Density Functional Study to Investigate the Ability of (ZnS)n (n = 1–12) Clusters Removing Hg0, HgCl, and HgCl2 via Electron Localization Function and Non−Covalent Interactions Analyses


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
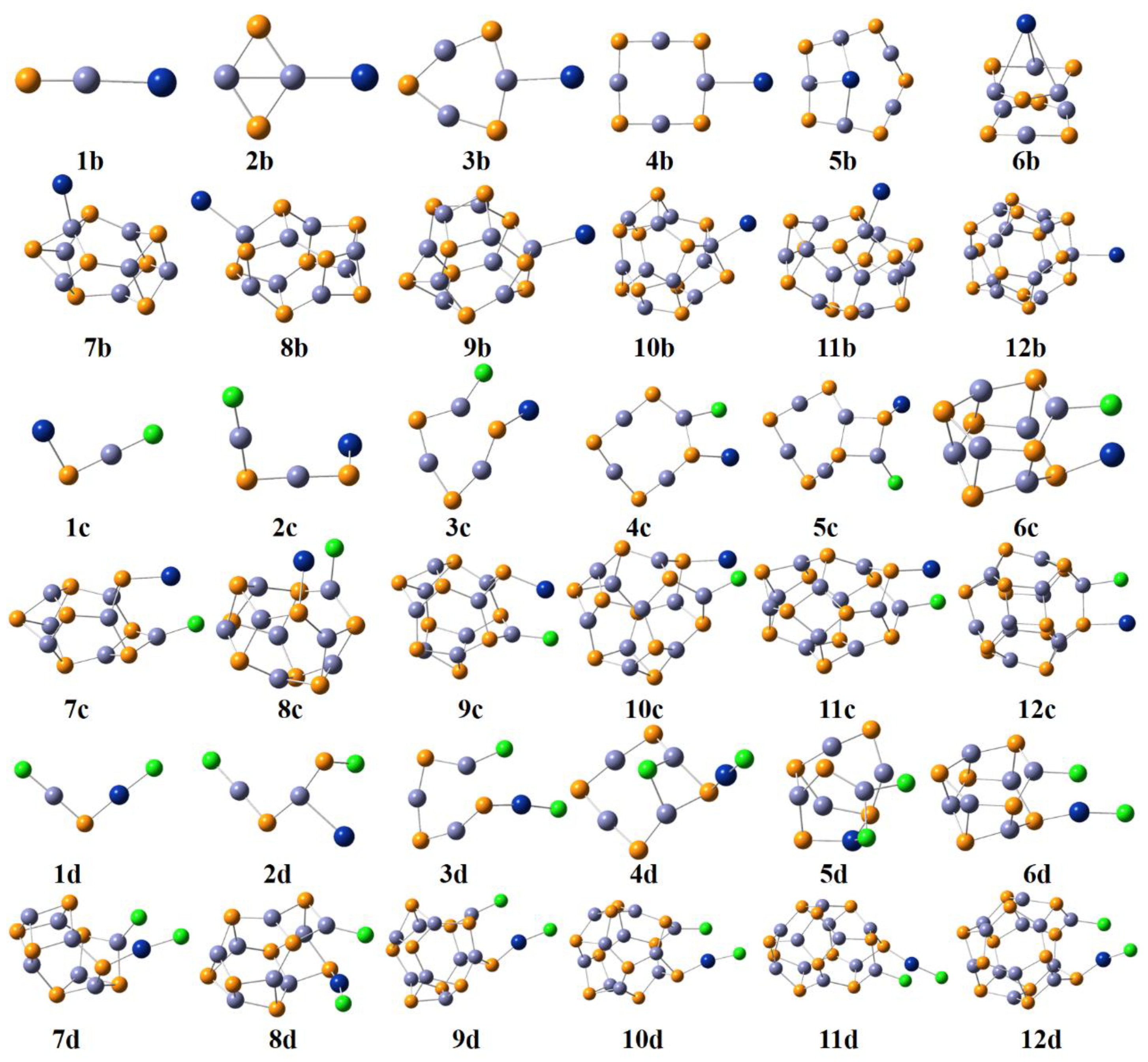
2.1. Geometries of (ZnS)n (n = 1–12) Clusters
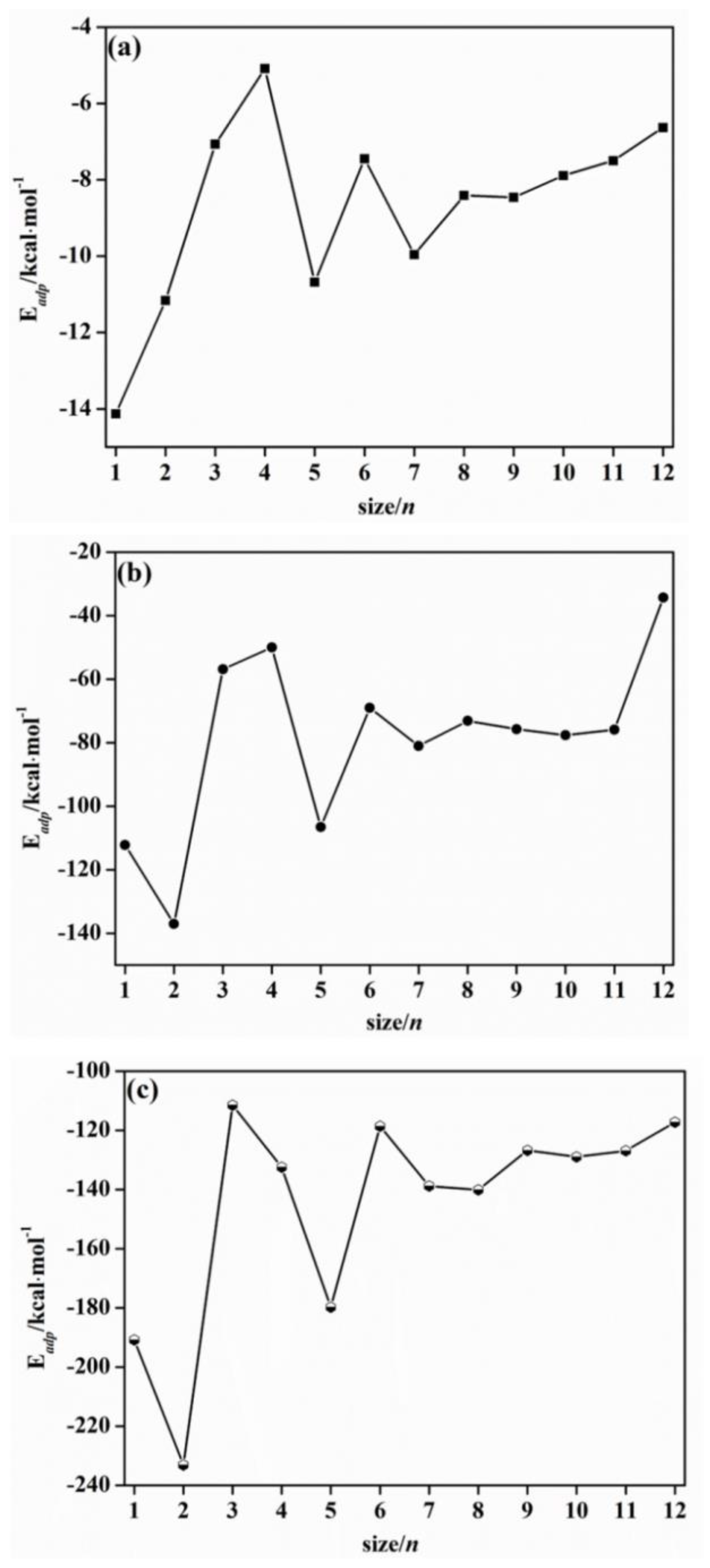
2.2. Adsorption Energy
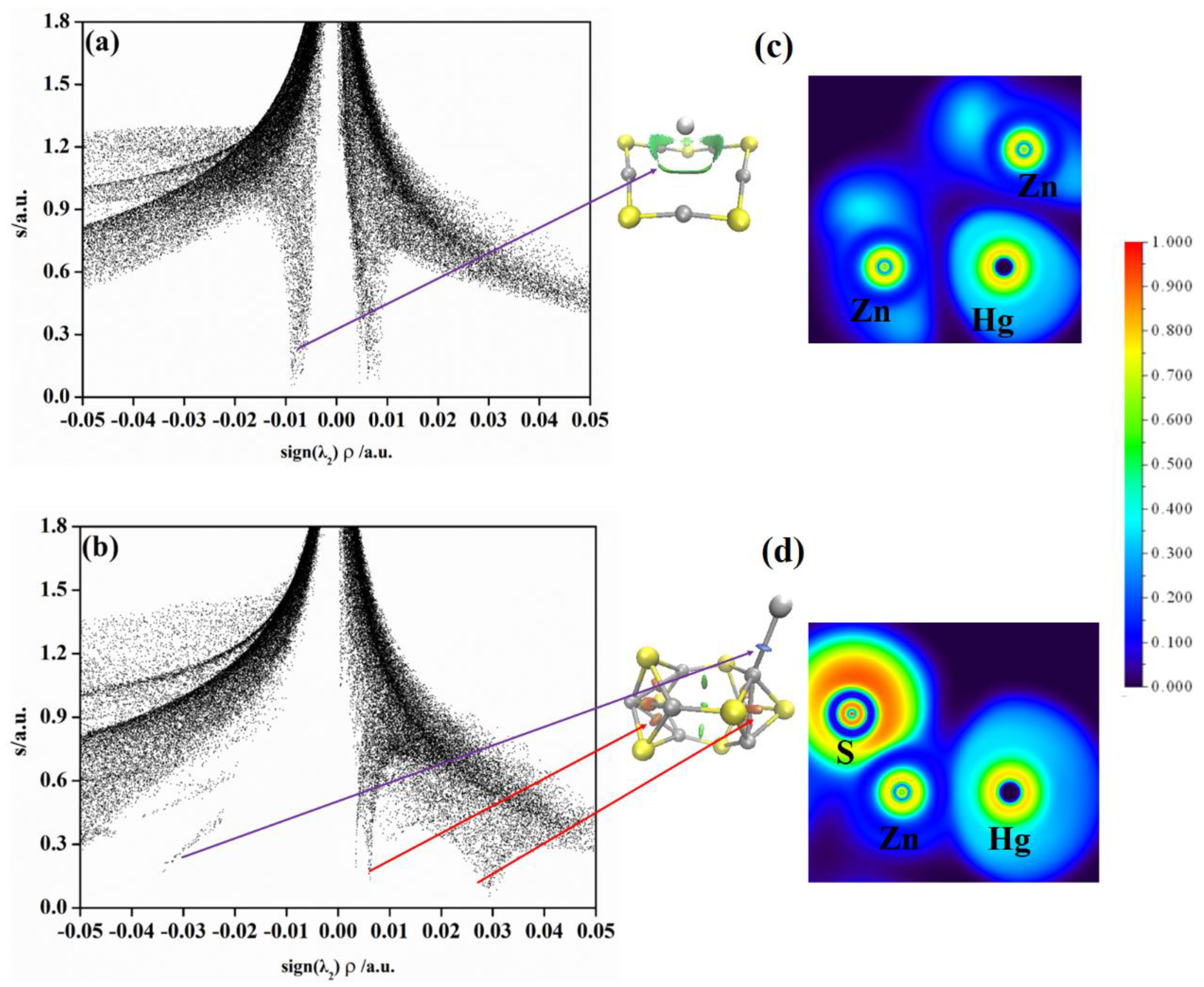
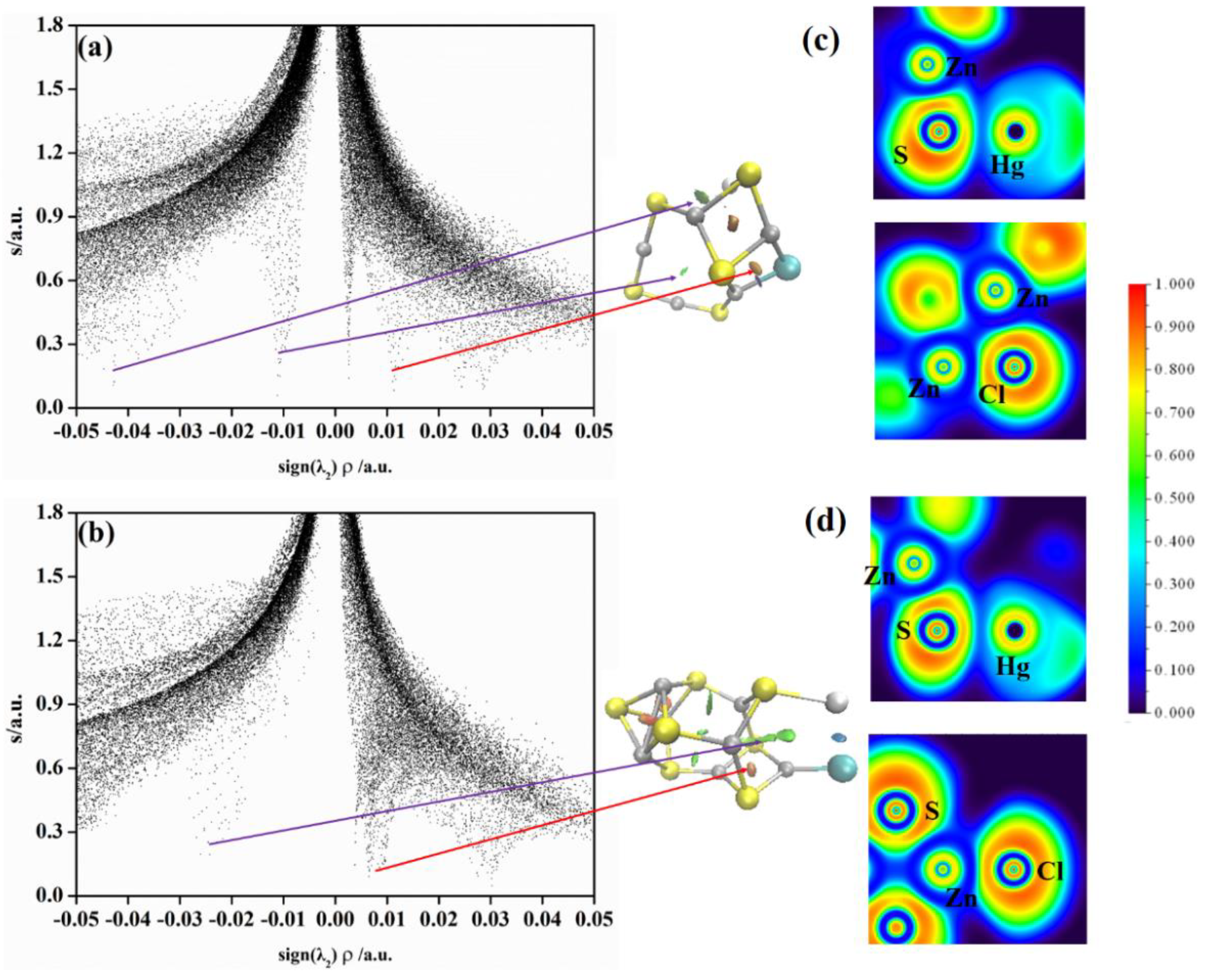
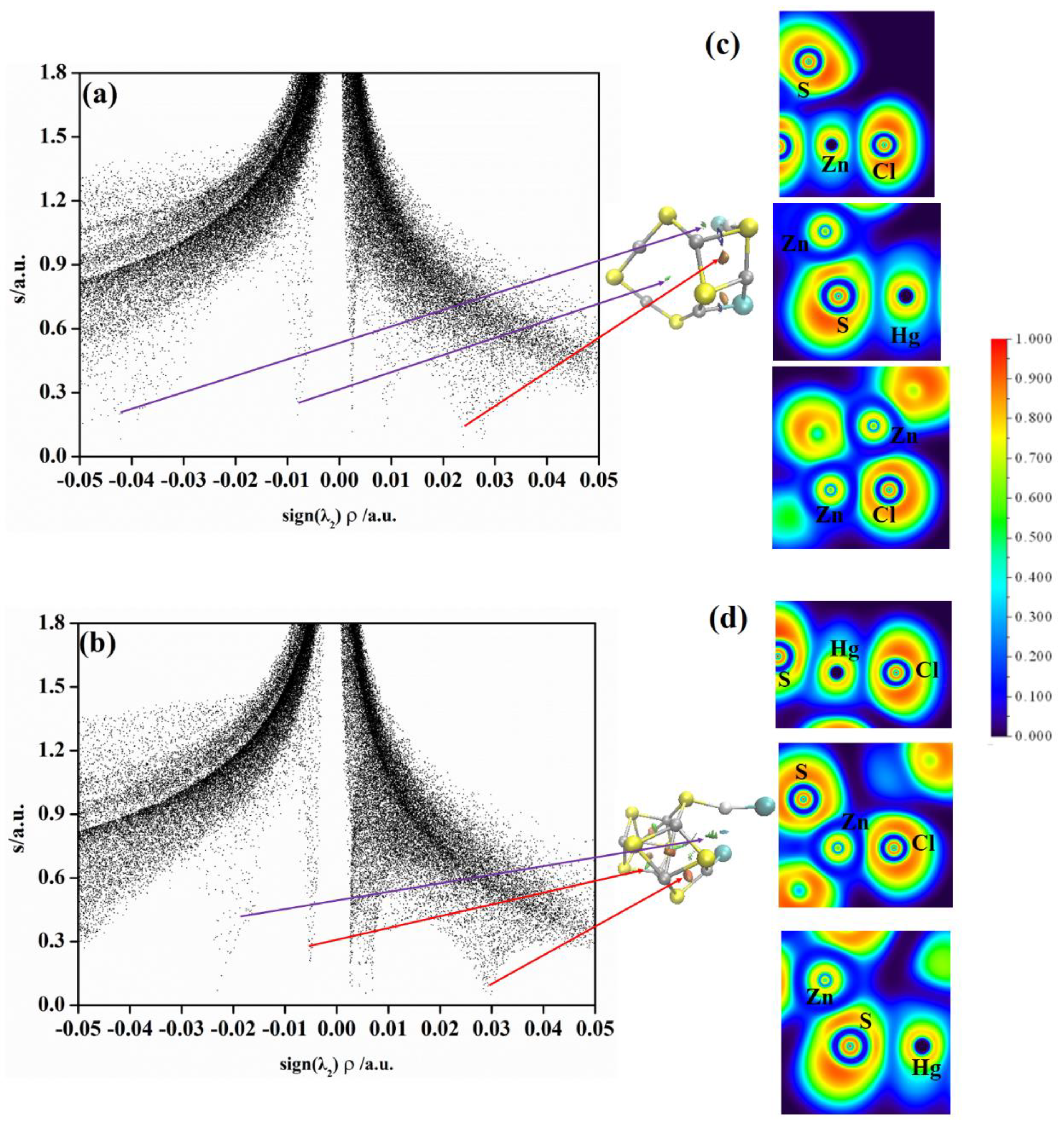
2.3. Non−Covalent Interactions (NCI) and Electron Local Function (ELF) Analyses
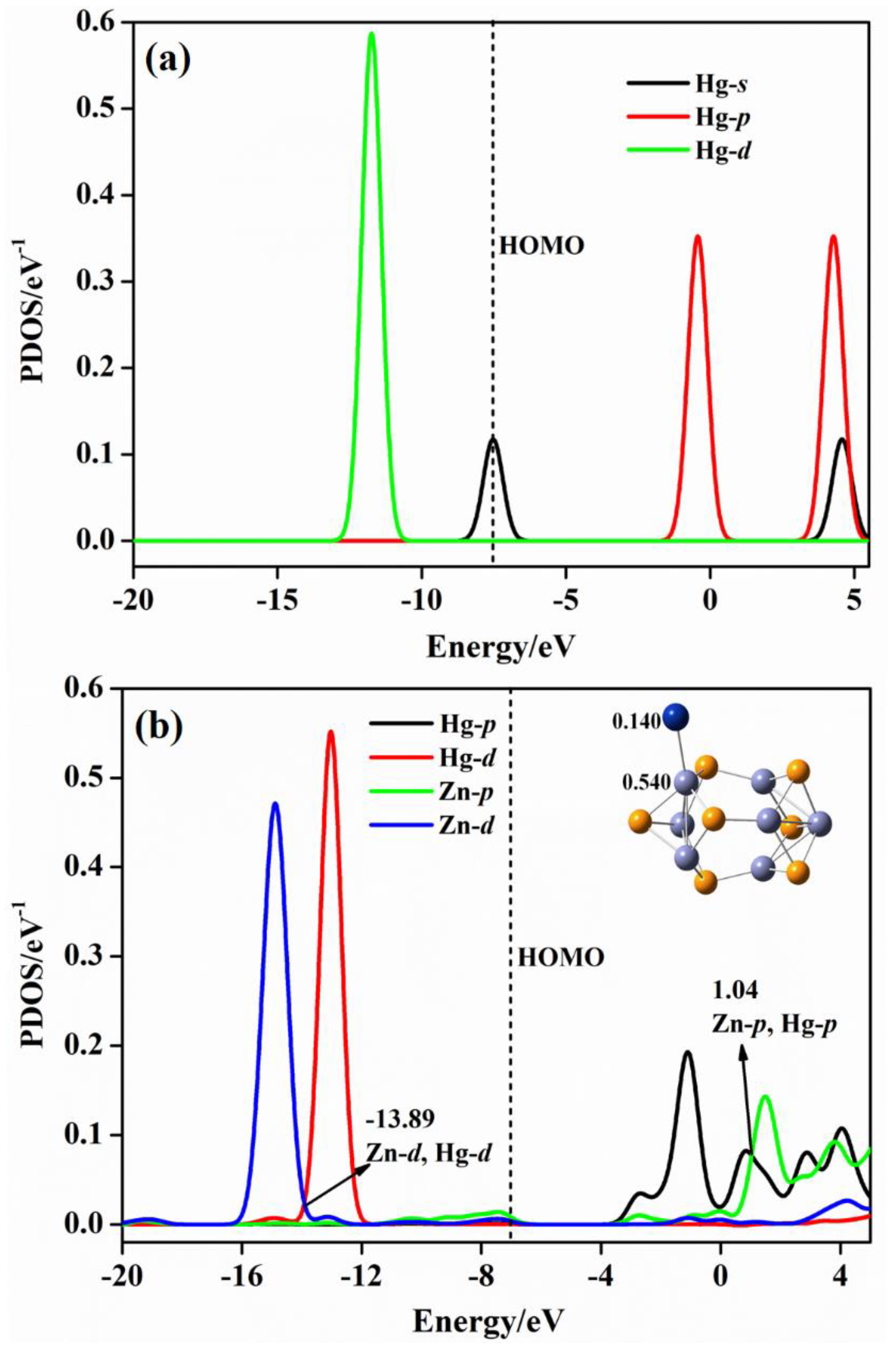
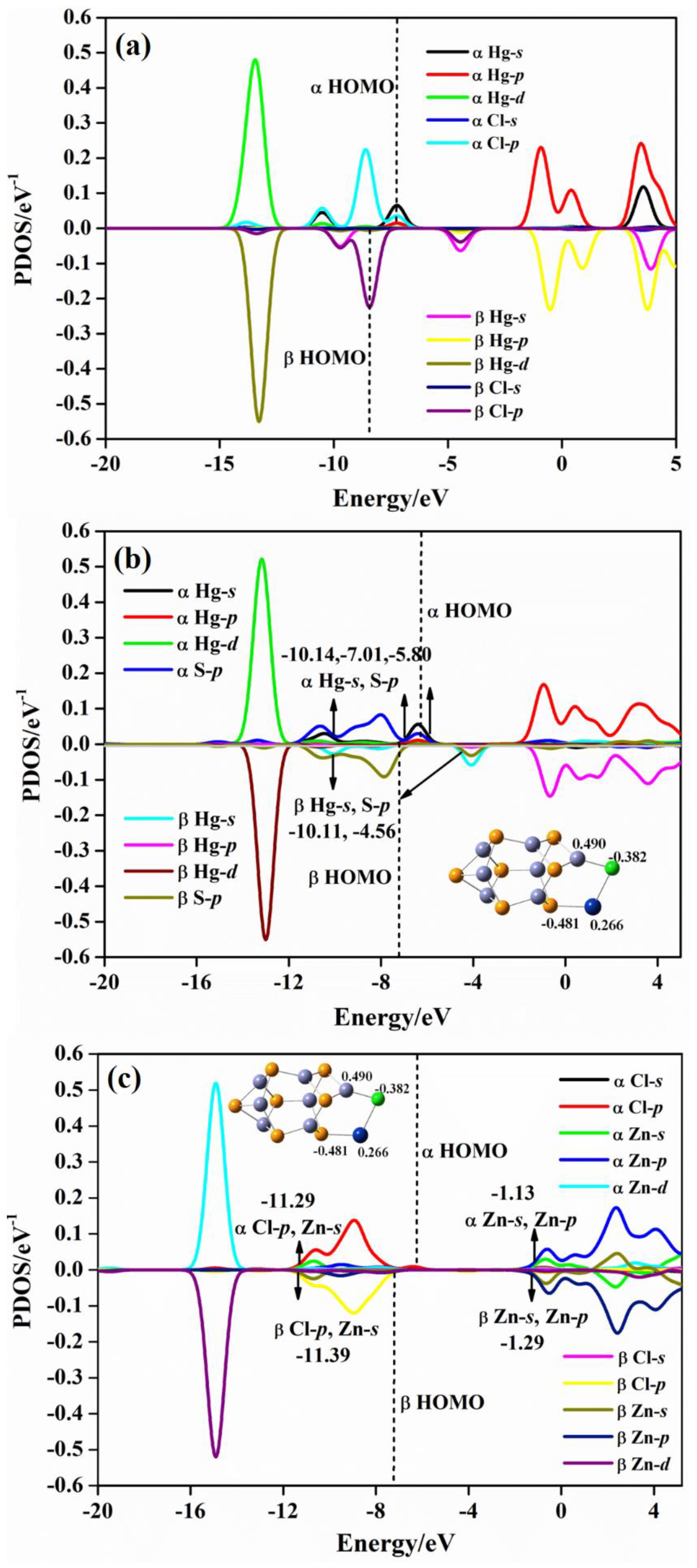
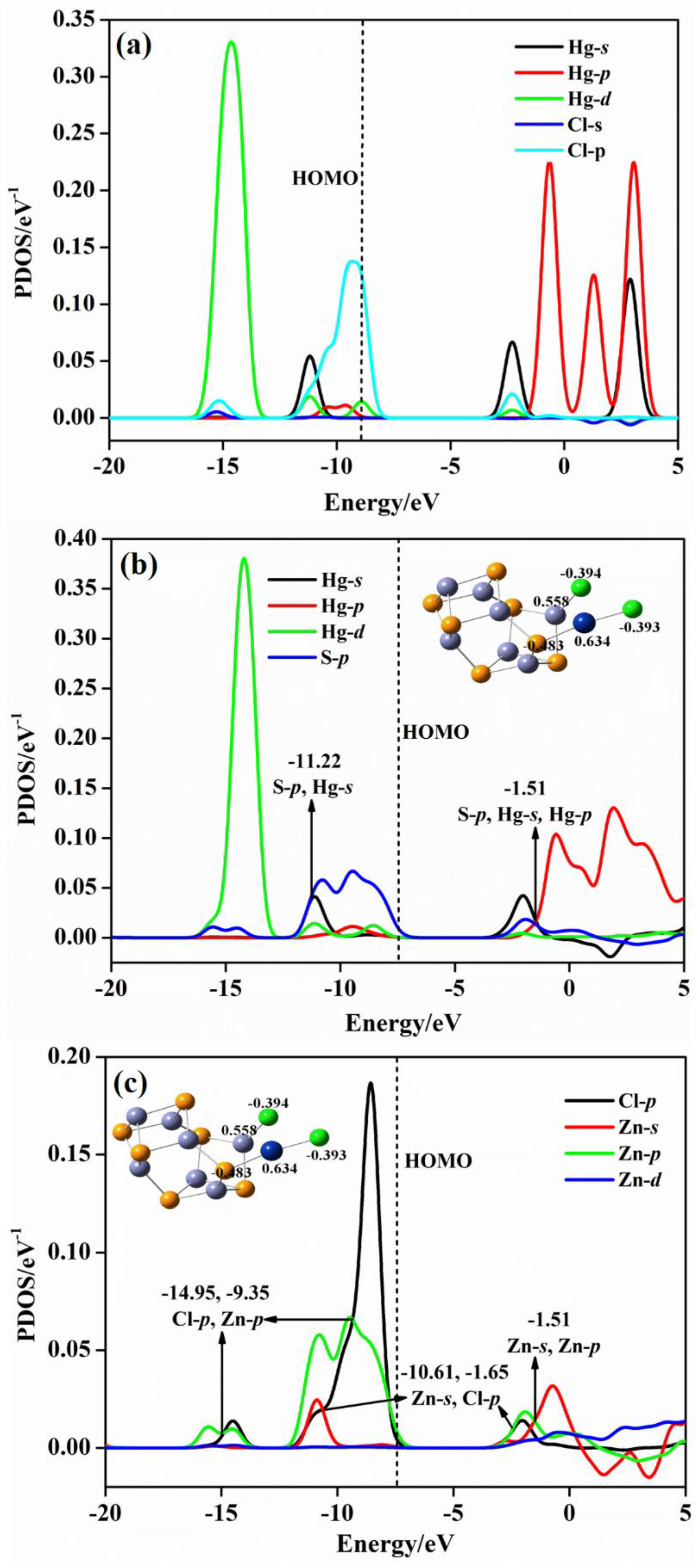
2.4. Projected Density of State (PDOS) Analysis
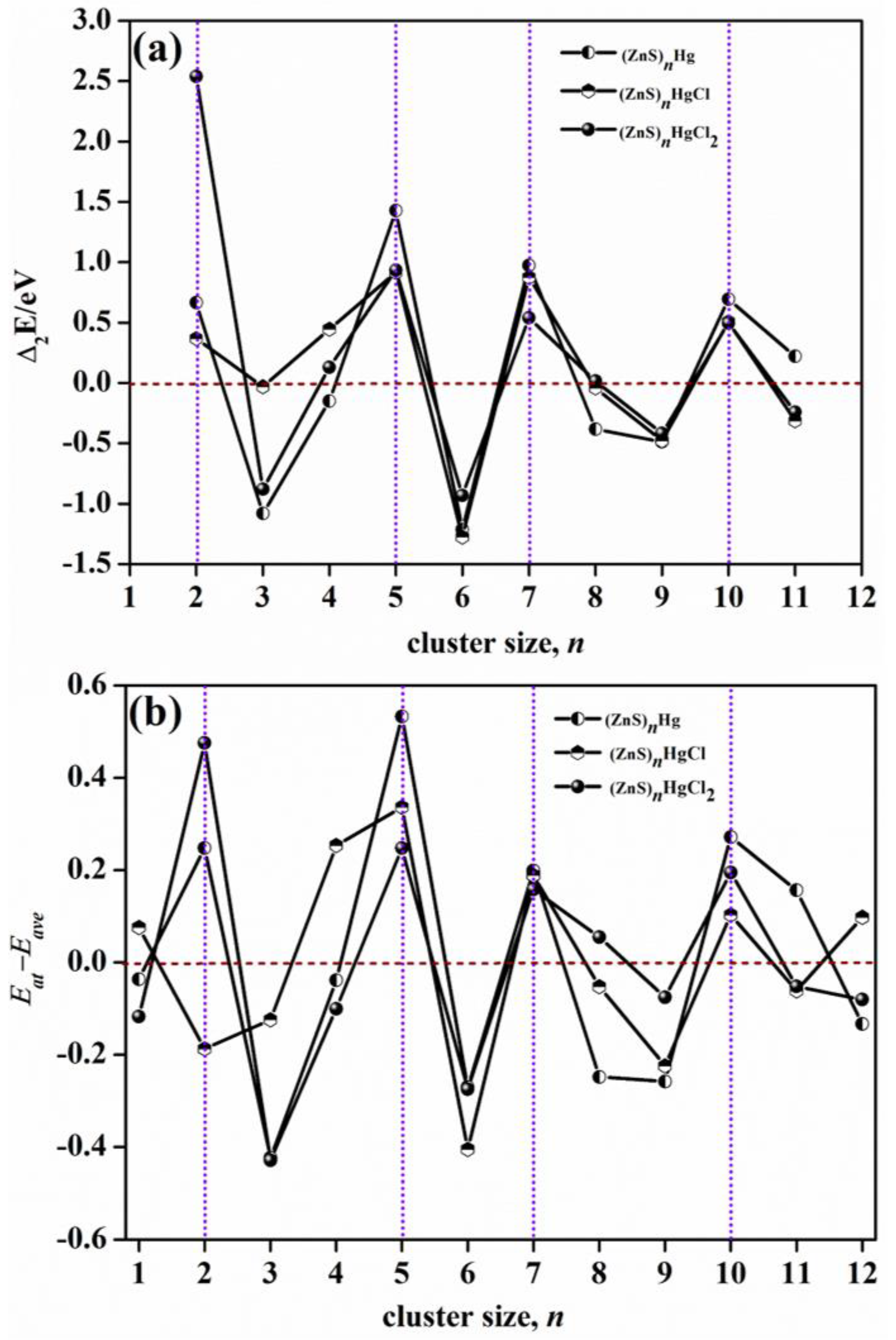
2.5. Stability
3. Materials and Methods
Computational Methods
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zheng, W.; Li, H.; Yang, Z.; Yang, J.; Qu, W.; Meng, F.; Feng, Y.; Xu, Z.; Guo, X. Advances in flue gas mercury abatement by mineral chalcogenides. Chem. Eng. J. 2021, 411, 128608. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhou, C.; Wu, H.; Liu, J.; Yang, H. Molecular study of heterogeneous mercury conversion mechanism over Cu−MOFs: Oxidation pathway and effect of halogen. Fuel 2021, 290, 120030. [Google Scholar] [CrossRef]
- Zhao, J.; Li, H.; Yang, Z.; Qu, W.; Yang, J.; Xu, Z.; Liu, H.; Shih, K. Light irradiation inhibits mercury adsorption by mineral sulfide sorbent. Fuel 2020, 288, 119663. [Google Scholar] [CrossRef]
