
The Molecular Basis of Heat-Stable Enterotoxin for Vaccine Development and Cancer Cell Detection

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
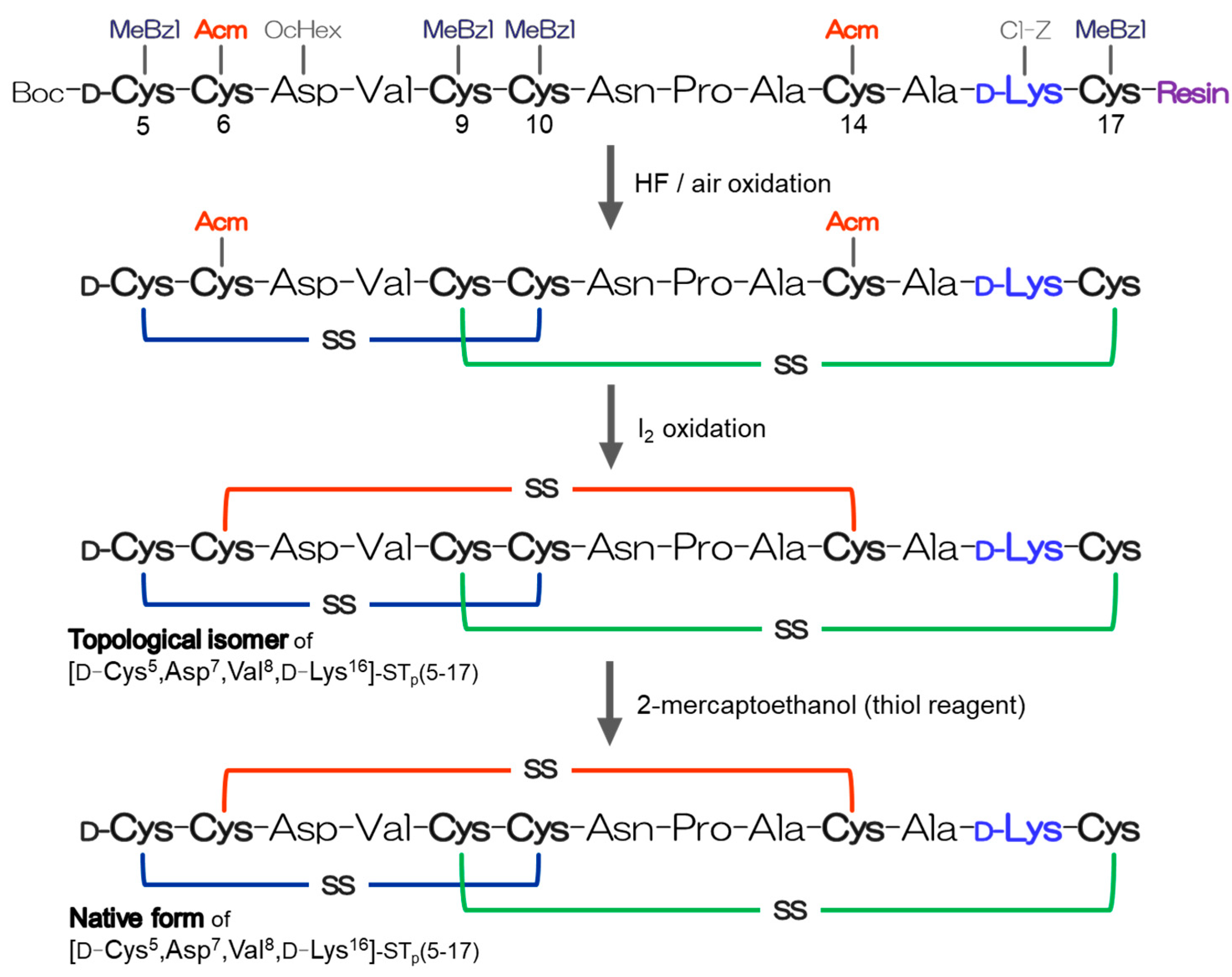
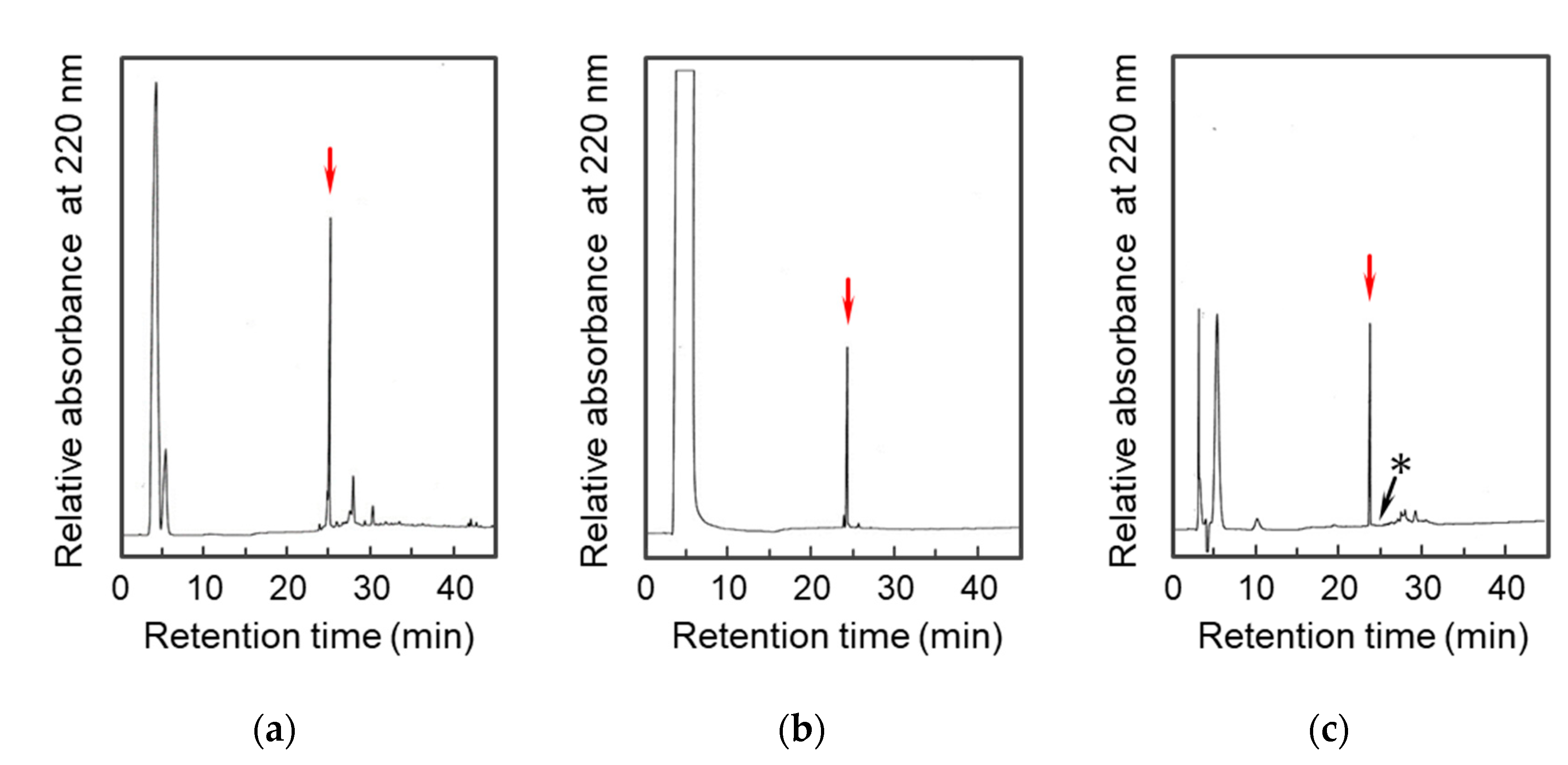
2.1. Preparation of the D-Lys-Substituted STa Peptides
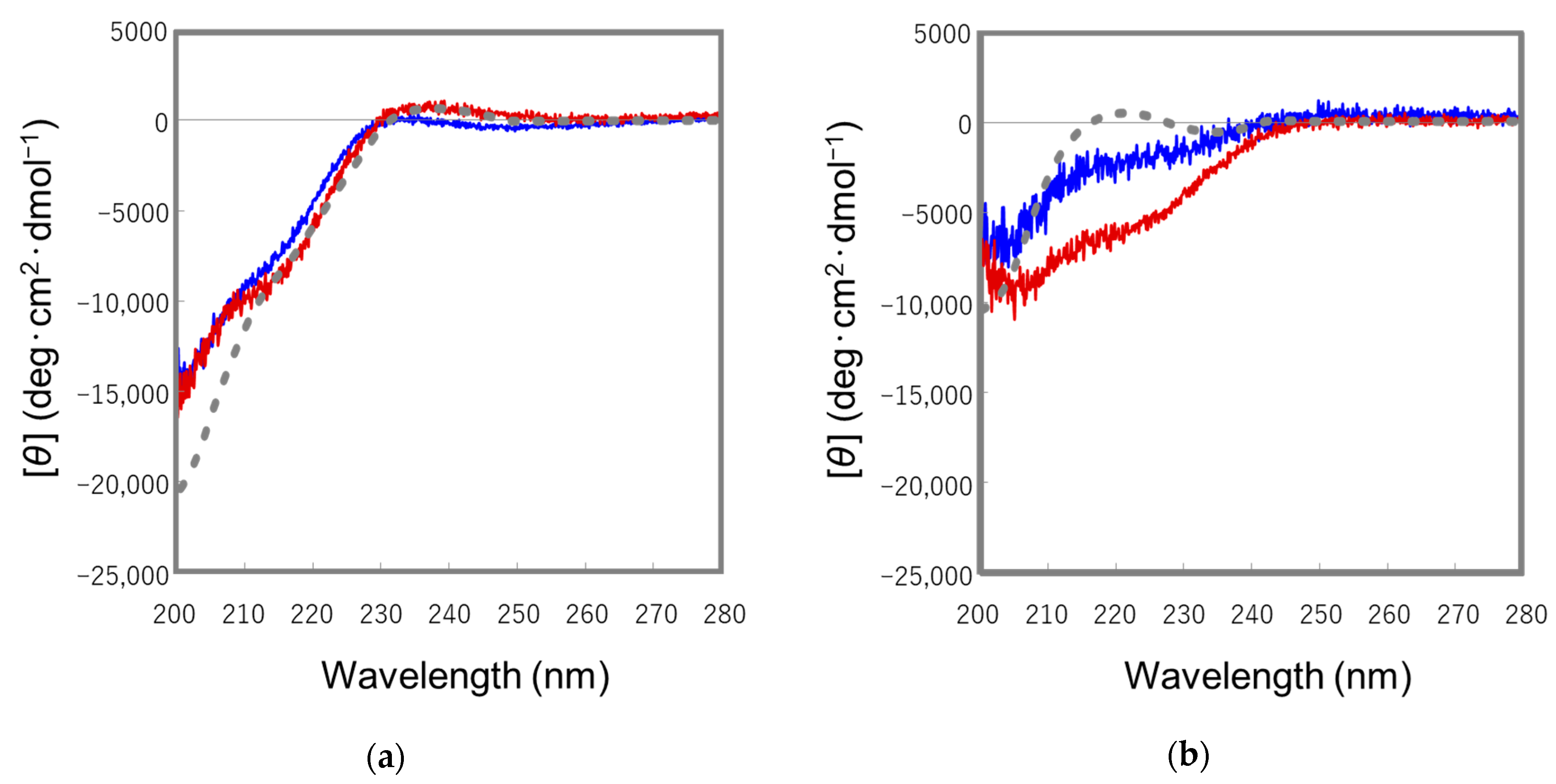
2.2. Conformational Analyses of the STp Peptides by Means of CD Spectroscopy
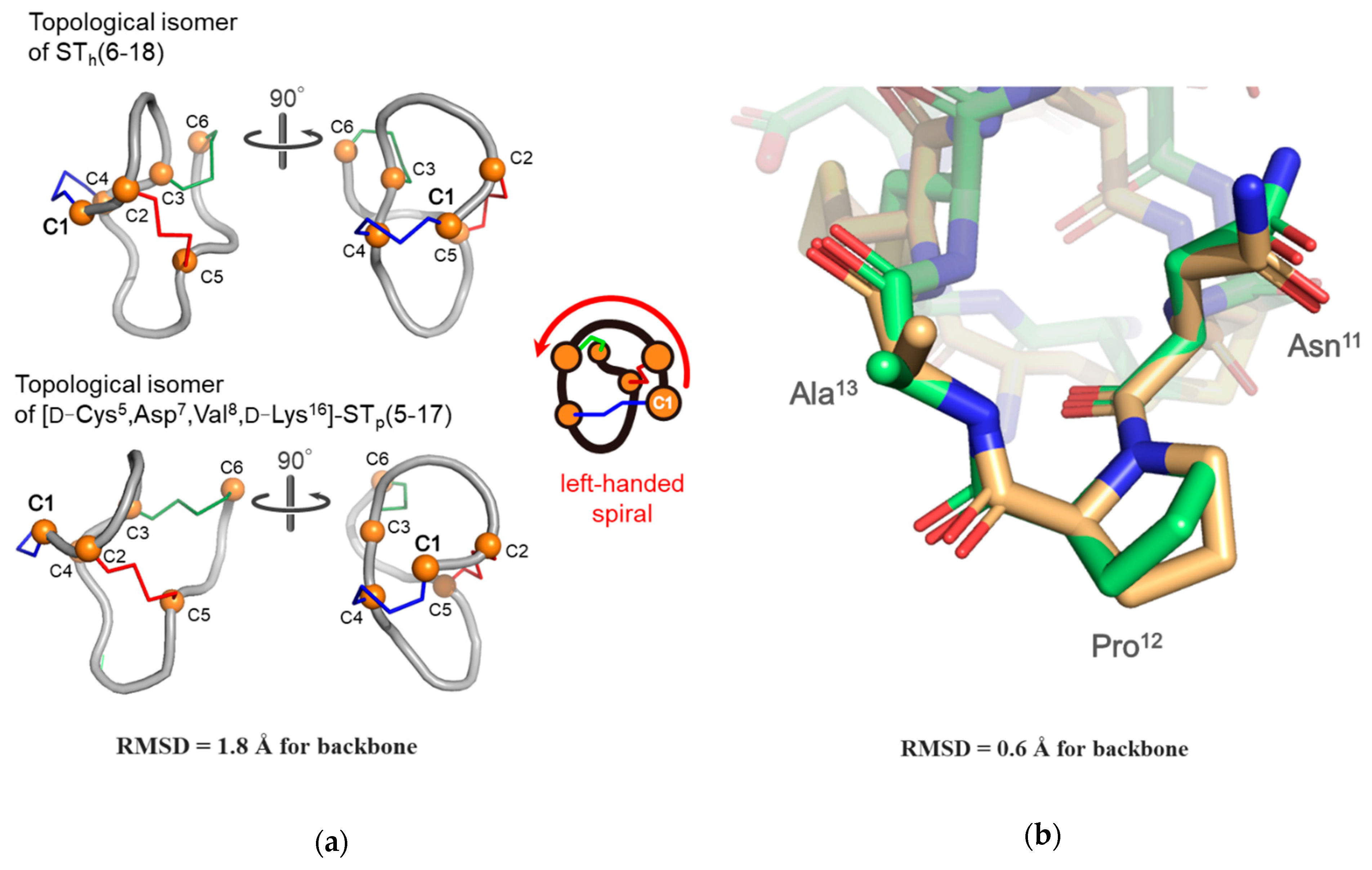
2.3. The Solution Structure of the Topological Isomer of the [D-Cys5,Asp7,Val8,D-Lys16]-STp(5-17) Peptide Determined by NMR Spectroscopy
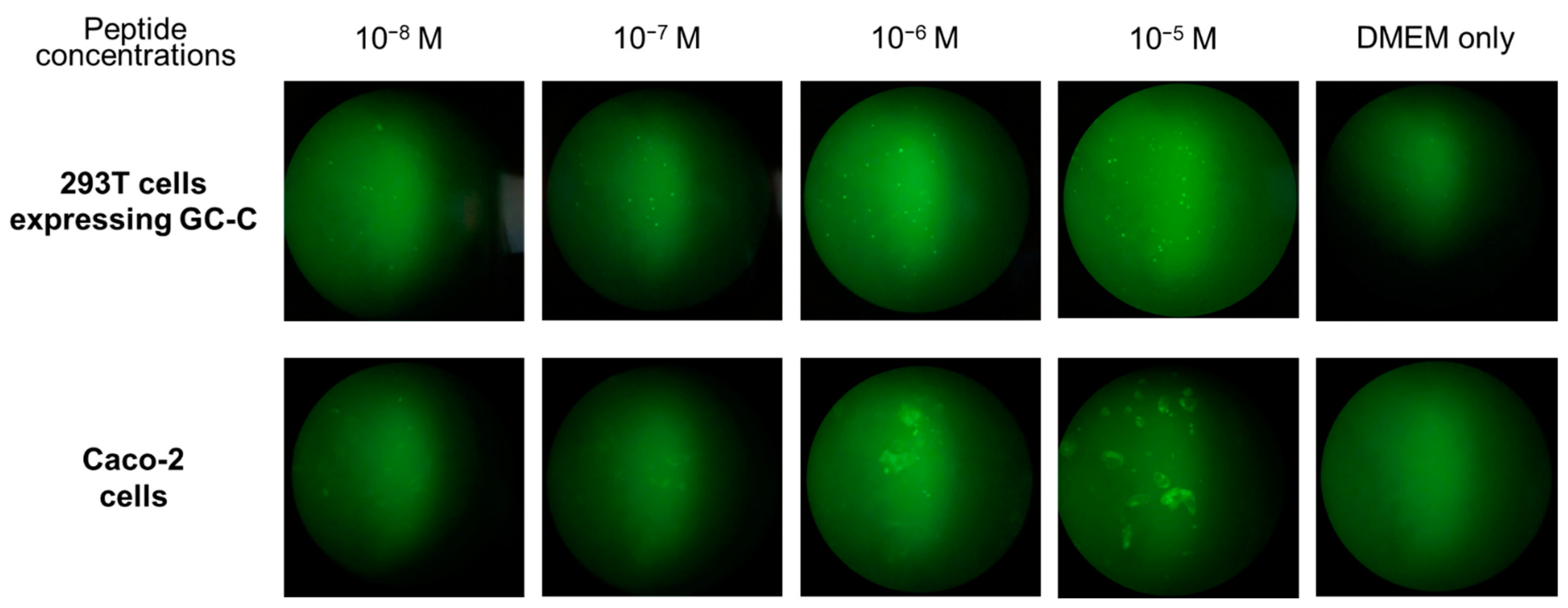
2.4. Detection of the GC-C Protein on Colon Cancer Cells
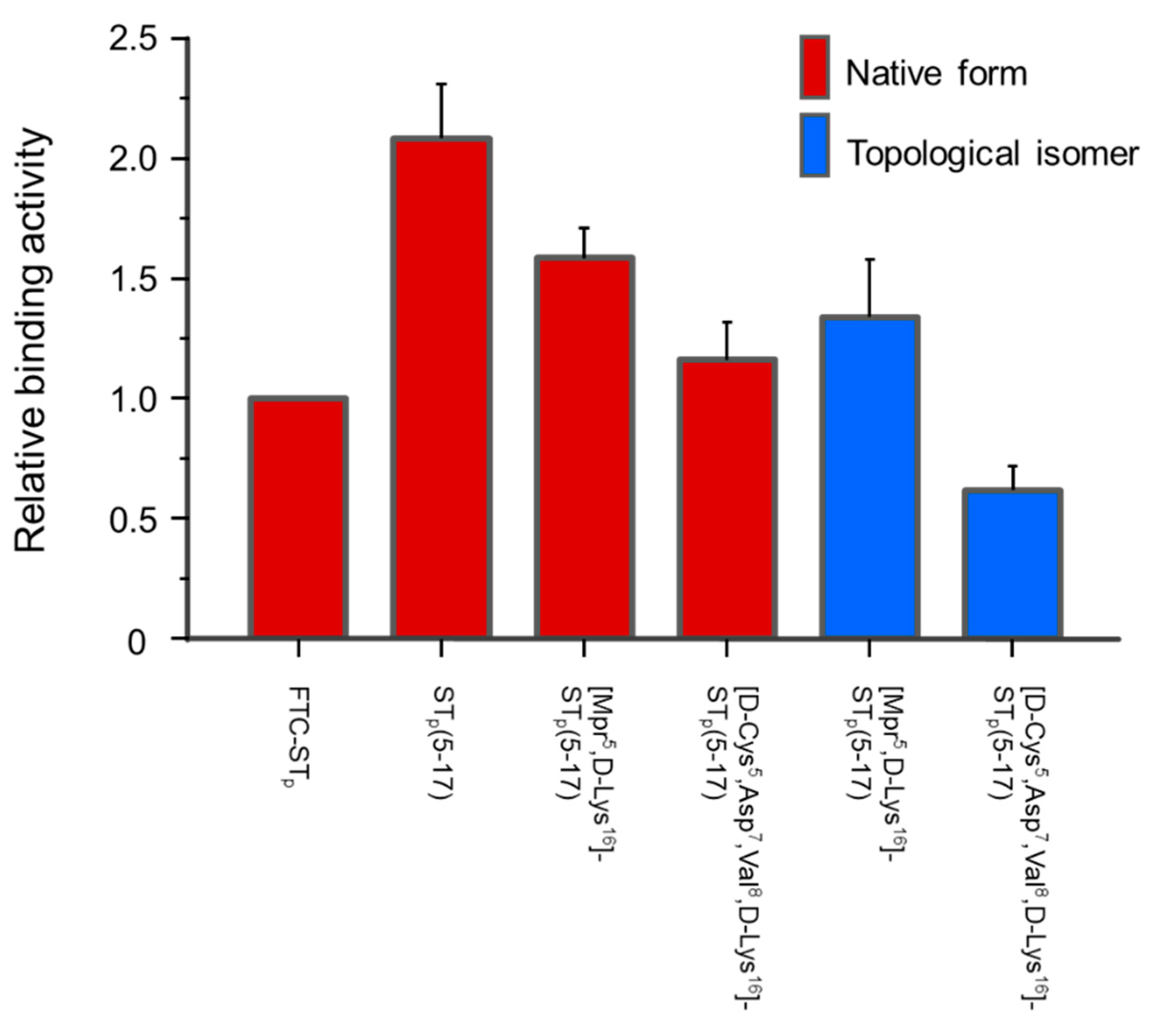
2.5. Receptor Binding Activity of the Synthetic STp Peptide
2.6. Design of STa Analogs Using the [D-Lys16]-STp(5-17) Peptide as a Molecular Basis for Vaccine Development
3. Materials and Methods
3.1. Materials
3.2. Peptide Synthesis
3.3. Reversed-Phase High Performance Liquid Chromatography (RP-HPLC)
3.4. Matrix Assisted Laser Desorption/Ionization Time of Flight Mass Spectrometry (MALDI-TOF/MS)
3.5. Circular Dichroism (CD) Measurement
3.6. NMR Measurements and Structure Calculations
3.7. Detection of the STa Receptor Using the Fluorescein Labeled STa Peptide
3.8. Competitive Binding Assay Using the Recombinant GC-C on 293T Cells
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Acm | acetamidomethyl |
| Boc | t-butyloxycarbonyl |
| CD | circular dichroism |
| Cl-Z | 2-chlorobenzyloxycarbonyl |
