
Recent Advances in the Discovery of Nicotinic Acetylcholine Receptor Allosteric Modulators
 , ,
, ,  , , and
, , and 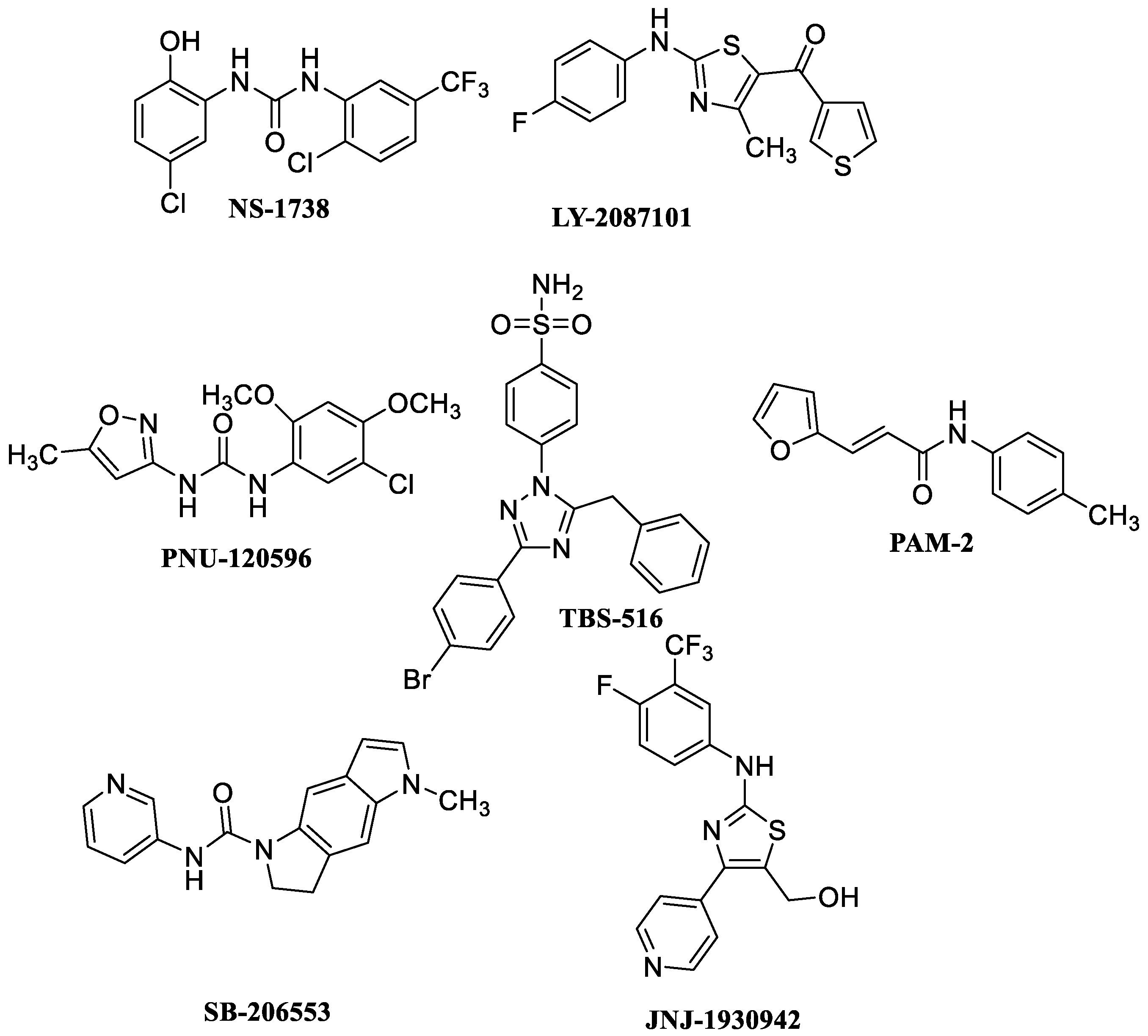{kind=link}
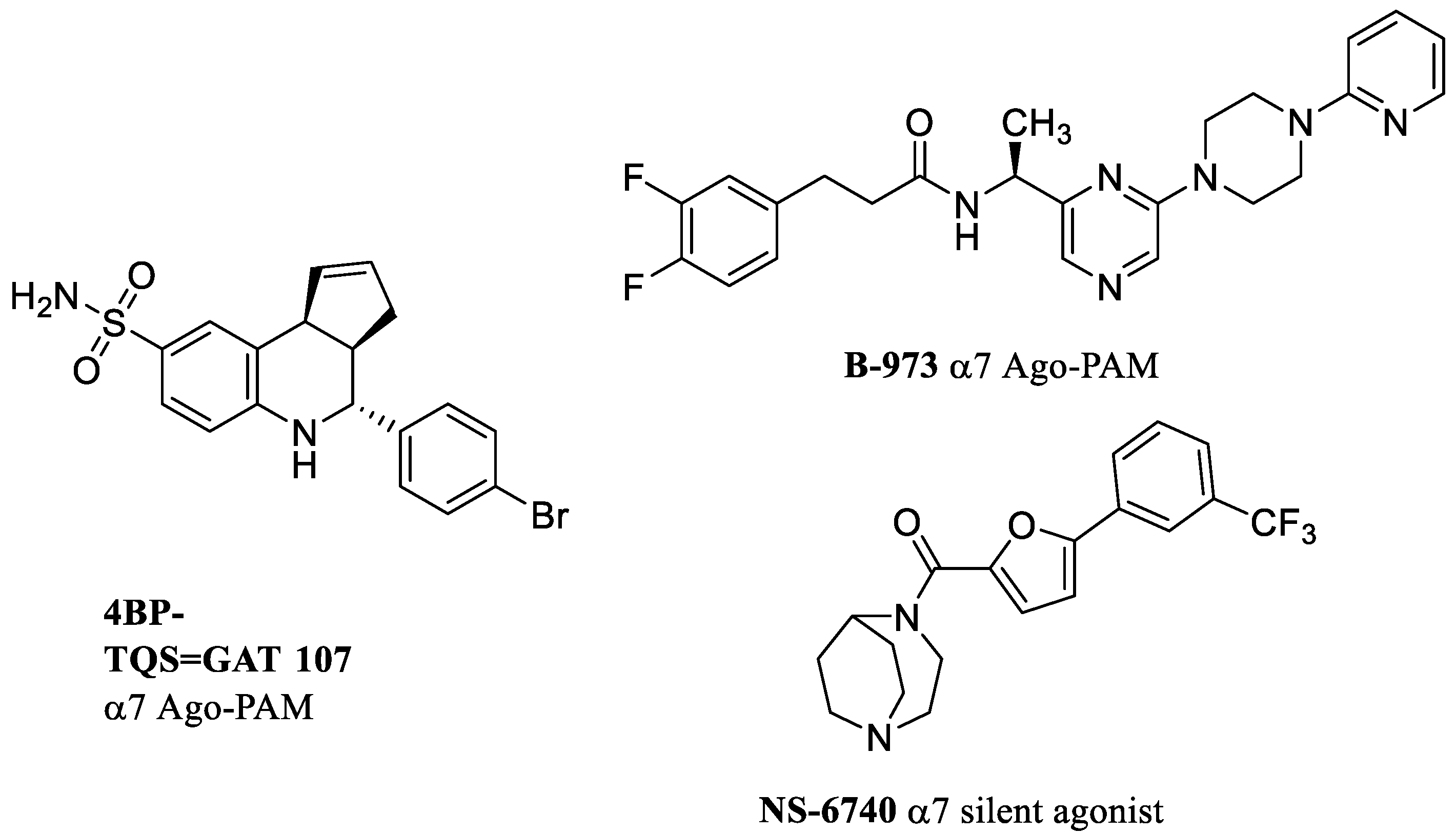{kind=link}
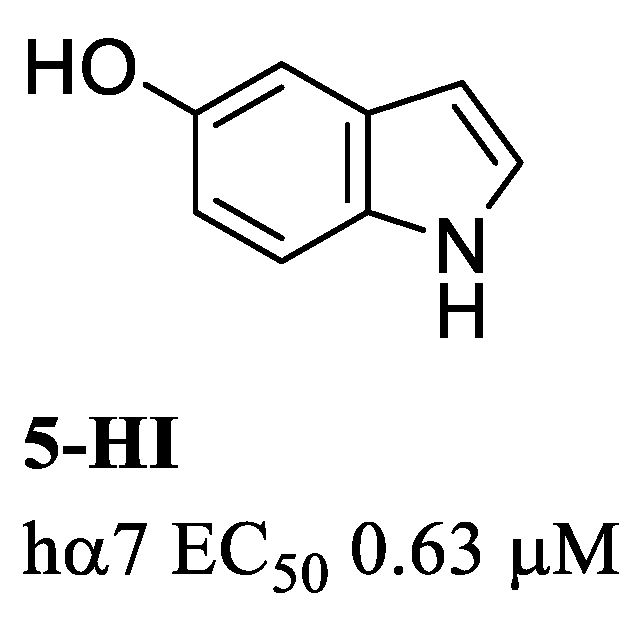{kind=link}
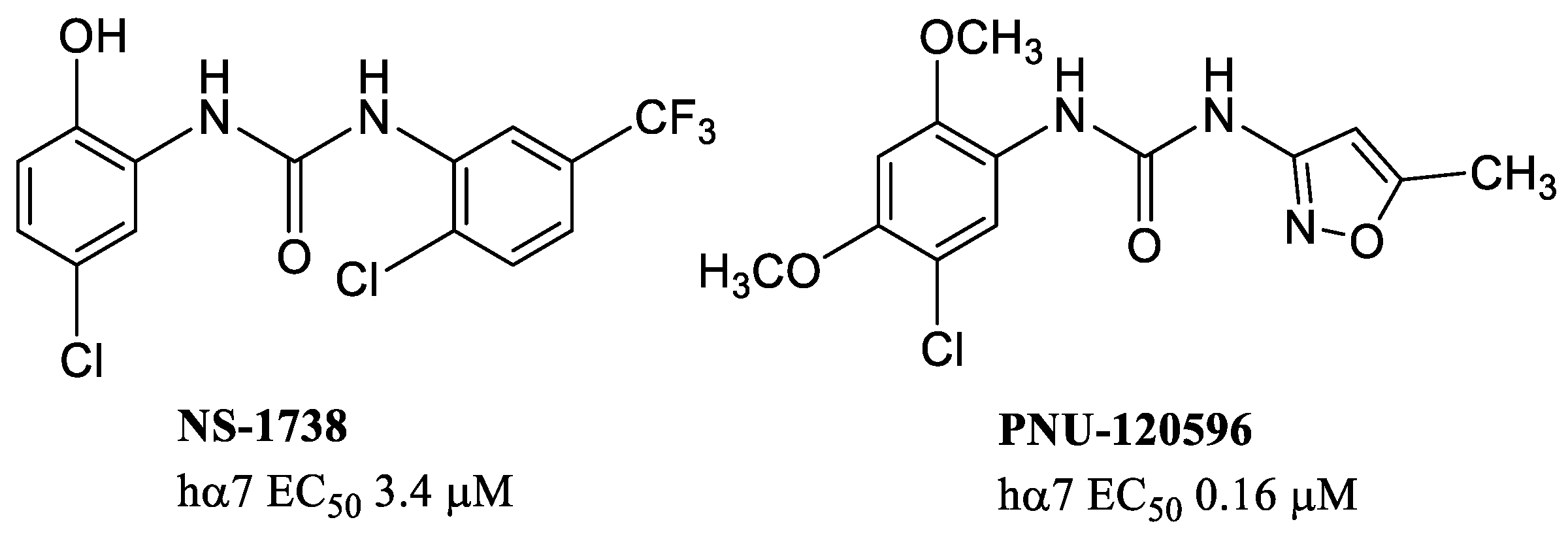{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. α7 Nicotinic Acetylcholine Receptors
3. The Positive Allosteric Modulation of α7 nAChRs
4. Allosteric Activating PAMs
5. Silent Allosteric Modulators
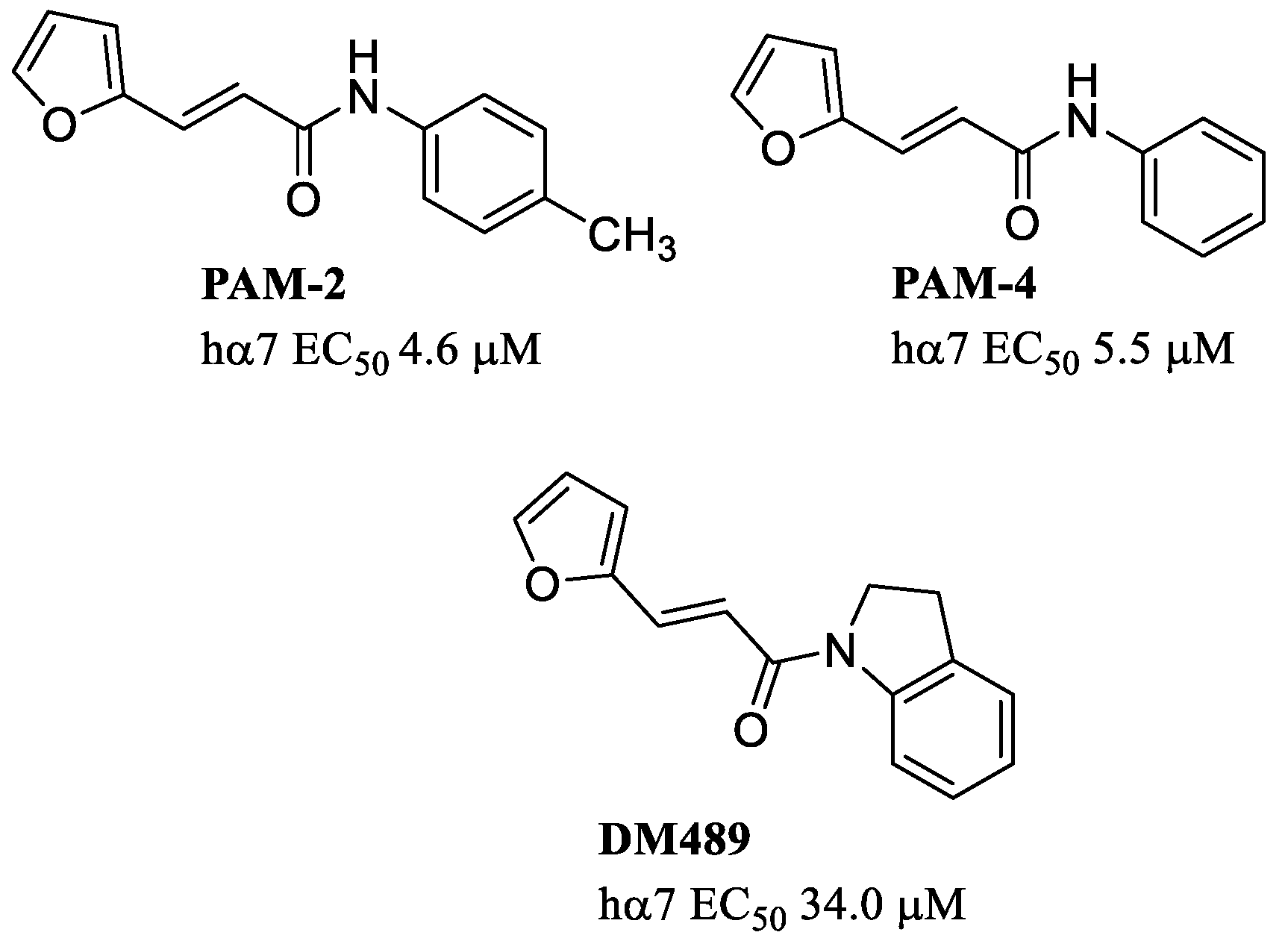
6. Silent Agonists
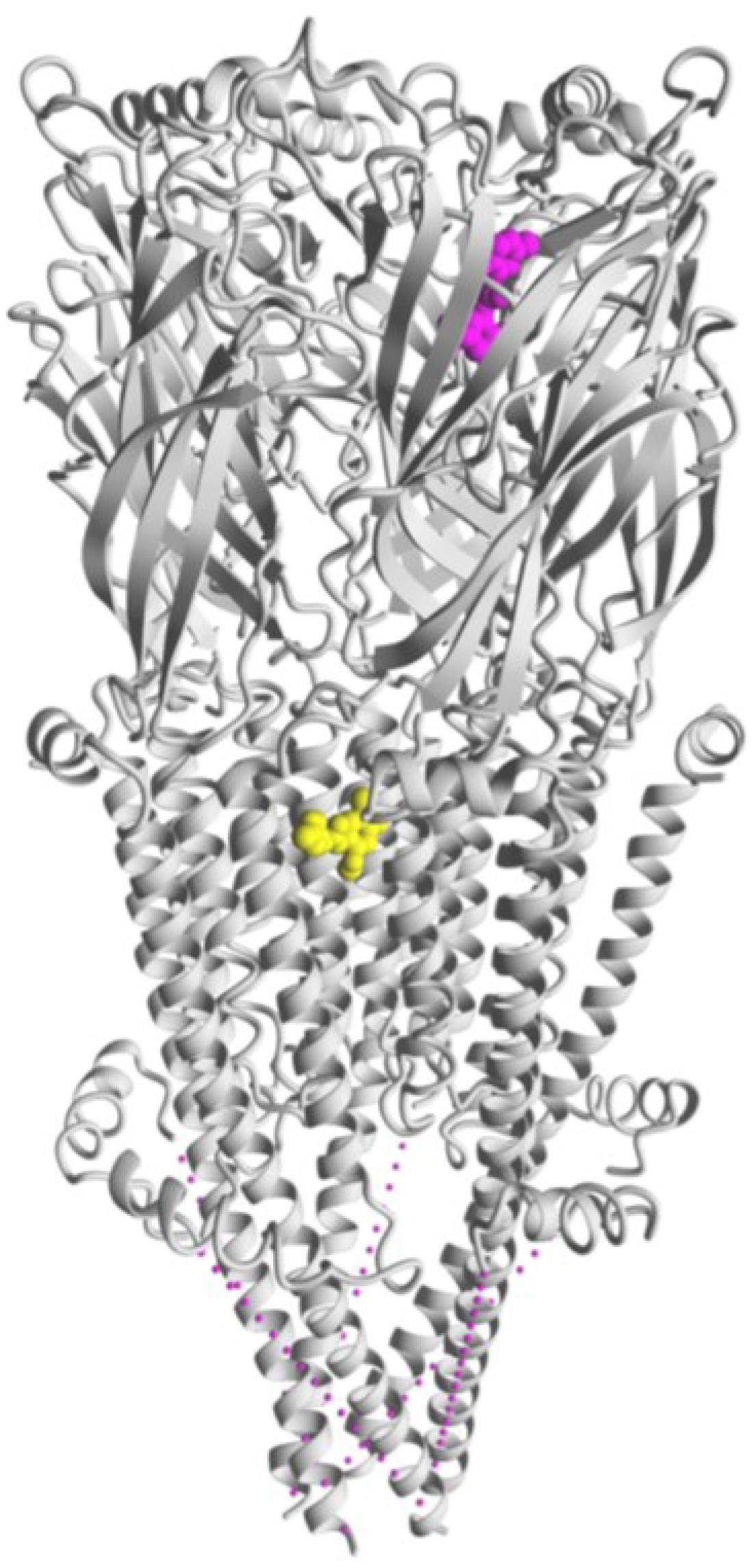
7. Allosteric Modulatory Sites at α7 nAChRs
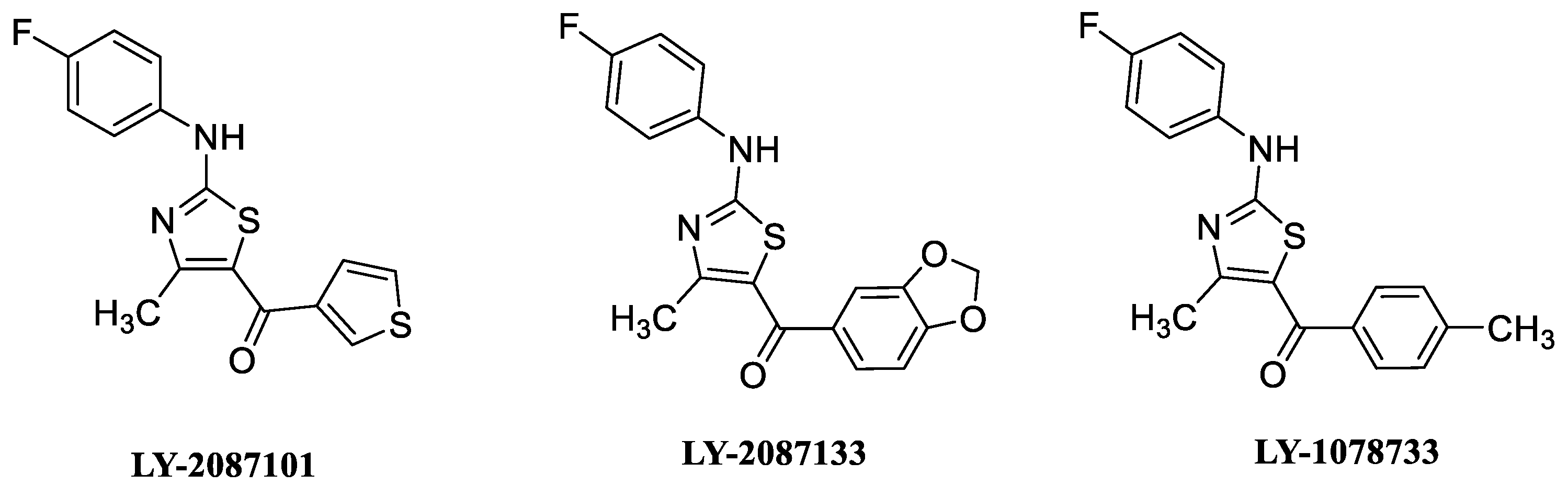
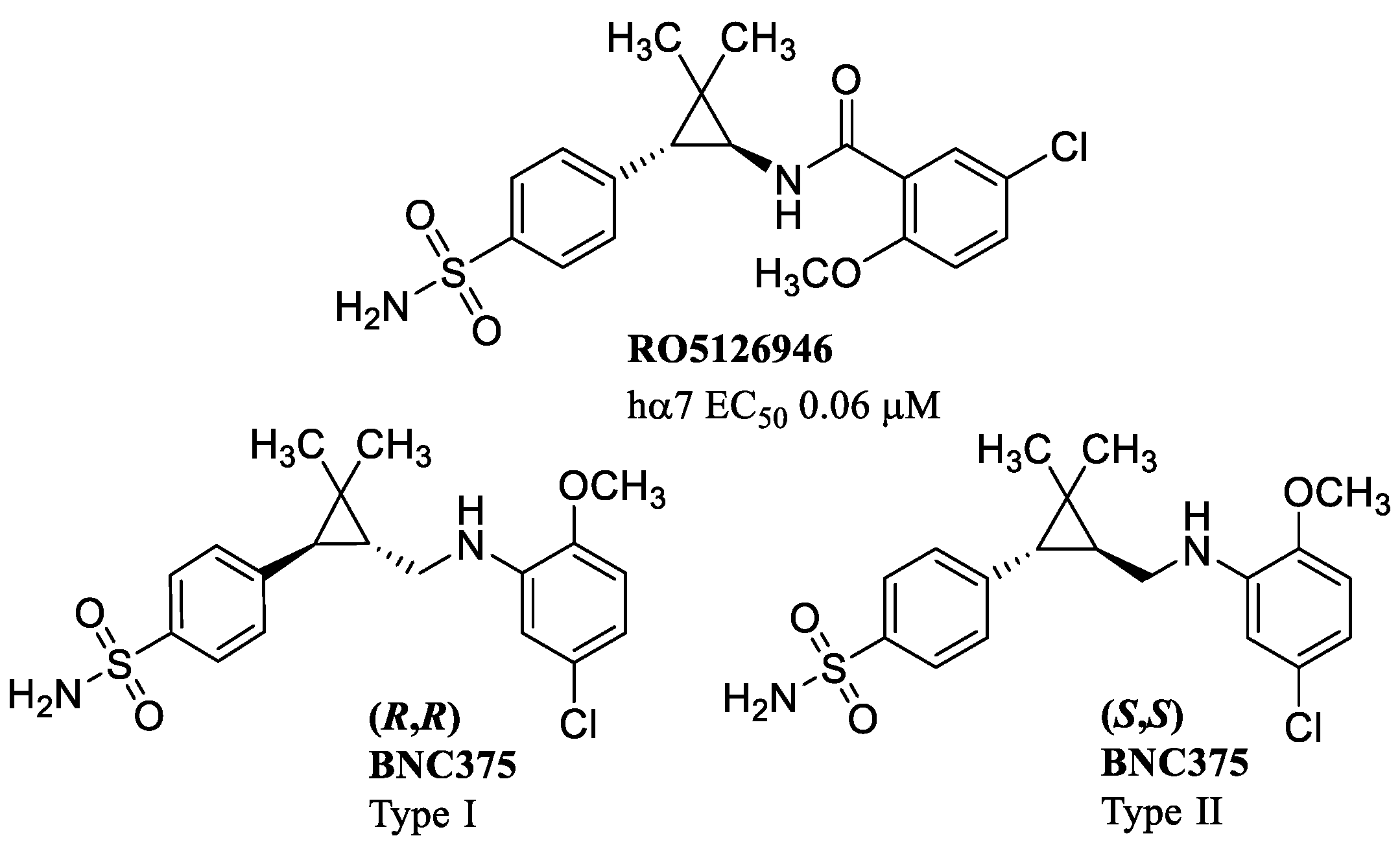
8. α7 PAMs
9. Positive Allosteric Modulators of the α4β2 nAChR
10. Allosteric Modulators at Other Non-α7 nAChR Subtypes
11. Negative Allosteric Modulators of nAChRs
12. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgement
Conflicts of Interest
References
- Romanelli, M.N.; Gratteri, P.; Guandalini, L.; Martini, E.; Bonaccini, C.; Gualtieri, F. Central Nicotinic Receptors: Structure, Function, Ligands, and Therapeutic Potential. ChemMedChem 2007, 2, 746–767. [Google Scholar] [CrossRef] [PubMed]
- Manetti, D.; Bellucci, C.; Chiaramonte, N.; Dei, S.; Teodori, E.; Romanelli, M.N. Designing selective modulators for the nicotinic receptor subtypes: Challenges and opportunities. Future Med. Chem. 2018, 10, 433–459. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.K.; Savolainen, M.; Young, G.T.; Zwart, R.; Sher, E.; Millar, N.S. Agonist activation of α7 nicotinic acetylcholine receptors via an allosteric transmembrane site. Proc. Natl. Acad. Sci. USA 2011, 108, 5867–5872. [Google Scholar] [CrossRef]
- Ng, H.J.; Whittemore, E.R.; Tran, M.B.; Hogenkamp, D.J.; Broide, R.S.; Johnstone, T.B.; Zheng, L.; Stevens, K.E.; Gee, K.W. Nootropic α7 nicotinic receptor allosteric modulator derived from GABAA receptor modulators. Proc. Natl. Acad. Sci. USA 2007, 104, 8059–8064. [Google Scholar] [CrossRef]
- Christensen, D.Z.; Mikkelsen, J.D.; Hansen, H.H.; Thomsen, M.S. Repeated administration of α7 nicotinic acetylcholine receptor (nAChR) agonists, but not positive allosteric modulators, increases α7 nAChR levels in the brain. J. Neurochem. 2010, 114, 1205–1216. [Google Scholar] [CrossRef]
- Wang, J.; Lindstrom, J. Orthosteric and allosteric potentiation of heteromeric neuronal nicotinic acetylcholine receptors. Br. J. Pharmacol. 2018, 175, 1805–1821. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.K.; Wang, J.; Papke, R.L. Positive allosteric modulators as an approach to nicotinic acetylcholine receptor-targeted therapeutics: Advantages and limitations. Biochem. Pharmacol. 2011, 82, 915–930. [Google Scholar] [CrossRef] [PubMed]
- Arias, H.R. Binding sites for exogenous and endogenous non-competitive inhibitors of the nicotinic acetylcholine receptor. Biochim. Et Biophys. Acta (BBA)-Rev. Biomembr. 1998, 1376, 173–220. [Google Scholar] [CrossRef]
- Wang, H.; Sun, X. Desensitized nicotinic receptors in brain. Brain Res. Rev. 2005, 48, 420–437. [Google Scholar] [CrossRef]
- Quick, M.W.; Lester, R.A.J. Desensitization of neuronal nicotinic receptors. J. Neurobiol. 2002, 53, 457–478. [Google Scholar] [CrossRef]
- Charpantier, E.; Wiesner, A.; Huh, K.-H.; Ogier, R.; Hoda, J.-C.; Allaman, G.; Raggenbass, M.; Feuerbach, D.; Bertrand, D.; Fuhrer, C. α7 Neuronal Nicotinic Acetylcholine Receptors Are Negatively Regulated by Tyrosine Phosphorylation and Src-Family Kinases. J. Neurosci. 2005, 25, 9836–9849. [Google Scholar] [CrossRef] [PubMed]
- Taly, A.; Corringer, P.-J.; Guedin, D.; Lestage, P.; Changeux, J.-P. Nicotinic receptors: Allosteric transitions and therapeutic targets in the nervous system. Nat Rev Drug Discov 2009, 8, 733–750. [Google Scholar] [CrossRef] [PubMed]
- Canning, B.J.; Liu, Q.; Tao, M.; Devita, R.; Perelman, M.; Hay, D.W.; Dicpinigaitis, P.V.; Liang, J. Evidence for Alpha7 Nicotinic Receptor Activation During the Cough Suppressing Effects Induced by Nicotine and Identification of ATA-101 as a Potential Novel Therapy for the Treatment of Chronic Cough. J. Pharmacol. Exp. Ther. 2022, 380, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Dajas-Bailador, F.A.; Mogg, A.J.; Wonnacott, S. Intracellular Ca2+ signals evoked by stimulation of nicotinic acetylcholine receptors in SH-SY5Y cells: Contribution of voltage-operated Ca2+ channels and Ca2+ stores. J. Neurochem. 2002, 81, 606–614. [Google Scholar] [CrossRef]
- Papke, R.L.; Bencherif, M.; Lippiello, P. An evaluation of neuronal nicotinic acetylcholine receptor activation by quaternary nitrogen compounds indicates that choline is selective for the α7 subtype. Neurosci. Lett. 1996, 213, 201–204. [Google Scholar] [CrossRef]
- Gulsevin, A.; Papke, R.L.; Stokes, C.; Garai, S.; Thakur, G.A.; Quadri, M.; Horenstein, N.A. Allosteric Agonism of α7 Nicotinic Acetylcholine Receptors: Receptor Modulation Outside the Orthosteric Site. Mol. Pharmacol. 2019, 95, 606–614. [Google Scholar] [CrossRef]