- Yu, Y.; Yang, Y.; Liu, J.; Wang, Z.; Ding, J. Nanosized Zn–In spinel−type sorbents for elemental mercury removal from flue gas. Energ Fuel 2021, 34, 12853–12859. [Google Scholar] [CrossRef]
- Yang, Z.; Li, H.; Feng, S.; Li, P.; Liao, C.; Liu, X.; Zhao, J.; Yang, J.; Lee, P.H.; Shih, K. Multiform sulfur adsorption centers and copper–terminated active sites of nano–CuS for efficient elemental mercury capture from coal combustion flue gas. Langmuir 2018, 34, 8739–8749. [Google Scholar] [CrossRef]
- He, W.; Ran, J.; Niu, J.; Yang, G.; Zhang, P. Mechanism insights into elemental mercury oxidation on RuO2(110) surface: A density functional study. Appl. Surf. Sci. 2019, 466, 920–927. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, Z.; Shen, F.; Zhou, Y.; Liu, J.; Yang, H. Two−dimensional WS2 as a new mercury removal material: Mercury conversion pathway and effect of defect. Fuel 2022, 307, 121864. [Google Scholar] [CrossRef]
- Li, H.; Feng, S.; Liu, Y.; Shih, K. Binding of mercury species and typical flue gas components on ZnS(110). Energy Fuel 2017, 31, 5355–5362. [Google Scholar] [CrossRef]
- Jiang, H.; Xing, Z.; Li, Z.; Pan, K.; Yang, Z.; Wang, K.; Guo, M.; Yang, S.; Zhou, W. Wide−spectrum response urchin−like Bi2S3 spheres and ZnS quantum dots co−decorated mesoporous g–C3N4 nanosheets heterojunctions for promoting charge separation and enhancing photothermal−photocatalytic performance. Appl. Surf. Sci. 2020, 527, 146653. [Google Scholar] [CrossRef]