| COSY | two dimensional correlation spectroscopy |
| DMEM | Dulbecco’s modified eagle medium |
| DQF | double quantum filtered |
| ETEC | Enterotoxigenic Escherichia coli |
| FBS | fetal bovine serum |
| FITC | fluorescein isothiocyanate |
| FTC | fluorescein thiocarbonate |
| GC-C | guanylyl cyclase C |
| MALDI-TOF/MS | matrix-assisted laser desorption/ionization time of flight mass spectrometry |
| MeBzl | methyl benzyl |
| MED | minimum effective dose |
| Mpr | mercaptopropionyl |
| NOE | nuclear Overhauser effect |
| NOESY | nuclear Overhauser effect spectroscopy |
| OcHex | O-cyclohexyl |
| PBS | phosphate buffered saline |
| RMSD | root mean square deviation |
| RP-HPLC | reversed-phase high performance liquid chromatography |
| STa | heat-stable enterotoxin |
| STh | heat-stable enterotoxin derived from an E. coli strain from human |
| STp | heat-stable enterotoxin derived from an E. coli strain from porcine |
| TFA | trifluoroacetic acid |
References
- World Health Organization. Future directions for research on enterotoxigenic Escherichia coli vaccines for developing countries. Wkly. Epidemiol. Rec. 2006, 81, 97–104. [Google Scholar]
- Chao, A.C.; de Sauvage, F.J.; Dong, Y.J.; Wagner, J.A.; Goeddel, D.V.; Gardner, P. Activation of intestinal CFTR Cl- channel by heat-stable enterotoxin and guanylin via cAMP-dependent protein kinase. EMBO J. 1994, 13, 1065–1072. [Google Scholar] [CrossRef] [PubMed]