- Khiroug, S.S.; Harkness, P.C.; Lamb, P.W.; Sudweeks, S.N.; Khiroug, L.; Millar, N.S.; Yakel, J.L. Rat nicotinic ACh receptor α7 and β2 subunits co-assemble to form functional heteromeric nicotinic receptor channels. J. Physiol. 2002, 540, 425–434. [Google Scholar] [CrossRef]
- Williams, D.K.; Stokes, C.; Horenstein, N.A.; Papke, R.L. The effective opening of nicotinic acetylcholine receptors with single agonist binding sites. J. Gen. Physiol. 2011, 137, 369–384. [Google Scholar] [CrossRef]
- Papke, R.L.; Meyer, E.; Nutter, T.; Uteshev, V.V. α7 Receptor-selective agonists and modes of α7 receptor activation. Eur. J. Pharmacol. 2000, 393, 179–195. [Google Scholar] [CrossRef]
- Uteshev, V.V.; Meyer, E.M.; Papke, R.L. Activation and inhibition of native neuronal alpha-bungarotoxin-sensitive nicotinic ACh receptors. Brain Res. 2002, 948, 33–46. [Google Scholar] [CrossRef]
- Andersen, N.; Corradi, J.; Sine, S.M.; Bouzat, C. Stoichiometry for activation of neuronal α7 nicotinic receptors. Proc. Natl. Acad. Sci. USA 2013, 110, 20819–20824. [Google Scholar] [CrossRef] [PubMed]
- Revah, F.; Bertrand, D.; Galzi, J.L.; Devillers-Thiéry, A.; Mulle, C.; Hussy, N.; Bertrand, S.; Ballivet, M.; Changeux, J.P. Mutations in the channel domain alter desensitization of a neuronal nicotinic receptor. Nature 1991, 353, 846–849. [Google Scholar] [CrossRef]
- Mazurov, A.; Hauser, T.; Miller, C.H. Selective α7 Nicotinic Acetylcholine Receptor Ligands. Curr. Med. Chem. 2006, 13, 1567–1584. [Google Scholar] [CrossRef]
- Mazurov, A.A.; Speake, J.D.; Yohannes, D. Discovery and Development of α7 Nicotinic Acetylcholine Receptor Modulators. J. Med. Chem. 2011, 54, 7943–7961. [Google Scholar] [CrossRef]
- Hatton, G.I.; Yang, Q.Z. Synaptic Potentials Mediated by α7 Nicotinic Acetylcholine Receptors in Supraoptic Nucleus. J. Neurosci. 2002, 22, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Uteshev, V.V. The therapeutic promise of positive allosteric modulation of nicotinic receptors. Eur. J. Pharmacol. 2014, 727, 181–185. [Google Scholar] [CrossRef]
- Uteshev, V.V. Allosteric Modulation of Nicotinic Acetylcholine Receptors: The Concept and Therapeutic Trends. Curr. Pharm. Des. 2016, 22, 1986–1997. [Google Scholar] [CrossRef]
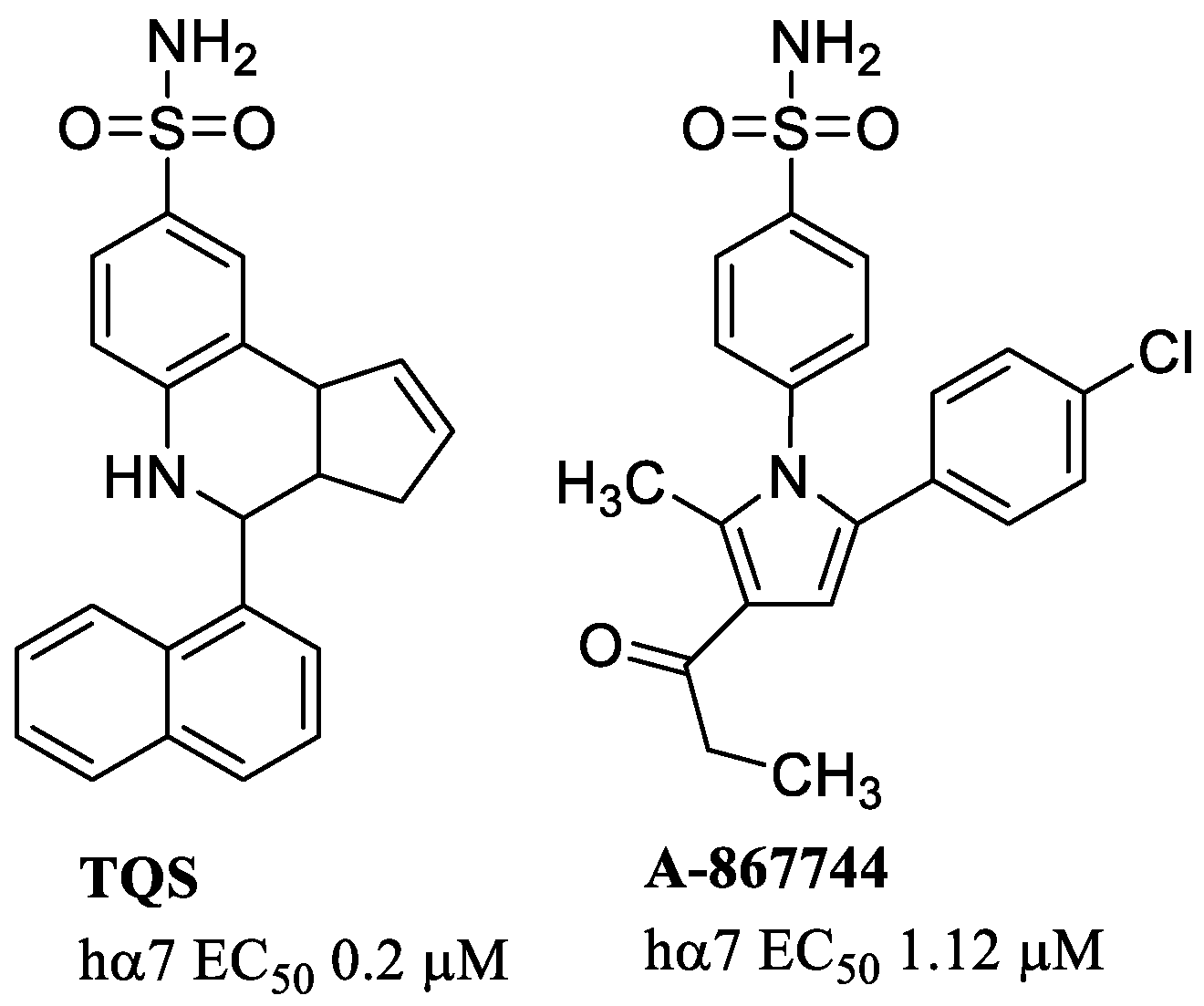
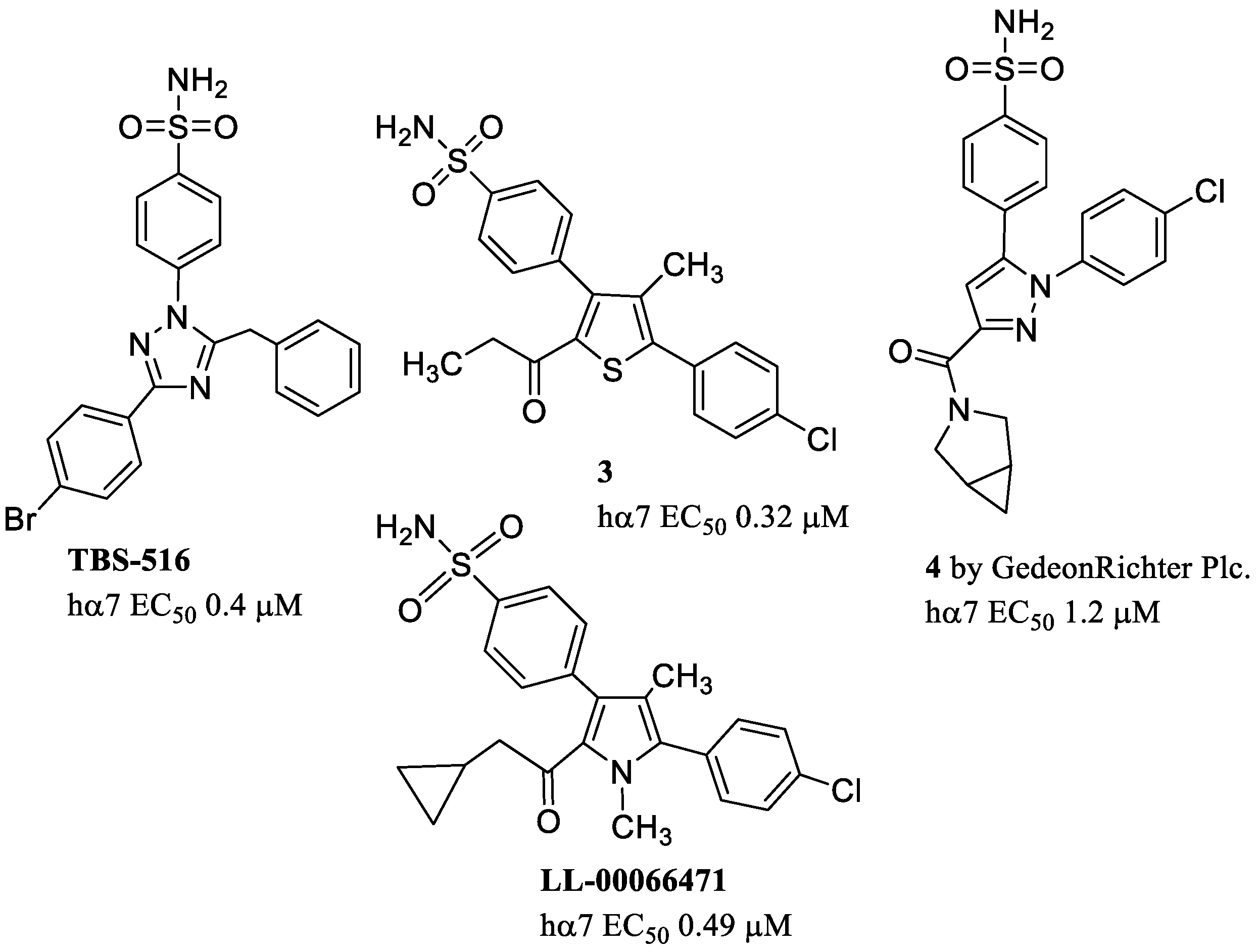
- Sinha, N.; Karche, N.P.; Verma, M.K.; Walunj, S.S.; Nigade, P.B.; Jana, G.; Kurhade, S.P.; Hajare, A.K.; Tilekar, A.R.; Jadhav, G.R.; et al. Discovery of Novel, Potent, Brain-Permeable, and Orally Efficacious Positive Allosteric Modulator of α7 Nicotinic Acetylcholine Receptor [4-(5-(4-Chlorophenyl)-4-methyl-2-propionylthiophen-3-yl)benzenesulfonamide]: Structure–Activity Relationship and Preclinical Characterization. J. Med. Chem. 2020, 63, 944–960. [Google Scholar] [CrossRef]
- Papke, R.L.; Horenstein, N.A. Therapeutic Targeting of α7 Nicotinic Acetylcholine Receptors. Pharmacol. Rev. 2021, 73, 1118–1149. [Google Scholar] [CrossRef]
- Bertrand, D.; Gopalakrishnan, M. Allosteric modulation of nicotinic acetylcholine receptors. Biochem. Pharmacol. 2007, 74, 1155–1163. [Google Scholar] [CrossRef]
- Targowska-Duda, K.M.; Kaczor, A.A.; Jozwiak, K.; Arias, H.R. Molecular interactions of type I and type II positive allosteric modulators with the human α7 nicotinic acetylcholine receptor: An in silico study. J. Biomol. Struct. Dyn. 2019, 37, 411–439. [Google Scholar] [CrossRef]
- Chatzidaki, A.; D’oyley, J.M.; Gill-Thind, J.K.; Sheppard, T.D.; Millar, N.S. The influence of allosteric modulators and transmembrane mutations on desensitisation and activation of α7 nicotinic acetylcholine receptors. Neuropharmacology 2015, 97, 75–85. [Google Scholar] [CrossRef]
- Dunlop, J.; Lock, T.; Jow, B.; Sitzia, F.; Grauer, S.; Jow, F.; Kramer, A.; Bowlby, M.R.; Randall, A.; Kowal, D.; et al. Old and New Pharmacology: Positive Allosteric Modulation of the α7 Nicotinic Acetylcholine Receptor by the 5-Hydroxytryptamine2B/C Receptor Antagonist SB-206553 (3,5-Dihydro-5-methyl-N-3-pyridinylbenzo[1,2-b:4,5-b′]di pyrrole-1(2H)-carboxamide). J. Pharmacol. Exp. Ther. 2009, 328, 766–776. [Google Scholar] [CrossRef] [PubMed]
- Dinklo, T.; Shaban, H.; Thuring, J.W.; Lavreysen, H.; Stevens, K.E.; Zheng, L.; Mackie, C.; Grantham, C.; Vandenberk, I.; Meulders, G.; et al. Characterization of 2-[[4-Fluoro-3-(trifluoromethyl)phenyl]amino]-4-(4-pyridinyl)-5-thiazolemethanol (JNJ-1930942), a Novel Positive Allosteric Modulator of the α7 Nicotinic Acetylcholine Receptor. J. Pharmacol. Exp. Ther. 2011, 336, 560–574. [Google Scholar] [CrossRef] [PubMed]
- Jindrichova, M.; Lansdell, S.J.; Millar, N.S. Changes in Temperature Have Opposing Effects on Current Amplitude in α7 and α4β2 Nicotinic Acetylcholine Receptors. PLoS ONE 2012, 7, e32073. [Google Scholar] [CrossRef] [PubMed]