- Meij, R. The fate of mercury in coal−fired power plants and the influence of wet flue−gas desulphurization. Water Air Soil Pollut. 1991, 56, 21–33. [Google Scholar] [CrossRef]
- Carpi, A. Mercury from combustion sources: A review of the chemical species emitted and their transport in the atmosphere. Water Air Soil Poll. 1997, 98, 241–254. [Google Scholar] [CrossRef]
- Presto, A.A.; Granite, E.J. Survey of catalysts for oxidation of mercury in flue gas. Environ. Sci. Technol. 2006, 40, 5601–5609. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Feng, S.; Qu, W.; Yang, J.; Liu, S.; Liu, Y. Adsorption and oxidation of elemental mercury on chlorinated ZnS surface. Energ Fuel 2018, 32, 7745–7751. [Google Scholar] [CrossRef]
- Pala, I.R.; Brock, S.L. ZnS nanoparticle gels for remediation of Pb2+ and Hg2+ polluted water. ACS Appl. Mater. Inter. 2012, 4, 2160–2167. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhu, L.; Wang, J.; Li, L.; Shih, K. Development of nano−sulfide sorbent for efficient removal of elemental mercury from coal combustion fuel gas. Environ. Sci. Technol. 2016, 50, 9551–9557. [Google Scholar] [CrossRef]
- Yang, Y.; Huang, R.; Xu, W.; Zhang, J.; Li, C.; Song, J.; Zhu, T. Different crystal forms of ZnS nanomaterials for the adsorption of elemental mercury. Environ. Sci. Technol. 2021, 55, 6965–6974. [Google Scholar] [CrossRef]
- Siddiqui, S.A.; Bouarissa, N. First principle study of the interaction of elemental Hg with small neutral, anionic and cationic Pdn (n = 1–6) clusters. J. Chem. Sci. 2013, 125, 1629–1637. [Google Scholar] [CrossRef]