- Garbers, D.L. Guanylyl cyclase receptors and their endocrine, paracrine, and autocrine ligands. Cell 1992, 71, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Forte, L.R.; Krause, W.J.; Freeman, R.H. Escherichia coli enterotoxin receptors: Localization in opossum kidney, intestine, and testis. Am. J. Physiol. 1989, 257 Pt 2, F874–F881. [Google Scholar] [CrossRef]
- Walter, U. Distribution of cyclic-GMP-dependent protein kinase in various rat tissues and cell lines determined by a sensitive and specific radioimmunoassay. Eur. J. Biochem. 1981, 118, 339–346. [Google Scholar] [CrossRef]
- Amarachintha, S.; Harmel-Laws, E.; Steinbrecher, K.A. Guanylate cyclase C reduces invasion of intestinal epithelial cells by bacterial pathogens. Sci. Rep. 2018, 8, 1521. [Google Scholar] [CrossRef]
- Visweswariah, S.S.; Ramachandran, V.; Ramamohan, S.; Das, G.; Ramachandran, J. Characterization and partial purification of the human receptor for the heat-stable enterotoxin. Eur. J. Biochem. 1994, 219, 727–736. [Google Scholar] [CrossRef]
- Shimonishi, Y.; Hidaka, Y.; Koizumi, M.; Hane, M.; Aimoto, S.; Takeda, T.; Miwatani, T.; Takeda, Y. Mode of disulfide bond formation of a heat-stable enterotoxin (STh) produced by a human strain of enterotoxigenic Escherichia coli. FEBS Lett. 1987, 215, 165–170. [Google Scholar] [CrossRef]
- Takao, T.; Shimonishi, Y.; Kobayashi, M.; Nishimura, O.; Arita, M.; Takeda, T.; Honda, T.; Miwatani, T. Amino acid sequence of heat-stable enterotoxin produced by Vibrio cholerae non-01. FEBS Lett. 1985, 193, 250–254. [Google Scholar] [CrossRef]