- Andersen, N.D.; Nielsen, B.E.; Corradi, J.; Tolosa, M.F.; Feuerbach, D.; Arias, H.R.; Bouzat, C. Exploring the positive allosteric modulation of human α7 nicotinic receptors from a single-channel perspective. Neuropharmacology 2016, 107, 189–200. [Google Scholar] [CrossRef]
- Gill, J.K.; Chatzidaki, A.; Ursu, D.; Sher, E.; Millar, N.S. Contrasting Properties of α7-Selective Orthosteric and Allosteric Agonists Examined on Native Nicotinic Acetylcholine Receptors. PLoS ONE 2013, 8, e55047. [Google Scholar] [CrossRef]
- Horenstein, N.A.; Papke, R.L.; Kulkarni, A.R.; Chaturbhuj, G.U.; Stokes, C.; Manther, K.; Thakur, G.A. Critical Molecular Determinants of α7 Nicotinic Acetylcholine Receptor Allosteric Activation: Separation of direct allosteric activation and positive allosteric modulation. J. Biol. Chem. 2016, 291, 5049–5067. [Google Scholar] [CrossRef]
- Antonio-Tolentino, K.; Hopkins, C.R. Selective α7 nicotinic receptor agonists and positive allosteric modulators for the treatment of schizophrenia—A review. Expert Opin. Investig. Drugs 2020, 29, 603–610. [Google Scholar] [CrossRef]
- Post-Munson, D.J.; Pieschl, R.L.; Molski, T.F.; Graef, J.D.; Hendricson, A.W.; Knox, R.J.; Mcdonald, I.M.; Olson, R.E.; Macor, J.E.; Weed, M.R.; et al. B-973, a novel piperazine positive allosteric modulator of the α7 nicotinic acetylcholine receptor. Eur. J. Pharmacol. 2017, 799, 16–25. [Google Scholar] [CrossRef]
- Conn, J.P.; Christopoulos, A.; Lindsley, C.W. Allosteric modulators of GPCRs: A novel approach for the treatment of CNS disorders. Nat. Rev. Drug Discov. 2009, 8, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Noblin, D.; Bertekap, R.J.; Burford, N.; Hendricson, A.; Zhang, L.; Knox, R.; Banks, M.; O’connell, J.A.A. Development of a High-Throughput Calcium Flux Assay for Identification of All Ligand Types Including Positive, Negative, and Silent Allosteric Modulators for G Protein-Coupled Receptors. ASSAY Drug Dev. Technol. 2012, 10, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Horenstein, N.A.; Papke, R.L. Anti-inflammatory Silent Agonists. ACS Med. Chem. Lett. 2017, 8, 989–991. [Google Scholar] [CrossRef] [PubMed]
- Papke, R.L.; Lindstrom, J.M. Nicotinic acetylcholine receptors: Conventional and unconventional ligands and signaling. Neuropharmacology 2020, 168, 108021. [Google Scholar] [CrossRef] [PubMed]
- Briggs, C.A.; Grønlien, J.H.; Curzon, P.; Timmermann, D.B.; Ween, H.; Thorin-Hagene, K.; Kerr, P.; Anderson, D.J.; Malysz, J.; Dyhring, T.; et al. Role of channel activation in cognitive enhancement mediated by α7 nicotinic acetylcholine receptors. Br. J. Pharmacol. 2009, 158, 1486–1494. [Google Scholar] [CrossRef] [PubMed]
- Blunt, C.E.W.; Dougherty, D.A. Binding Interactions of NS6740, a Silent Agonist of the α7 Nicotinic Acetylcholine Receptor. Mol. Pharmacol. 2019, 96, 212–218. [Google Scholar] [CrossRef]
- Pismataro, M.C.; Horenstein, N.A.; Stokes, C.; Quadri, M.; De Amici, M.; Papke, R.L.; Dallanoce, C. Design, synthesis, and electrophysiological evaluation of NS6740 derivatives: Exploration of the structure-activity relationship for alpha7 nicotinic acetylcholine receptor silent activation. Eur. J. Med. Chem. 2020, 205, 112669. [Google Scholar] [CrossRef]
- Pismataro, M.C.; Horenstein, N.A.; Stokes, C.; Dallanoce, C.; Thakur, G.A.; Papke, R.L. Stable desensitization of α7 nicotinic acetylcholine receptors by NS6740 requires interaction with S36 in the orthosteric agonist binding site. Eur. J. Pharmacol. 2021, 905, 174179. [Google Scholar] [CrossRef]
- Bertrand, D.; Bertrand, S.; Cassar, S.; Gubbins, E.; Li, J.; Gopalakrishnan, M. Positive Allosteric Modulation of the α7 Nicotinic Acetylcholine Receptor: Ligand Interactions with Distinct Binding Sites and Evidence for a Prominent Role of the M2-M3 Segment. Mol. Pharmacol. 2008, 74, 1407–1416. [Google Scholar] [CrossRef]
- Young, G.T.; Zwart, R.; Walker, A.S.; Sher, E.; Millar, N.S. Potentiation of alpha7 nicotinic acetylcholine receptors via an allosteric transmembrane site. Proc. Natl. Acad. Sci. USA 2008, 105, 14686–14691. [Google Scholar] [CrossRef]
- Cecchini, M.; Changeux, J.-P. The nicotinic acetylcholine receptor and its prokaryotic homologues: Structure, conformational transitions & allosteric modulation. Neuropharmacology 2015, 96, 137–149. [Google Scholar] [PubMed]
- Mclaughlin, J.T.; Barron, S.C.; See, J.A.; Rosenberg, R.L. Conformational changes in α7 acetylcholine receptors underlying allosteric modulation by divalent cations. BMC Pharmacol. 2009, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- Eddins, D.; Lyford, L.K.; Lee, J.W.; Desai, S.A.; Rosenberg, R.L. Permeant but not impermeant divalent cations enhance activation of nondesensitizing α7 nicotinic receptors. Am. J. Physiol.-Cell Physiol. 2002, 282, C796–C804. [Google Scholar] [CrossRef]