- Ling, L.; Fan, L.; Feng, X.; Wang, B.; Zhang, R. Effects of the size and Cu modulation of Pdn (n ⩽ 38) clusters on Hg0 adsorption. Chem. Engin. J. 2017, 308, 289–298. [Google Scholar] [CrossRef]
- Siddiqui, S.A.; Bouarissa, N.; Rasheed, T.; Al-Assiri, M. Quantum chemical study of the interaction of elemental Hg with small neutral, anionic and cationic Aun (n = 1–6) clusters. Mater. Res. Bull. 2013, 48, 995–1002. [Google Scholar] [CrossRef]
- Sun, L.; Zhang, A.; Su, S.; Wang, H.; Liu, J.; Xiang, J. A DFT study of the interaction of elemental mercury with small neutral and charged silver clusters. Chem. Phys. Lett. 2011, 517, 227–233. [Google Scholar] [CrossRef]
- Wang, Y.; Huo, Q.; Fan, H.; Wang, J.; Chang, L. A theoretical study on structures of neutral (CuS)n (n = 1–10) clusters and their interaction with Hg0. Fuel 2022, 321, 123972. [Google Scholar] [CrossRef]
- Chen, H.; Shi, D.; Wang, B.; Qi, J. An unbiased structural optimization of zinc sulfide clusters (ZnS)n (n = 2–18). J. Comput. Theor. Nanosci. 2011, 8, 2454–2461. [Google Scholar] [CrossRef]
- Hamad, S.; Catlow, C.R.A.; Spanó, E.; Matxain, J.M.; Ugalde, J.M. Structure and properties of ZnS nanoclusters. J. Phys. Chem. B 2005, 109, 2703–2709. [Google Scholar] [CrossRef]
- Li, H.; Zu, H.; Deng, Y.; He, W.; Yang, Z.; Yang, J.; Zhao, S.; Qu, W. Mechanisms of gas−phase mercury immobilized by metal sulfides from combustion flue gas: A mini review. Energ Fuel 2022, 36, 6027–6037. [Google Scholar] [CrossRef]