- Takao, T.; Tominaga, N.; Yoshimura, S.; Shimonishi, Y.; Hara, S.; Inoue, T.; Miyama, A. Isolation, primary structure and synthesis of heat-stable enterotoxin produced by Yersinia enterocolitica. Eur. J. Biochem. 1985, 152, 199–206. [Google Scholar] [CrossRef]
- Yoshimura, S.; Ikemura, H.; Watanabe, H.; Aimoto, S.; Shimonishi, Y.; Hara, S.; Takeda, T.; Miwatani, T.; Takeda, Y. Essential Structure for Full Entero-Toxigenic Activity of Heat-Stable Entero-Toxin Produced by Entero-Toxigenic Escherichia-Coli. Febs. Lett. 1985, 181, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Hidaka, Y.; Kubota, H.; Yoshimura, S.; Ito, H.; Takeda, Y.; Shimonishi, Y. Disulfide linkages in a heat-stable enterotoxin (STp) produced by a porcine strain of enterotoxigenic Escherichia Coli. Bull. Chem. Soc. Jpn. 1988, 61, 1265–1271. [Google Scholar] [CrossRef]
- Aimoto, S.; Takao, T.; Shimonishi, Y.; Hara, S.; Takeda, T.; Takeda, Y.; Miwatani, T. Amino-acid sequence of a heat-stable enterotoxin produced by human enterotoxigenic Escherichia coli. Eur. J. Biochem. 1982, 129, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Takao, T.; Hitouji, T.; Aimoto, S.; Shimonishi, Y.; Hara, S.; Takeda, T.; Takeda, Y.; Miwatani, T. Amino acid sequence of a heat-stable enterotoxin isolated from enterotoxigenic Escherichia coli strain 18D. FEBS Lett. 1983, 152, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, H.; Sato, T.; Kubota, H.; Hata, Y.; Katsube, Y.; Shimonishi, Y. Molecular structure of the toxin domain of heat-stable enterotoxin produced by a pathogenic strain of Escherichia coli. A putative binding site for a binding protein on rat intestinal epithelial cell membranes. J. Biol. Chem. 1991, 266, 5934–5941. [Google Scholar] [CrossRef]