- Zwart, R.; De Filippi, G.; Broad, L.M.; Mcphie, G.I.; Pearson, K.H.; Baldwinson, T.; Sher, E. 5-Hydroxyindole potentiates human α7 nicotinic receptor-mediated responses and enhances acetylcholine-induced glutamate release in cerebellar slices. Neuropharmacology 2002, 43, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Timmermann, D.B.; Grønlien, J.H.; Kohlhaas, K.L.; Nielsen, E.Ø.; Dam, E.; Jørgensen, T.D.; Ahring, P.K.; Peters, D.; Holst, D.; Chrsitensen, J.K.; et al. An Allosteric Modulator of the α7 Nicotinic Acetylcholine Receptor Possessing Cognition-Enhancing Properties in Vivo. J. Pharmacol. Exp. Ther. 2007, 323, 294–307. [Google Scholar] [CrossRef] [PubMed]
- Chatzidaki, A.; Millar, N.S. Allosteric modulation of nicotinic acetylcholine receptors. Biochem. Pharmacol. 2015, 97, 408–417. [Google Scholar] [CrossRef]
- Guerra-Álvarez, M.; Moreno-Ortega, A.J.; Navarro, E.; Fernández-Morales, J.C.; Egea, J.; López, M.G.; Cano-Abad, M.F. Positive allosteric modulation of alpha-7 nicotinic receptors promotes cell death by inducing Ca2+ release from the endoplasmic reticulum. J. Neurochem. 2015, 133, 309–319. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, S.; Zhou, Y.; Zhang, M.; Chen, H.; Eric Xu, H.; Sun, D.; Liu, L.; Tian, C. Structural basis of human α7 nicotinic acetylcholine receptor activation. Cell Res. 2021, 31, 713–716. [Google Scholar] [CrossRef] [PubMed]
- Broad, L.M.; Zwart, R.; Pearson, K.H.; Lee, M.; Wallace, L.; Mcphie, G.I.; Emkey, R.; Hollinshead, S.P.; Dell, C.P.; Baker, S.R.; et al. Identification and pharmacological profile of a new class of selective nicotinic acetylcholine receptor potentiators. J. Pharmacol. Exp. Ther. 2006, 318, 1108–1117. [Google Scholar] [CrossRef]
- Sahdeo, S.; Wallace, T.; Hirakawa, R.; Knoflach, F.; Bertrand, D.; Maag, H.; Misner, D.; Tombaugh, G.C.; Santarelli, L.; Brameld, K.; et al. Characterization of RO5126946, a Novel α7 Nicotinic Acetylcholine Receptor–Positive Allosteric Modulator. J. Pharmacol. Exp. Ther. 2014, 350, 455–468. [Google Scholar] [CrossRef]
- Harvey, A.J.; Avery, T.D.; Schaeffer, L.; Joseph, C.; Huff, B.C.; Singh, R.; Morice, C.; Giethlen, B.; Grishin, A.A.; Coles, C.J.; et al. Discovery of BNC375, a Potent, Selective, and Orally Available Type I Positive Allosteric Modulator of α7 nAChRs. ACS Med. Chem. Lett. 2019, 10, 754–760. [Google Scholar] [CrossRef] [PubMed]
- Arias, H.R.; Gu, R.-X.; Feuerbach, D.; Guo, B.-B.; Ye, Y.; Wei, D.-Q. Novel Positive Allosteric Modulators of the Human α7 Nicotinic Acetylcholine Receptor. Biochemistry 2011, 50, 5263–5278. [Google Scholar] [CrossRef] [PubMed]
- Targowska-Duda, K.M.; Budzynska, B.; Michalak, A.; Jozwiak, K.; Biala, G.; Arias, H.R. 3-Furan-2-yl-N-p-tolyl-acrylamide, a highly selective positive allosteric modulator of α7 nicotinic receptors, produces anxiolytic-like activity in mice. J. Psychopharmacol. 2019, 33, 558–567. [Google Scholar] [CrossRef] [PubMed]
- Targowska-Duda, K.M.; Budzynska, B.; Michalak, A.; Wnorowski, A.; Loland, C.J.; Maj, M.; Manetti, D.; Romanelli, M.N.; Jozwiak, K.; Biala, G.; et al. Type I and type II positive allosteric modulators of α7 nicotinic acetylcholine receptors induce antidepressant-like activity in mice by a mechanism involving receptor potentiation but not neurotransmitter reuptake inhibition. Correlation with mTOR intracellular pathway activation. Eur. Neuropsychopharmacol. 2021, 52, 31–47. [Google Scholar] [CrossRef]
- Potasiewicz, A.; Hołuj, M.; Kos, T.; Popik, P.; Arias, H.R.; Nikiforuk, A. 3-Furan-2-yl-N-p-tolyl-acrylamide, a positive allosteric modulator of the α7 nicotinic receptor, reverses schizophrenia-like cognitive and social deficits in rats. Neuropharmacology 2017, 113 (Pt A), 188–197. [Google Scholar] [CrossRef]
- Phenis, D.; Vunck, S.A.; Valentini, V.; Arias, H.; Schwarcz, R.; Bruno, J.P. Activation of alpha7 nicotinic and NMDA receptors is necessary for performance in a working memory task. Psychopharmacology 2020, 237, 1723–1735. [Google Scholar] [CrossRef] [PubMed]
- Bagdas, D.; Sevdar, G.; Gul, Z.; Younis, R.; Cavun, S.; Tae, H.-S.; Ortells, M.O.; Arias, H.R.; Gurun, M.S. (E)-3-furan-2-yl-N-phenylacrylamide (PAM-4) decreases nociception and emotional manifestations of neuropathic pain in mice by α7 nicotinic acetylcholine receptor potentiation. Neurol. Res. 2021, 43, 1056–1068. [Google Scholar] [CrossRef]
- Arias, H.R.; Ghelardini, C.; Lucarini, E.; Tae, H.-S.; Yousuf, A.; Marcovich, I.; Manetti, D.; Romanelli, M.N.; Elgoyhen, A.B.; Adams, D.J.; et al. (E)-3-Furan-2-yl-N-p-tolyl-acrylamide and its Derivative DM489 Decrease Neuropathic Pain in Mice Predominantly by α7 Nicotinic Acetylcholine Receptor Potentiation. ACS Chem. Neurosci. 2020, 11, 3603–3614. [Google Scholar] [CrossRef]
- Faghih, R.; Gopalakrishnan, M.; Briggs, C.A. Allosteric Modulators of the α7 Nicotinic Acetylcholine Receptor. J. Med. Chem. 2008, 51, 701–712. [Google Scholar] [CrossRef]
- Grønlien, J.H.; Håkerud, M.; Ween, H.; Thorin-Hagene, K.; Briggs, C.A.; Gopalakrishnan, M.; Malysz, J. Distinct Profiles of α7 nAChR Positive Allosteric Modulation Revealed by Structurally Diverse Chemotypes. Mol. Pharmacol. 2007, 72, 715–724. [Google Scholar] [CrossRef]