- Lalsare, D.; Kshirsagar, A. First principles results of structural and electronic properties of ZnS clusters. Bull. Mater. Sci. 2012, 35, 1055–1062. [Google Scholar] [CrossRef]
- Matxain, J.M.; Fowler, J.E.; Ugalde, J.M. Small clusters of II−VI materials: ZniSi, i = 1−9. Phys. Rev. A 2000, 61, 053201. [Google Scholar] [CrossRef]
- Sanville, E.; Burnin, A.; BelBruno, J.J. Experimental and computational study of small (n = 1−16) stoichiometric zinc and cadmium chalcogenide clusters. J. Phys. Chem. A 2006, 110, 2378–2386. [Google Scholar] [CrossRef]
- Nguyen, K.A.; Pachter, R.; Day, P.N. Computational prediction of structures and optical excitations for nanoscale ultrasmall ZnS and CdSe clusters. J. Chem. Theory Comput. 2013, 9, 3581–3596. [Google Scholar] [CrossRef]
- Wang, B.; Nagase, S.; Zhao, J.; Wang, G. Structural growth sequences and electronic properties of zinc oxide clusters (ZnO)n (n = 2−18). J. Phys. Chem. C 2007, 111, 4956–4963. [Google Scholar] [CrossRef]
- Spanó, E.; Hamad, S.; Catlow, C.R.A. ZnS bubble clusters with onion−like structures. Chem. Commun. 2004, 864–865. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Feng, S.; Yang, Z.; Yang, J.; Liu, S.; Hu, Y.; Zhong, L.; Qu, W. Density functional theory study of mercury adsorption on CuS Surface: Effect of typical flue gas components. Energ Fuel 2019, 33, 1540–1546. [Google Scholar] [CrossRef]