- Ikemura, H.; Watanabe, H.; Aimoto, S.; Shimonishi, Y.; Hara, S.; Takeda, T.; Takeda, Y.; Miwatani, T. Heat-stable Enterotoxin (STh) of Human Enterotoxigenic Escherichia coli (Strain SK-1). Structure-activity Relationship. B Chem. Soc. Jpn. 1984, 57, 2550–2556. [Google Scholar] [CrossRef]
- Yoshimura, S.; Hidaka, Y.; Aimoto, S.; Shimonishi, Y.; Takeda, T.; Miwatani, T.; Takeda, Y. Structure-Activity Relationship of a Heat-Stable Enterotoxin Produced by Yersinia enterocolitica. B Chem. Soc. Jpn. 1987, 60, 2481–2489. [Google Scholar] [CrossRef]
- Kubota, H.; Hidaka, Y.; Ozaki, H.; Ito, H.; Hirayama, T.; Takeda, Y.; Shimonishi, Y. A long-acting heat-stable enterotoxin analog of enterotoxigenic Escherichia coli with a single D-amino acid. Biochem. Biophys. Res. Commun. 1989, 161, 229–235. [Google Scholar] [CrossRef]
- Yamasaki, S.; Sato, T.; Hidaka, Y.; Ozaki, H.; Ito, H.; Hirayama, T.; Takeda, Y.; Sugimura, T.; Tai, A.; Shimonishi, Y. Structure-Activity Relationship of Escherichia coli Heat-Stable Enterotoxin: Role of Ala Residue at Position 14 in Toxin-Receptor Interaction. B Chem. Soc. Jpn. 1990, 63, 2063–2070. [Google Scholar] [CrossRef]
- Leavens, M.J.; Cherney, M.M.; Finnegan, M.L.; Bowler, B.E. Probing Denatured State Conformational Bias in a Three-Helix Bundle, UBA(2), Using a Cytochrome c Fusion Protein. Biochemistry 2018, 57, 1711–1721. [Google Scholar] [CrossRef]