- Malysz, J.; Grønlien, J.H.; Anderson, D.J.; Håkerud, M.; Thorin-Hagene, K.; Ween, H.; Wetterstrand, C.; Briggs, C.A.; Faghih, R.; Bunnelle, W.H.; et al. In Vitro Pharmacological Characterization of a Novel Allosteric Modulator of α7 Neuronal Acetylcholine Receptor, 4-(5-(4-Chlorophenyl)-2-methyl-3-propionyl-1H-pyrrol-1-yl)benzenesulfonamide (A-867744), Exhibiting Unique Pharmacological Profile. J. Pharmacol. Exp. Ther. 2009, 330, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Pesti, K.; Lukacs, P.; Mike, A. Type I-like behavior of the type II α7 nicotinic acetylcholine receptor positive allosteric modulator A-867744. PeerJ 2019, 7, e7542. [Google Scholar] [CrossRef]
- Gill-Thind, J.K.; Dhankher, P.; D’oyley, J.M.; Sheppard, T.D.; Millar, N.S. Structurally Similar Allosteric Modulators of α7 Nicotinic Acetylcholine Receptors Exhibit Five Distinct Pharmacological Effects. J. Biol. Chem. 2015, 290, 3552–3562. [Google Scholar] [CrossRef] [PubMed]
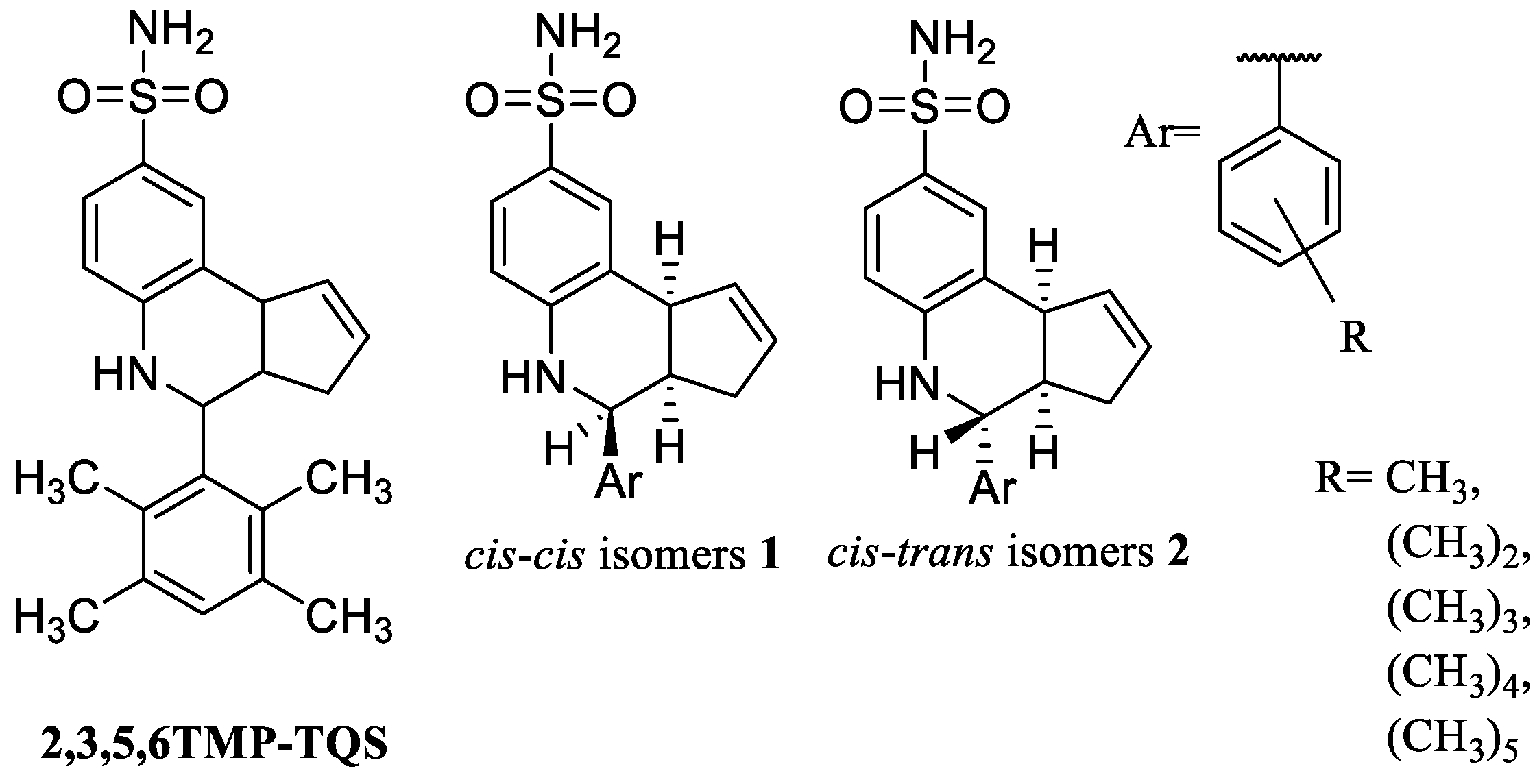
- Papke, R.L.; Garai, S.; Stokes, C.; Horenstein, N.A.; Zimmerman, A.D.; Abboud, K.A.; Thakur, G.A. Differing Activity Profiles of the Stereoisomers of 2,3,5,6TMP-TQS, a Putative Silent Allosteric Modulator of α7 nAChR. Mol. Pharmacol. 2020, 98, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Newcombe, J.; Chatzidaki, A.; Sheppard, T.D.; Topf, M.; Millar, N.S. Diversity of Nicotinic Acetylcholine Receptor Positive Allosteric Modulators Revealed by Mutagenesis and a Revised Structural Model. Mol. Pharmacol. 2018, 93, 128–140. [Google Scholar] [CrossRef]
- Verma, M.K.; Goel, R.N.; Bokare, A.M.; Dandekar, M.P.; Koul, S.; Desai, S.; Tota, S.; Singh, N.; Nigade, P.B.; Patil, V.B.; et al. LL-00066471, a novel positive allosteric modulator of α7 nicotinic acetylcholine receptor ameliorates cognitive and sensorimotor gating deficits in animal models: Discovery and preclinical characterization. Eur. J. Pharmacol. 2021, 891, 173685. [Google Scholar] [CrossRef]
- Sinha, N.; Karche, N.P.; Tilekar, A.R.; Palle, V.P.; Kamboj, R.K. 4-(5-(4-chlorophenyl)-2-(2-cycloprylacetyl)-1,4-dimethyl-1H-pyrrol-3-yl)benzenesufonamide as alpha 7 nAChR Modulator. WO 2014195848 A1. 2014. [Google Scholar]
- Ledneczki, I.; Tapolcsányi, P.; Gábor, E.; Visegrády, A.; Vass, M.; Éles, J.; Holm, P.; Horváth, A.; Pocsai, A.; Mahó, S.; et al. Discovery of novel positive allosteric modulators of the α7 nicotinic acetylcholine receptor: Scaffold hopping approach. Eur. J. Med. Chem. 2021, 214, 113189. [Google Scholar] [CrossRef]
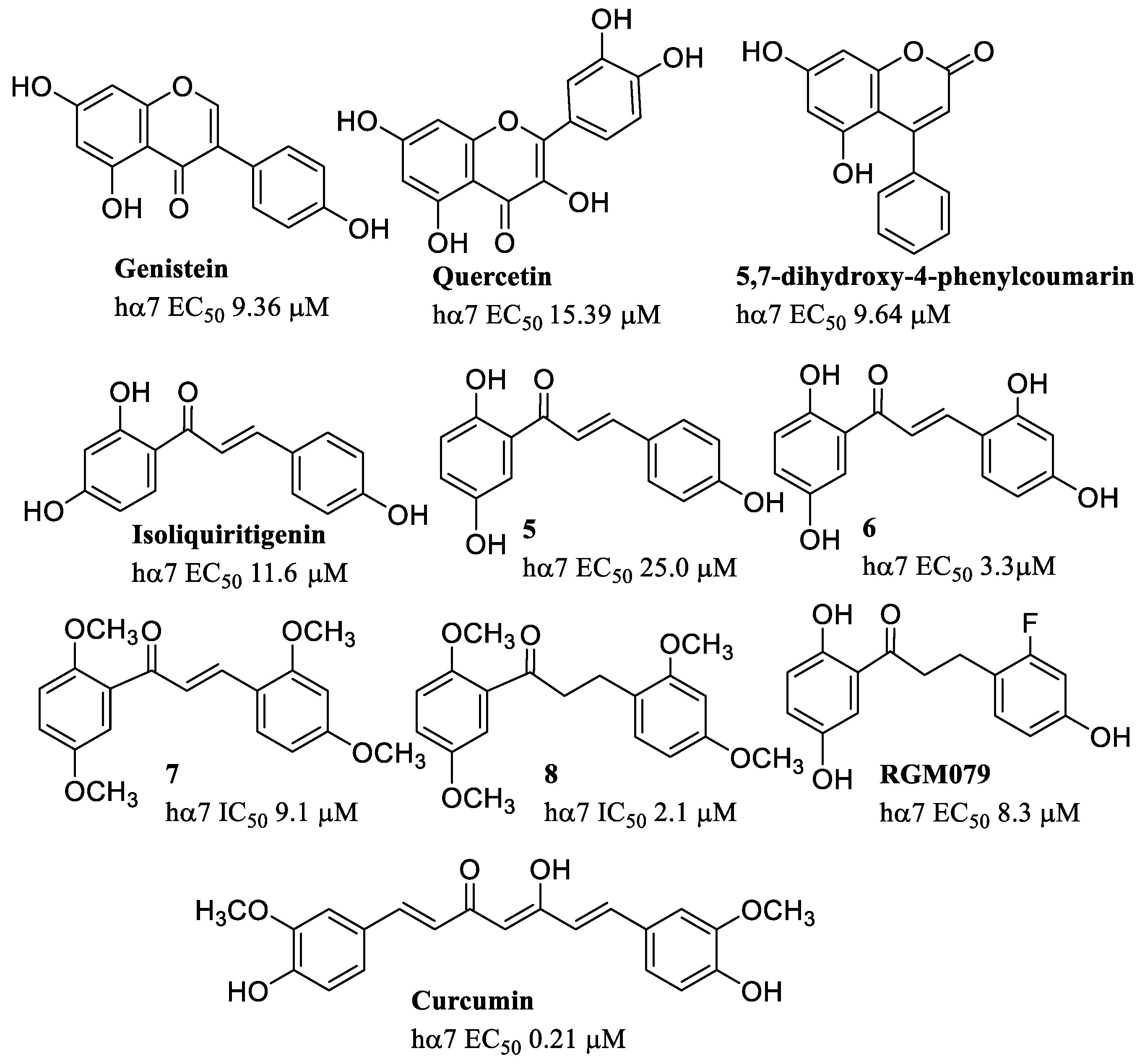
- Nielsen, B.E.; Bermudez, I.; Bouzat, C. Flavonoids as positive allosteric modulators of α7 nicotinic receptors. Neuropharmacology 2019, 160, 107794. [Google Scholar] [CrossRef]
- Balsera, B.; Mulet, J.; Fernández-Carvajal, A.; Torre-Martínez, R.D.L.; Ferrer-Montiel, A.; Hernández-Jiménez, J.G.; Estévez-Herrera, J.; Borges, R.; Freitas, A.E.; López, M.G.; et al. Chalcones as positive allosteric modulators of α7 nicotinic acetylcholine receptors: A new target for a privileged structure. Eur. J. Med. Chem. 2014, 86, 724–739. [Google Scholar] [CrossRef]
- Criado, M.; Balsera, B.; Mulet, J.; Sala, S.; Sala, F.; De La Torre-Martínez, R.; Fernández-Carvajal, A.; Ferrer-Montie, L.; Moreno-Fernández, S.; Miguel, M.; et al. 1,3-diphenylpropan-1-ones as allosteric modulators of α7 nACh receptors with analgesic and antioxidant properties. Future Med. Chem. 2016, 8, 731–749. [Google Scholar] [CrossRef]
- Pérez De Vega, M.J.; Fernandez-Mendivil, C.; De La Torre Martínez, R.; González-Rodríguez, S.; Mullet, J.; Sala, F.; Sala, S.; Criado, M.; Moreno-Fernández, S.; Miguel, M.; et al. 1-(2′,5′-Dihydroxyphenyl)-3-(2-fluoro-4-hydroxyphenyl)-1-propanone (RGM079): A Positive Allosteric Modulator of α7 Nicotinic Receptors with Analgesic and Neuroprotective Activity. ACS Chem. Neurosci. 2019, 10, 3900–3909. [Google Scholar] [CrossRef]
- Nelson, K.M.; Dahlin, J.L.; Bisson, J.; Graham, J.; Pauli, G.F.; Walters, M.A. The Essential Medicinal Chemistry of Curcumin. J. Med. Chem. 2017, 60, 1620–1637. [Google Scholar] [CrossRef] [PubMed]
- El Nebrisi, E.G.; Bagdas, D.; Toma, W.; Al Samri, H.; Brodzik, A.; Alkhlaif, Y.; Yang, K.-H.S.; Howarth, F.C.; Damaj, I.M.; Oz, M. Curcumin Acts as a Positive Allosteric Modulator of α7-Nicotinic Acetylcholine Receptors and Reverses Nociception in Mouse Models of Inflammatory Pain. J. Pharmacol. Exp. Ther. 2018, 365, 190–200. [Google Scholar] [CrossRef]
- Perkins, K.A.; Roy Chengappa, K.N.; Karelitz, J.L.; Boldry, M.C.; Michael, V.; Herb, T.; Gannon, J.; Brar, J.; Ford, L.; Rassnick, S.; et al. Initial Cross-Over Test of A Positive Allosteric Modulator of Alpha-7 Nicotinic Receptors to Aid Cessation in Smokers With Or Without Schizophrenia. Neuropsychopharmacology 2018, 43, 1334–1342. [Google Scholar] [CrossRef] [PubMed]
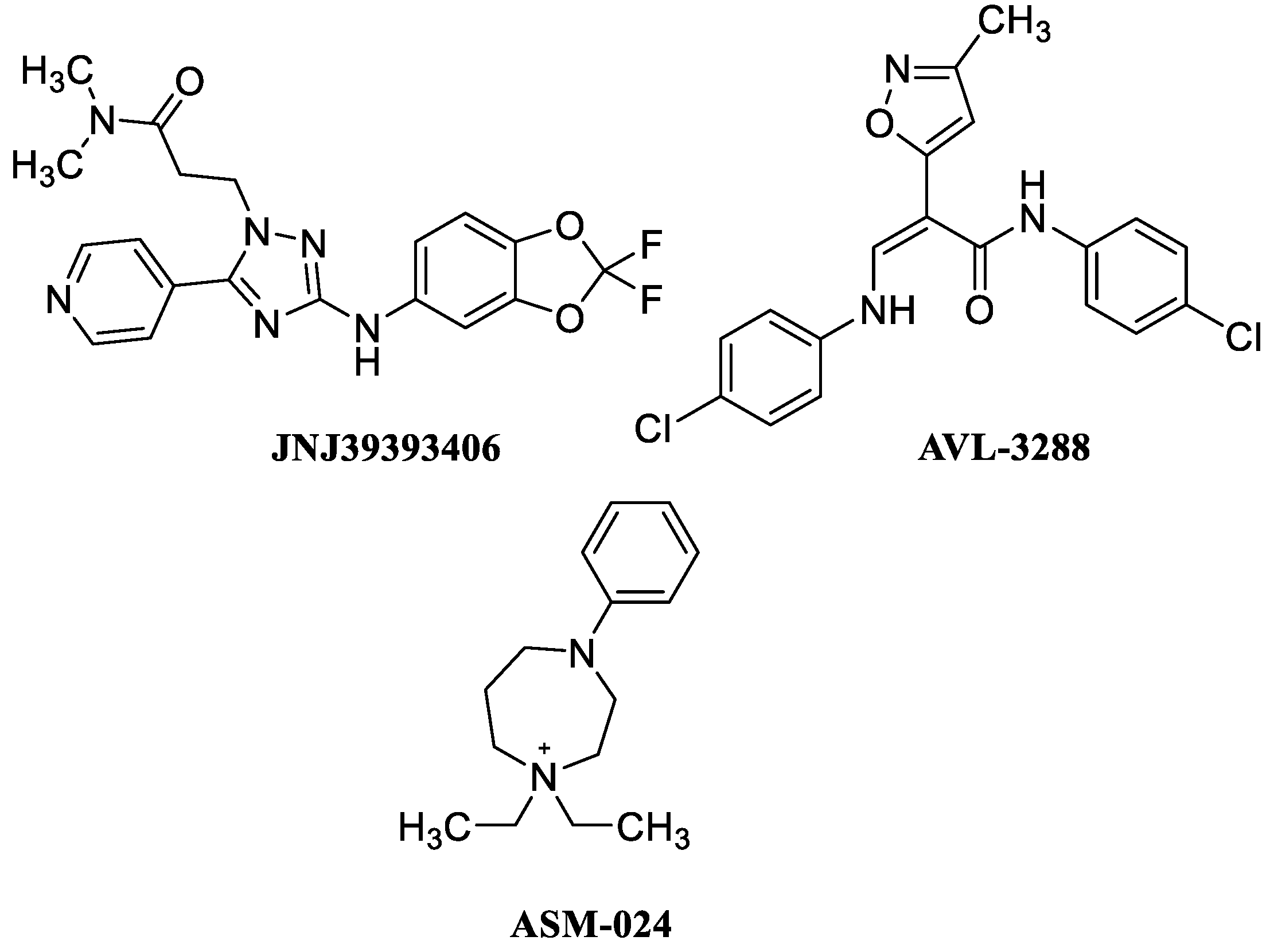