- Chen, J.; Zhu, W.; Chang, X.; Ding, D.; Zhang, T.; Zhou, C.; Wu, H.; Yang, H.; Sun, L. DFT insights to mercury species mechanism on pure and Mn doped Fe3O4(111) surfaces. Appl. Surf. Sci. 2020, 514, 145876. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, J.; Zhang, A.; Zhou, Y. Theoretical investigation of arsenic and selenium species adsorption behavior on different mineral adsorbents. Ind. Eng. Chem. Res. 2019, 58, 23559–23566. [Google Scholar] [CrossRef]
- Bondi, A. Van der Waals volumes and radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori−Sánchez, P.; Contreras−García, J.; Cohen, A.J.; Yang, W. Revealing noncovalent interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef]
- Johnston, R.L. Evolving better nanoparticles: Genetic algorithms for optimising cluster geometries. Dalton Trans. 2003, 22, 4193–4207. [Google Scholar] [CrossRef]
- Tian, Z.; Song, C.; Wang, C. First principle study on the structures and properties of Agm(Ag2S)6 (m = 3–12) clusters. J. Nanoparticle Res. 2022, 24, 1–12. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Kulichenko, M.; Chen, W.J.; Zhang, Y.Y.; Xu, C.Q.; Li, J.; Wang, L.S. Double σ−aromaticity in a planar zinc−doped gold cluster: Au9Zn−. J. Phys. Chem. A 2021, 125, 4606–4613. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Yan, G.; Liu, Y.; Cui, Z. Two–layer molecular rotors: A zinc dimer rotating over planar hypercoordinate motifs. J. Comput. Chem. 2023, 44, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Barroso, J.; Wang, M.; Liang, W.; Chen, C.; Zarate, X.; Orozco–Ic, M.; Cui, Z.; Merino, G. Structure and bonding of molecular stirrers with formula B7M2− and B8M2 (M = Zn, Cd, Hg). Phys. Chem. Chem. Phys. 2020, 22, 12312–12320. [Google Scholar] [CrossRef] [PubMed]