- Shimamoto, S.; Fukutsuji, M.; Osumi, T.; Goto, M.; Toyoda, H.; Hidaka, Y. Topological Regulation of the Bioactive Conformation of a Disulfide-Rich Peptide, Heat-Stable Enterotoxin. Molecules 2020, 25, 4798. [Google Scholar] [CrossRef] [PubMed]
- Snook, A.E.; Stafford, B.J.; Li, P.; Tan, G.; Huang, L.; Birbe, R.; Schulz, S.; Schnell, M.J.; Thakur, M.; Rothstein, J.L.; et al. Guanylyl cyclase C-induced immunotherapeutic responses opposing tumor metastases without autoimmunity. J. Natl. Cancer Inst. 2008, 100, 950–961. [Google Scholar] [CrossRef]
- Snook, A.E.; Li, P.; Stafford, B.J.; Faul, E.J.; Huang, L.; Birbe, R.C.; Bombonati, A.; Schulz, S.; Schnell, M.J.; Eisenlohr, L.C.; et al. Lineage-specific T-cell responses to cancer mucosa antigen oppose systemic metastases without mucosal inflammatory disease. Cancer Res. 2009, 69, 3537–3544. [Google Scholar] [CrossRef] [PubMed]
- Mattiuzzi, C.; Lippi, G. Current Cancer Epidemiology. J. Epidemiol. Glob. Health 2019, 9, 217–222. [Google Scholar] [CrossRef]
- Hidaka, Y.; Ohno, M.; Hemmasi, B.; Hill, O.; Forssmann, W.G.; Shimonishi, Y. In vitro disulfide-coupled folding of guanylyl cyclase-activating peptide and its precursor protein. Biochemistry 1998, 37, 8498–8507. [Google Scholar] [CrossRef] [PubMed]
- de Brevern, A.G. Extension of the classical classification of beta-turns. Sci. Rep. 2016, 6, 33191. [Google Scholar] [CrossRef] [PubMed]
- Venkatachalam, C.M. Stereochemical criteria for polypeptides and proteins. V. Conformation of a system of three linked peptide units. Biopolymers 1968, 6, 1425–1436. [Google Scholar] [CrossRef]