- Davidson, M.; Levi, L.; Park, J.; Nastas, I.; Ford, L.; Rassnick, S.; Canuso, C.; Davis, J.M.; Weiser, M. The effects of JNJ-39393406 a positive allosteric nicotine modulator on mood and cognition in patients with unipolar depression: A double-blind, add-on, placebo-controlled trial. Eur. Neuropsychopharmacol. 2021, 51, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Kantrowitz, J.T.; Javitt, D.C.; Freedman, R.; Sehatpour, P.; Kegeles, L.S.; Carlson, M.; Sobeih, T.; Wall, M.M.; Choo, T.-H.; Vail, B.; et al. Double blind, two dose, randomized, placebo-controlled, cross-over clinical trial of the positive allosteric modulator at the alpha7 nicotinic cholinergic receptor AVL-3288 in schizophrenia patients. Neuropsychopharmacology 2020, 45, 1339–1345. [Google Scholar] [CrossRef]
- Boulet, L.-P.; Gauvreau, G.M.; Cockcroft, D.W.; Davis, B.; Vachon, L.; Cormier, Y.; O’byrne, P.M. Effects of ASM-024, a modulator of acetylcholine receptor function, on airway responsiveness and allergen-induced responses in patients with mild asthma. Can. Respir. J. 2015, 22, 230–23489. [Google Scholar] [CrossRef]
- Gotti, C.; Clementi, F.; Fornari, A.; Gaimarri, A.; Guiducci, S.; Manfredi, I. Structural and functional diversity of native brain nicotinic receptors. Biochem. Pharmacol. 2009, 78, 703–711. [Google Scholar] [CrossRef]
- Buisson, B.; Bertrand, D. Nicotine addiction: The possible role of functional upregulation. Trends Pharmacol. Sci. 2002, 23, 130–136. [Google Scholar] [CrossRef]
- Nelson, M.E.; Kuryatov, A.; Choi, C.H.; Zhou, Y.; Lindstrom, J. Alternate Stoichiometries of α4β2 Nicotinic Acetylcholine Receptors. Mol. Pharmacol. 2003, 63, 332–341. [Google Scholar] [CrossRef]
- Fedorov, N.B.; Benson, L.C.; Graef, J.; Lippiello, P.M.; Bencherif, M. Differential Pharmacologies of Mecamylamine Enantiomers: Positive Allosteric Modulation and Noncompetitive Inhibition. J. Pharmacol. Exp. Ther. 2009, 328, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Bondarenko, V.; Targowska-Duda, K.M.; Jozwiak, K.; Tang, P.; Arias, H.R. Molecular Interactions between Mecamylamine Enantiomers and the Transmembrane Domain of the Human α4β2 Nicotinic Receptor. Biochemistry 2014, 53, 908–918. [Google Scholar] [CrossRef] [PubMed]
- Grupe, M.; Grunnet, M.; Bastlund, J.F.; Jensen, A.A. Targeting α4β2 Nicotinic Acetylcholine Receptors in Central Nervous System Disorders: Perspectives on Positive Allosteric Modulation as a Therapeutic Approach. Basic Clin. Pharmacol. Toxicol. 2015, 116, 187–200. [Google Scholar] [CrossRef]
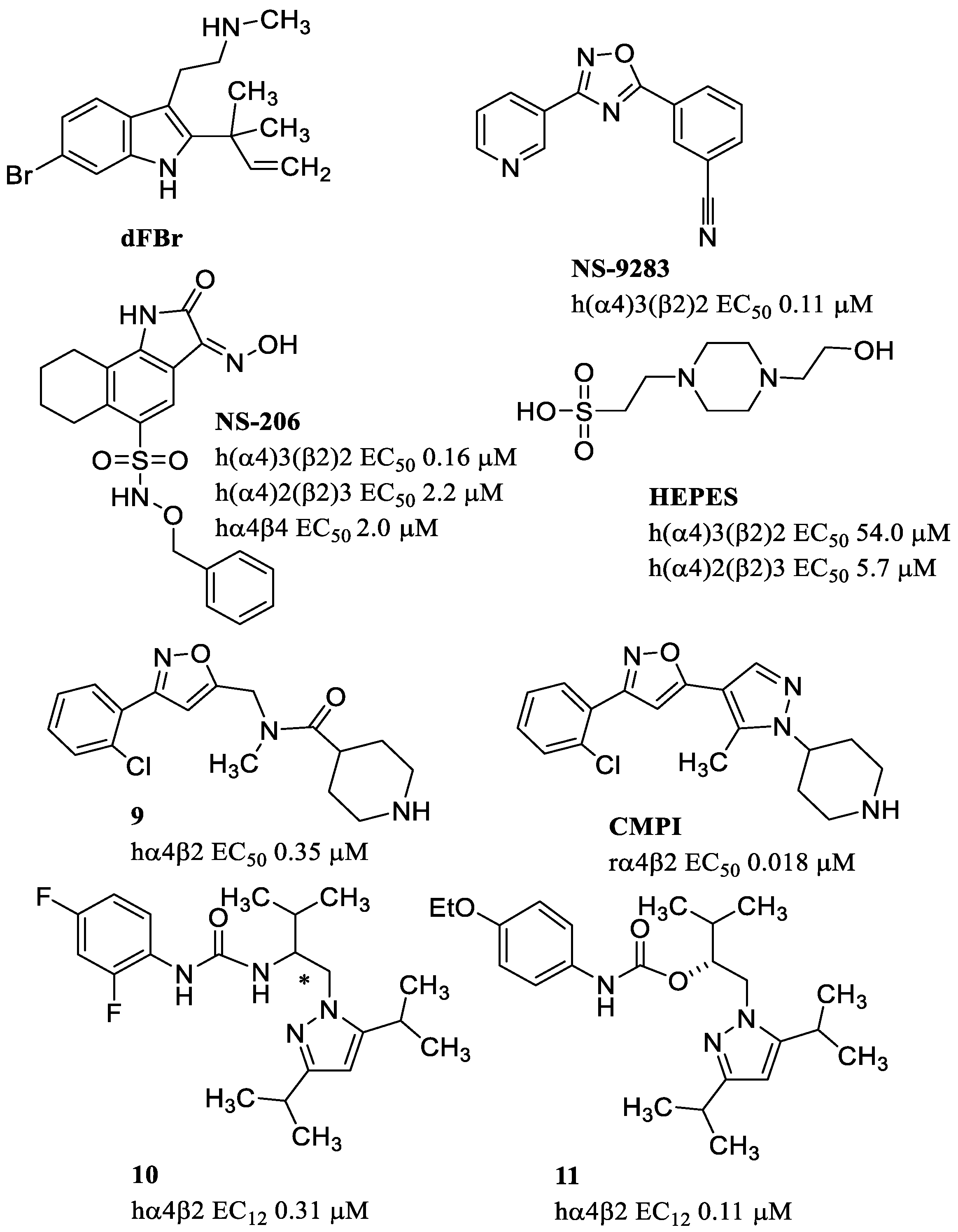
- Kim, J.-S.; Padnya, A.; Weltzin, M.; Edmonds, B.W.; Schulte, M.K.; Glennon, R.A. Synthesis of desformylflustrabromine and its evaluation as an α4β2 and α7 nACh receptor modulator. Bioorg. Med. Chem. Lett. 2007, 17, 4855–4860. [Google Scholar] [CrossRef] [PubMed]
- Nikiforuk, A.; Litwa, E.; Krawczyk, M.; Popik, P.; Arias, H. Desformylflustrabromine, a positive allosteric modulator of α4β2-containing nicotinic acetylcholine receptors, enhances cognition in rats. Pharmacol. Rep. 2020, 72, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Moroni, M.; Vijayan, R.; Carbone, A.; Zwart, R.; Biggin, P.C.; Bermudez, I. Non-Agonist-Binding Subunit Interfaces Confer Distinct Functional Signatures to the Alternate Stoichiometries of the α4β2 Nicotinic Receptor: An α4– α4 Interface Is Required for Zn2+ Potentiation. J. Neurosci. 2008, 28, 6884–6894. [Google Scholar] [CrossRef] [PubMed]
- Eiselé, J.-L.; Bertrand, S.; Galzi, J.-L.; Devillers-Thiéry, A.; Changeux, J.-P.; Bertrand, D. Chimaeric nicotinic–serotonergic receptor combines distinct ligand binding and channel specificities. Nature 1993, 366, 479–483. [Google Scholar] [CrossRef]
- Hsiao, B.; Dweck, D.; Luetje, C.W. Subunit-Dependent Modulation of Neuronal Nicotinic Receptors by Zinc. J. Neurosci. 2001, 21, 1848–1856. [Google Scholar] [CrossRef]
- Arias, H.R. Chapter 5 - Positive and negative modulation of nicotinic receptors. In Advances in Protein Chemistry and Structural Biology; Rossen, D., Ed.; Academic Press: Cambridge, MA, USA, 2010; Volume 80, pp. 153–203. [Google Scholar] [CrossRef]
- Olsen, J.A.; Kastrup, J.S.; Peters, D.; Gajhede, M.; Balle, T.; Ahring, P.K. Two Distinct Allosteric Binding Sites at α4β2 Nicotinic Acetylcholine Receptors Revealed by NS206 and NS9283 Give Unique Insights to Binding Activity-associated Linkage at Cys-loop Receptors. J. Biol. Chem. 2013, 288, 35997–36006. [Google Scholar] [CrossRef]
- Timmermann, D.B.; Sandager-Nielsen, K.; Dyhring, T.; Smith, M.; Jacobsen, A.M.; Nielsen, E.; Grunnet, M.; Christensen, J.K.; Peters, D.; Kohlhaas, K.; et al. Augmentation of cognitive function by NS9283, a stoichiometry-dependent positive allosteric modulator of α2- and α4-containing nicotinic acetylcholine receptors. Br. J. Pharmacol. 2012, 167, 164–182. [Google Scholar] [CrossRef]