- Tsantis, S.T.; Bekiari, V.; Tzimopoulos, D.I.; Raptopoulou, C.P.; Psycharis, V.; Tsipis, A.; Perlepes, S.P. Reactivity of coordinated 2−pyridyl oximes: Synthesis, structure, spectroscopic characterization and theoretical studies of dichlorodi zinc (II) obtained from the reaction between zinc(II) nitrate and pyridine−2−chloroxime. Inorganics 2020, 8, 47. [Google Scholar] [CrossRef]
- Aguirre, A.R.; Parrilha, G.L.; Louro, S.R.; Alves, O.C.; Diniz, R.; Durval, F.; Rocha, W.; Beraldo, H. Structural and theoretical studies on copper(II) and zinc(II) complexes with a 9−anthraldehyde−derived thiosemicarbazone. Polyhedron 2022, 217, 115724. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT−D) for the 94 elements H−Pu. J. Chem. Phys. 2010, 132, 154104–154119. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Petersson, G.; Nakatsuji, H.; et al. Gaussian 16; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView 6.0.; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Song, C.; Tian, Z. Systematic study on the structures and properties of (Ag2S)n (n = 1–8) clusters. J. Mol. Model. 2019, 25, 310. [Google Scholar] [CrossRef]
- Emamian, S.; Lu, T.; Kruse, H.; Emamian, H. Exploring nature and predicting strength of hydrogen bonds: A correlation analysis between atoms–in–molecules descriptors, binding energies, and energy components of symmetry-adapted perturbation theory. J. Comput. Chem. 2019, 40, 2868–2881. [Google Scholar] [CrossRef]