- Imperiali, B.; Moats, R.A.; Fisher, S.L.; Prins, T.J. A conformational study of peptides with the general structure Ac-L-Xaa-Pro-D-Xaa-L-Xaa-NH2: Spectroscopic evidence for a peptide with significant.beta.-turn character in water and in dimethyl sulfoxide. J. Am. Chem. Soc. 1992, 114, 3182–3188. [Google Scholar] [CrossRef]
- Wada, A.; Hirayama, T.; Kitaura, H.; Fujisawa, J.; Hasegawa, M.; Hidaka, Y.; Shimonishi, Y. Identification of ligand recognition sites in heat-stable enterotoxin receptor, membrane-associated guanylyl cyclase C by site-directed mutational analysis. Infect. Immun. 1996, 64, 5144–5150. [Google Scholar] [CrossRef]
- Govasli, M.L.; Diaz, Y.; Zegeye, E.D.; Darbakk, C.; Taxt, A.M.; Puntervoll, P. Purification and Characterization of Native and Vaccine Candidate Mutant Enterotoxigenic Escherichia coli Heat-Stable Toxins. Toxins 2018, 10, 274. [Google Scholar] [CrossRef]
- Merrifield, R.B. Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149–2154. [Google Scholar] [CrossRef]
- Brunger, A.T.; Adams, P.D.; Clore, G.M.; DeLano, W.L.; Gros, P.; Grosse-Kunstleve, R.W.; Jiang, J.S.; Kuszewski, J.; Nilges, M.; Pannu, N.S.; et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Cryst. D Biol Cryst. 1998, 54, 905–921. [Google Scholar]