- Weltzin, M.M.; Huang, Y.; Schulte, M.K. Allosteric modulation of alpha4beta2 nicotinic acetylcholine receptors by HEPES. Eur. J. Pharmacol. 2014, 732, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, B.K.; Berry, V.; Boezio, A.A.; Cao, L.; Clarkin, K.; Guo, W.; Harmange, J.-C.; Hierl, M.; Huang, L.; Janosky, B.; et al. Discovery and optimization of substituted piperidines as potent, selective, CNS-penetrant α4β2 nicotinic acetylcholine receptor potentiators. Bioorg. Med. Chem. Lett. 2008, 18, 5209–5212. [Google Scholar] [CrossRef] [PubMed]
- Wilkerson, J.L.; Deba, F.; Crowley, M.L.; Hamouda, A.K.; Mcmahon, L.R. Advances in the In vitro and In vivo pharmacology of Alpha4beta2 nicotinic receptor positive allosteric modulators. Neuropharmacology 2020, 168, 108008. [Google Scholar] [CrossRef] [PubMed]
- Deba, F.; Munoz, K.; Peredia, E.; Akk, G.; Hamouda, A.K. Assessing potentiation of the (α4)3(β2)2 nicotinic acetylcholine receptor by the allosteric agonist CMPI. J. Biol. Chem. 2022, 298, 101455. [Google Scholar] [CrossRef]
- Springer, S.K.; Woodin, K.S.; Berry, V.; Boezio, A.A.; Cao, L.; Clarkin, K.; Harmange, J.-C.; Hierl, M.; Knop, J.; Malmberg, A.B.; et al. Synthesis and activity of substituted carbamates as potentiators of the α4β2 nicotinic acetylcholine receptor. Bioorg. Med. Chem. Lett. 2008, 18, 5643–5647. [Google Scholar] [CrossRef]
- Hahn, B.; Reneski, C.H.; Lane, M.; Elmer, G.I.; Pereira, E.F.R. Evidence for positive allosteric modulation of cognitive-enhancing effects of nicotine by low-dose galantamine in rats. Pharmacol. Biochem. Behav. 2020, 199, 173043. [Google Scholar] [CrossRef]
- Iorga, I.; Herlem, D.; Barré, E.; Guillou, C. Acetylcholine nicotinic receptors: Finding the putative binding site of allosteric modulators using the “blind docking” approach. J. Mol. Mod. 2006, 12, 366–372. [Google Scholar] [CrossRef]
- Ludwig, J.; Höffle-Maas, A.; Samochocki, M.; Luttmann, E.; Albuquerque, E.X.; Fels, G.; Maelicke, A. Localization by site-directed mutagenesis of a galantamine binding site on α7 nicotinic acetylcholine receptor extracellular domain. J. Recept. Signal Transduct. 2010, 30, 469–483. [Google Scholar] [CrossRef]
- Hsiao, B.; Mihalak, K.B.; Repicky, S.E.; Everhart, D.; Mederos, A.H.; Malhotra, A.; Luetje, C.W. Determinants of Zinc Potentiation on the α4 Subunit of Neuronal Nicotinic Receptors. Mol. Pharmacol. 2006, 69, 27–36. [Google Scholar] [CrossRef]
- Hales, T.G.; Dunlop, J.I.; Deeb, T.Z.; Carland, J.E.; Kelley, S.P.; Lambert, J.J.; Peters, J.A. Common Determinants of Single Channel Conductance within the Large Cytoplasmic Loop of 5-Hydroxytryptamine Type 3 and α4β2 Nicotinic Acetylcholine Receptors*. J. Biol. Chem. 2006, 281, 8062–8071. [Google Scholar] [CrossRef]
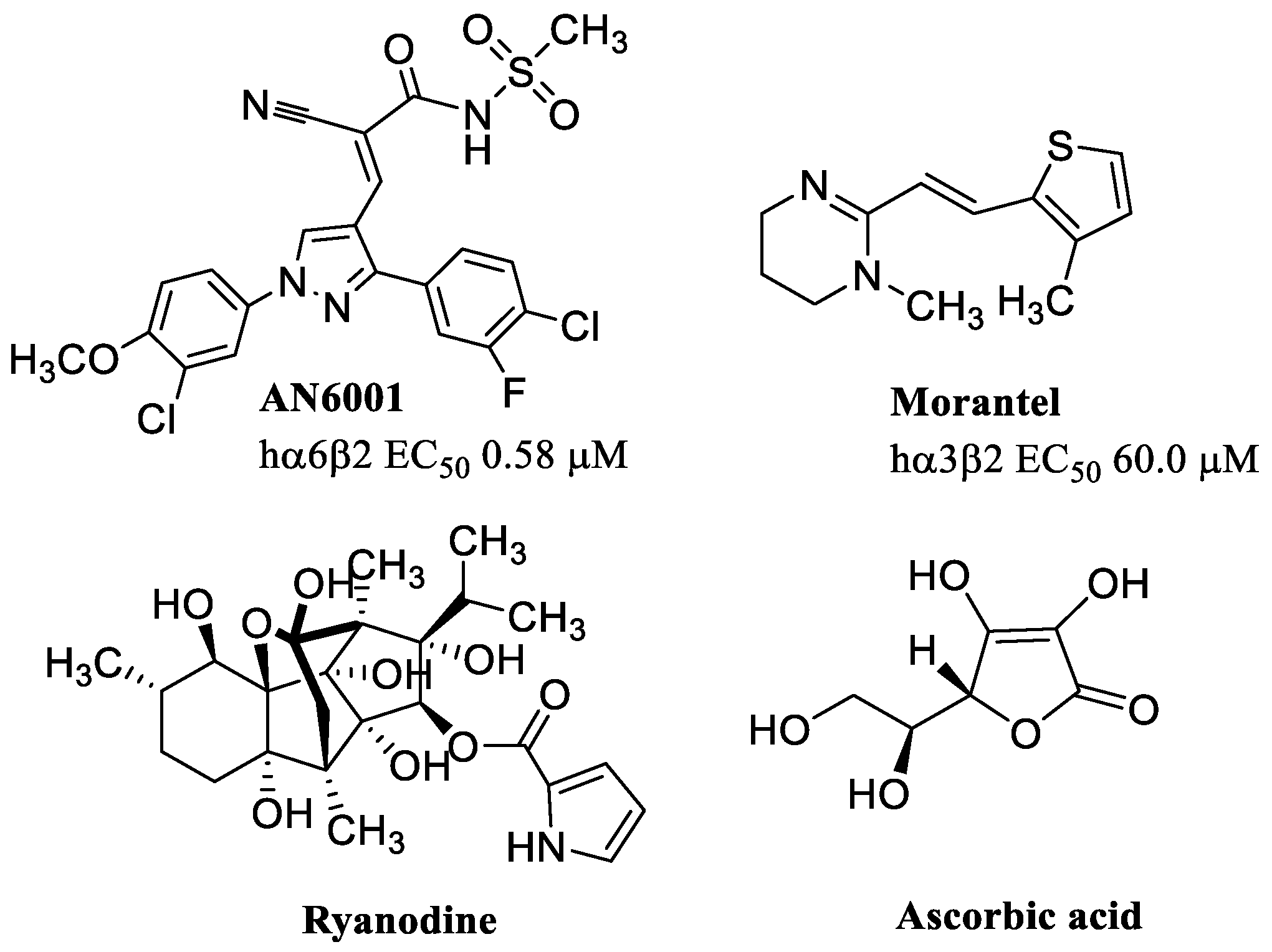
- Van Hout, M.; Klein, J.; Ahring, P.K.; Brown, D.T.; Thaneshwaran, S.; Dos Santos, A.B.; Jensen, A.A.; Kohlmeier, K.A.; Christophersen, P.; Dyhring, T. Characterization of AN6001, a positive allosteric modulator of α6β2-containing nicotinic acetylcholine receptors. Biochem. Pharmacol. 2020, 174, 113788. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.-Y.; Smith, C.M.; Sine, S.M.; Levandoski, M.M. Morantel Allosterically Enhances Channel Gating of Neuronal Nicotinic Acetylcholine α3β2 Receptors. Mol. Pharmacol. 2008, 74, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.; Henry, J.T.; Lewis, A.H.; Wang, N.; Levandoski, M.M. The Positive Allosteric Modulator Morantel Binds at Noncanonical Subunit Interfaces of Neuronal Nicotinic Acetylcholine Receptors. J. Neurosci. 2009, 29, 8734–8742. [Google Scholar] [CrossRef] [PubMed]
- Cohen, B.N.; Figl, A.; Quick, M.W.; Labarca, C.; Davidson, N.; Lester, H.A. Regions of beta 2 and beta 4 responsible for differences between the steady state dose-response relationships of the alpha 3 beta 2 and alpha 3 beta 4 neuronal nicotinic receptors. J. Gen. Physiol. 1995, 105, 745–764. [Google Scholar] [CrossRef] [PubMed]
- Elgoyhen, A.B. The α9α10 nicotinic acetylcholine receptor: A compelling drug target for hearing loss? Expert Opin. Ther. Targets 2022, 26, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Zouridakis, M.; Papakyriakou, A.; Ivanov, I.A.; Kasheverov, I.E.; Tsetlin, V.; Tzartos, S.; Giastas, P. Crystal Structure of the Monomeric Extracellular Domain of α9 Nicotinic Receptor Subunit in Complex With α-Conotoxin RgIA: Molecular Dynamics Insights Into RgIA Binding to α9α10 Nicotinic Receptors. Front. Pharmacol. 2019, 10, 474. [Google Scholar] [CrossRef]
- Gómez-Casati, M.E.; Fuchs, P.A.; Elgoyhen, A.B.; Katz, E. Biophysical and pharmacological characterization of nicotinic cholinergic receptors in rat cochlear inner hair cells. J. Physiol. 2005, 566, 103–118. [Google Scholar] [CrossRef]
- Mcintosh, J.M.; Absalom, N.; Chebib, M.; Elgoyhen, A.B.; Vincler, M. Alpha9 nicotinic acetylcholine receptors and the treatment of pain. Biochem. Pharmacol. 2009, 78, 693–702. [Google Scholar] [CrossRef]