- Deng, J.H.; Luo, J.; Mao, Y.L.; Lai, S.; Gong, Y.N.; Zhong, D.C.; Lu, T.B. π−π stacking interactions: Non−negligible forces for stabilizing porous supramolecular frameworks. Sci. Adv. 2020, 6, eaax9976. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, Z.; Song, C.; Wu, H. Density Functional Study to Investigate the Ability of (ZnS)n (n = 1–12) Clusters Removing Hg0, HgCl, and HgCl2 via Electron Localization Function and Non−Covalent Interactions Analyses. Molecules 2023, 28, 1214. https://doi.org/10.3390/molecules28031214
Tian Z, Song C, Wu H. Density Functional Study to Investigate the Ability of (ZnS)n (n = 1–12) Clusters Removing Hg0, HgCl, and HgCl2 via Electron Localization Function and Non−Covalent Interactions Analyses. Molecules. 2023; 28(3):1214. https://doi.org/10.3390/molecules28031214
Chicago/Turabian StyleTian, Zhimei, Chongfu Song, and Hai Wu. 2023. "Density Functional Study to Investigate the Ability of (ZnS)n (n = 1–12) Clusters Removing Hg0, HgCl, and HgCl2 via Electron Localization Function and Non−Covalent Interactions Analyses" Molecules 28, no. 3: 1214. https://doi.org/10.3390/molecules28031214
APA StyleTian, Z., Song, C., & Wu, H. (2023). Density Functional Study to Investigate the Ability of (ZnS)n (n = 1–12) Clusters Removing Hg0, HgCl, and HgCl2 via Electron Localization Function and Non−Covalent Interactions Analyses. Molecules, 28(3), 1214. https://doi.org/10.3390/molecules28031214