- Koradi, R.; Billeter, M.; Wuthrich, K. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graph. 1996, 14, 51–55. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Rullmannn, J.A.; MacArthur, M.W.; Kaptein, R.; Thornton, J.M. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 1996, 8, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Wada, A.; Hirayama, T.; Kitao, S.; Fujisawa, J.; Hidaka, Y.; Shimonishi, Y. Pig intestinal membrane-bound receptor (guanylyl cyclase) for heat-stable enterotoxin: cDNA cloning, functional expression, and characterization. Microbiol. Immunol. 1994, 38, 535–541. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goto, M.; Yoshino, S.; Hiroshima, K.; Kawakami, T.; Murota, K.; Shimamoto, S.; Hidaka, Y. The Molecular Basis of Heat-Stable Enterotoxin for Vaccine Development and Cancer Cell Detection. Molecules 2023, 28, 1128. https://doi.org/10.3390/molecules28031128
Goto M, Yoshino S, Hiroshima K, Kawakami T, Murota K, Shimamoto S, Hidaka Y. The Molecular Basis of Heat-Stable Enterotoxin for Vaccine Development and Cancer Cell Detection. Molecules. 2023; 28(3):1128. https://doi.org/10.3390/molecules28031128
Chicago/Turabian StyleGoto, Masaya, Shinya Yoshino, Kyona Hiroshima, Toru Kawakami, Kaeko Murota, Shigeru Shimamoto, and Yuji Hidaka. 2023. "The Molecular Basis of Heat-Stable Enterotoxin for Vaccine Development and Cancer Cell Detection" Molecules 28, no. 3: 1128. https://doi.org/10.3390/molecules28031128
APA StyleGoto, M., Yoshino, S., Hiroshima, K., Kawakami, T., Murota, K., Shimamoto, S., & Hidaka, Y. (2023). The Molecular Basis of Heat-Stable Enterotoxin for Vaccine Development and Cancer Cell Detection. Molecules, 28(3), 1128. https://doi.org/10.3390/molecules28031128