- Zorrilla De San Martín, J.; Ballestero, J.; Katz, E.; Elgoyhen, A.B.; Fuchs, P.A. Ryanodine is a Positive Modulator of Acetylcholine Receptor Gating in Cochlear Hair Cells. J. Assoc. Res. Otolaryngol. 2007, 8, 474–483. [Google Scholar] [CrossRef]
- Boffi, J.; Wedemeyer, C.; Lipovsek, M.; Katz, E.; Calvo, D.; Elgoyhen, A. Positive modulation of the α9α10 nicotinic cholinergic receptor by ascorbic acid. Br. J. Pharmacol. 2013, 168, 954–965. [Google Scholar] [CrossRef]
- Moaddel, R.; Abdrakhmanova, G.; Kozak, J.; Jozwiak, K.; Toll, L.; Jimenez, L.; Rosenberg, A.; Tran, T.; Xiao, Y.; Zarate, C.A.; et al. Sub-anesthetic concentrations of (R,S)-ketamine metabolites inhibit acetylcholine-evoked currents in α7 nicotinic acetylcholine receptors. Eur. J. Pharmacol. 2013, 698, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Rodd, Z.A.; Hauser, S.R.; Swartzwelder, H.S.; Waeiss, R.A.; Lahiri, D.K.; Bell, R.L. Regulation of the deleterious effects of binge-like exposure to alcohol during adolescence by α7 nicotinic acetylcholine receptor agents: Prevention by pretreatment with a α7 negative allosteric modulator and emulation by a α7 agonist in alcohol-preferring (P) male and female rats. Psychopharmacology 2020, 237, 2601–2611. [Google Scholar] [CrossRef] [PubMed]
- Glassco, W.; Suchocki, J.; George, C.; Martin, B.R.; May, E.L. Synthesis, optical resolution, absolute configuration, and preliminary pharmacology of (+)- and (−)-cis-2,3,3a,4,5,9b-hexahydro-1-methyl-1H-pyrrolo[3,2-h]isoquinoline, a structural analog of nicotine. J. Med. Chem. 1993, 36, 3381–3385. [Google Scholar] [CrossRef] [PubMed]
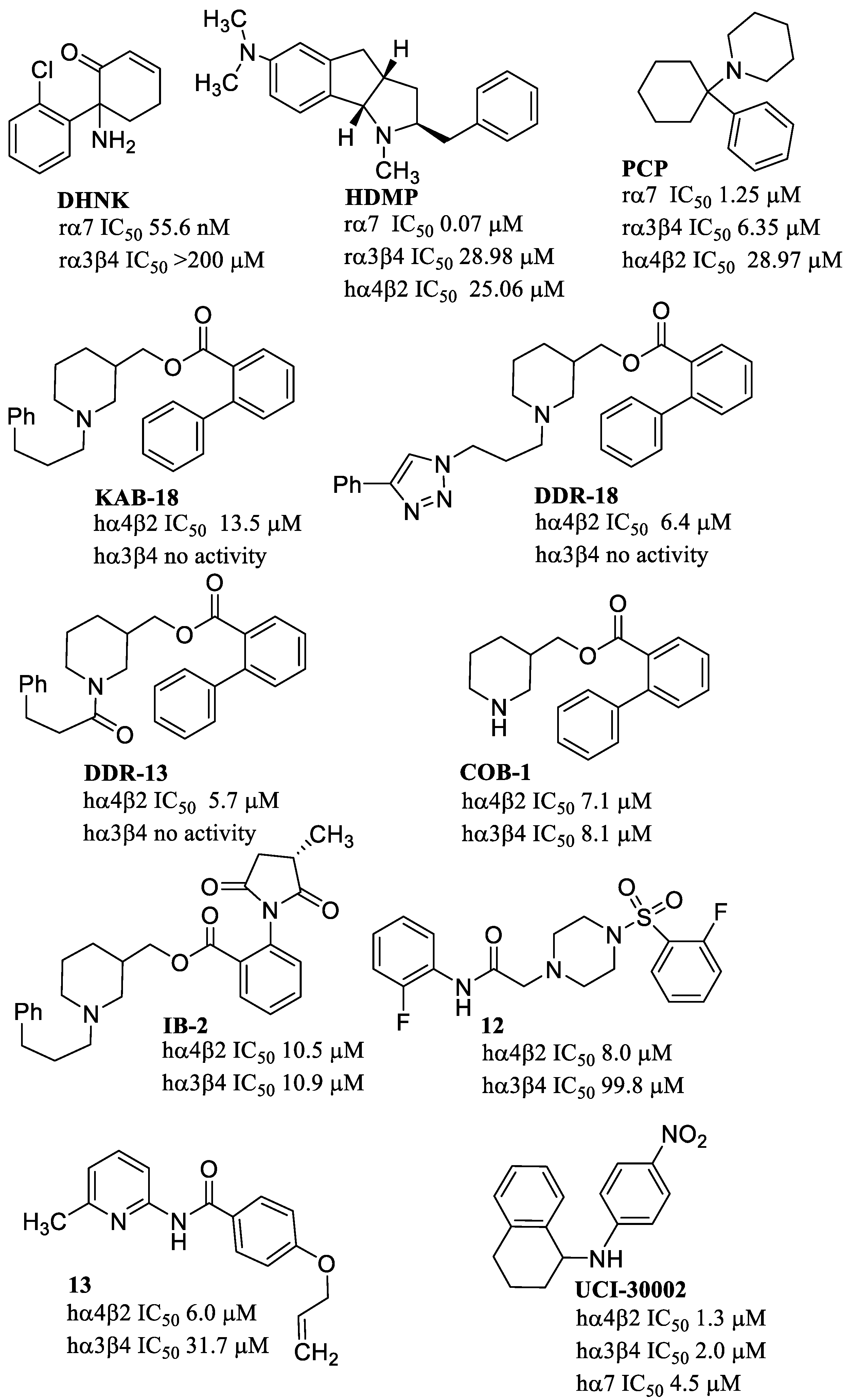
- Abdrakhmanova, G.R.; Blough, B.E.; Nesloney, C.; Navarro, H.A.; Damaj, M.I.; Carroll, F.I. In vitro and in vivo characterization of a novel negative allosteric modulator of neuronal nAChRs. Neuropharmacology 2010, 59, 511–517. [Google Scholar] [CrossRef] [PubMed]
- González-Cestari, T.F.; Henderson, B.J.; Pavlovicz, R.E.; Mckay, S.B.; El-Hajj, R.A.; Pulipaka, A.B.; Orac, C.M.; Reed, D.D.; Boyd, R.T.; Zhu, M.X.; et al. Effect of Novel Negative Allosteric Modulators of Neuronal Nicotinic Receptors on Cells Expressing Native and Recombinant Nicotinic Receptors: Implications for Drug Discovery. J. Pharmacol. Exp. Ther. 2009, 328, 504–515. [Google Scholar] [CrossRef]
- Henderson, B.J.; Pavlovicz, R.E.; Allen, J.D.; González-Cestari, T.F.; Orac, C.M.; Bonnell, A.B.; Zhu, M.X.; Boyd, R.T.; Li, C.; Bergmeier, S.C.; et al. Negative Allosteric Modulators That Target Human α4β2 Neuronal Nicotinic Receptors. J. Pharmacol. Exp. Ther. 2010, 334, 761–774. [Google Scholar] [CrossRef]
- Henderson, B.J.; González-Cestari, T.F.; Yi, B.; Pavlovicz, R.E.; Boyd, R.T.; Li, C.; Bergmeier, S.C.; Mckay, D.B. Defining the Putative Inhibitory Site for a Selective Negative Allosteric Modulator of Human α4β2 Neuronal Nicotinic Receptors. ACS Chem. Neurosci. 2012, 3, 682–692. [Google Scholar] [CrossRef]
- Pavlovicz, R.E.; Henderson, B.J.; Bonnell, A.B.; Boyd, R.T.; Mckay, D.B.; Li, C. Identification of a Negative Allosteric Site on Human α4β2 and α3β4 Neuronal Nicotinic Acetylcholine Receptors. PLoS ONE 2011, 6, e24949. [Google Scholar] [CrossRef]
- Yi, B.; Long, S.; González-Cestari, T.F.; Henderson, B.J.; Pavlovicz, R.E.; Werbovetz, K.; Li, C.; Mckay, D.B. Discovery of benzamide analogs as negative allosteric modulators of human neuronal nicotinic receptors: Pharmacophore modeling and structure–activity relationship studies. Bioorg. Med. Chem. 2013, 21, 4730–4743. [Google Scholar] [CrossRef]
- Henderson, B.J.; Carper, D.J.; González-Cestari, T.F.; Yi, B.; Mahasenan, K.; Pavlovicz, R.E.; Dalefield, M.L.; Coleman, R.S.; Li, C.; Mckay, D.B. Structure–Activity Relationship Studies of Sulfonylpiperazine Analogues as Novel Negative Allosteric Modulators of Human Neuronal Nicotinic Receptors. J. Med. Chem. 2011, 54, 8681–8692. [Google Scholar] [CrossRef]
















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manetti, D.; Dei, S.; Arias, H.R.; Braconi, L.; Gabellini, A.; Teodori, E.; Romanelli, M.N. Recent Advances in the Discovery of Nicotinic Acetylcholine Receptor Allosteric Modulators. Molecules 2023, 28, 1270. https://doi.org/10.3390/molecules28031270
Manetti D, Dei S, Arias HR, Braconi L, Gabellini A, Teodori E, Romanelli MN. Recent Advances in the Discovery of Nicotinic Acetylcholine Receptor Allosteric Modulators. Molecules. 2023; 28(3):1270. https://doi.org/10.3390/molecules28031270
Chicago/Turabian StyleManetti, Dina, Silvia Dei, Hugo R. Arias, Laura Braconi, Alessio Gabellini, Elisabetta Teodori, and Maria Novella Romanelli. 2023. "Recent Advances in the Discovery of Nicotinic Acetylcholine Receptor Allosteric Modulators" Molecules 28, no. 3: 1270. https://doi.org/10.3390/molecules28031270
APA StyleManetti, D., Dei, S., Arias, H. R., Braconi, L., Gabellini, A., Teodori, E., & Romanelli, M. N. (2023). Recent Advances in the Discovery of Nicotinic Acetylcholine Receptor Allosteric Modulators. Molecules, 28(3), 1270. https://doi.org/10.3390/molecules28031270