org/10.3390/molecules28031340
APA StyleMummadi, S., & Krempner, C. (2023). Triphenylborane in Metal-Free Catalysis. Molecules, 28(3), 1340. https://doi.org/10.3390/molecules28031340