


Biotransformation of Ursonic Acid by Aspergillus ochraceus and Aspergillus oryzae to Discover Anti-Neuroinflammatory Derivatives

 ,
,
Abstract
:1. Introduction
2. Results
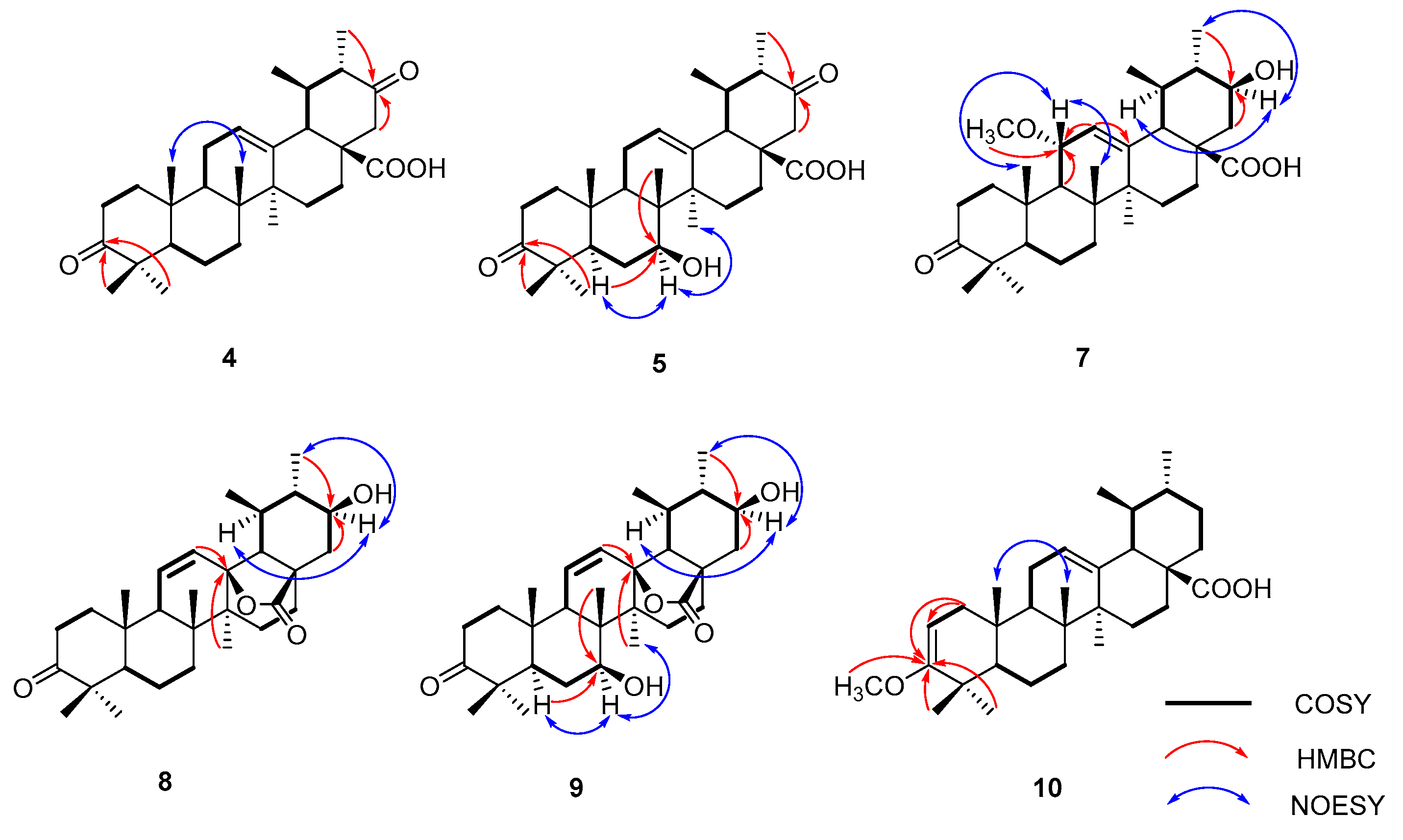
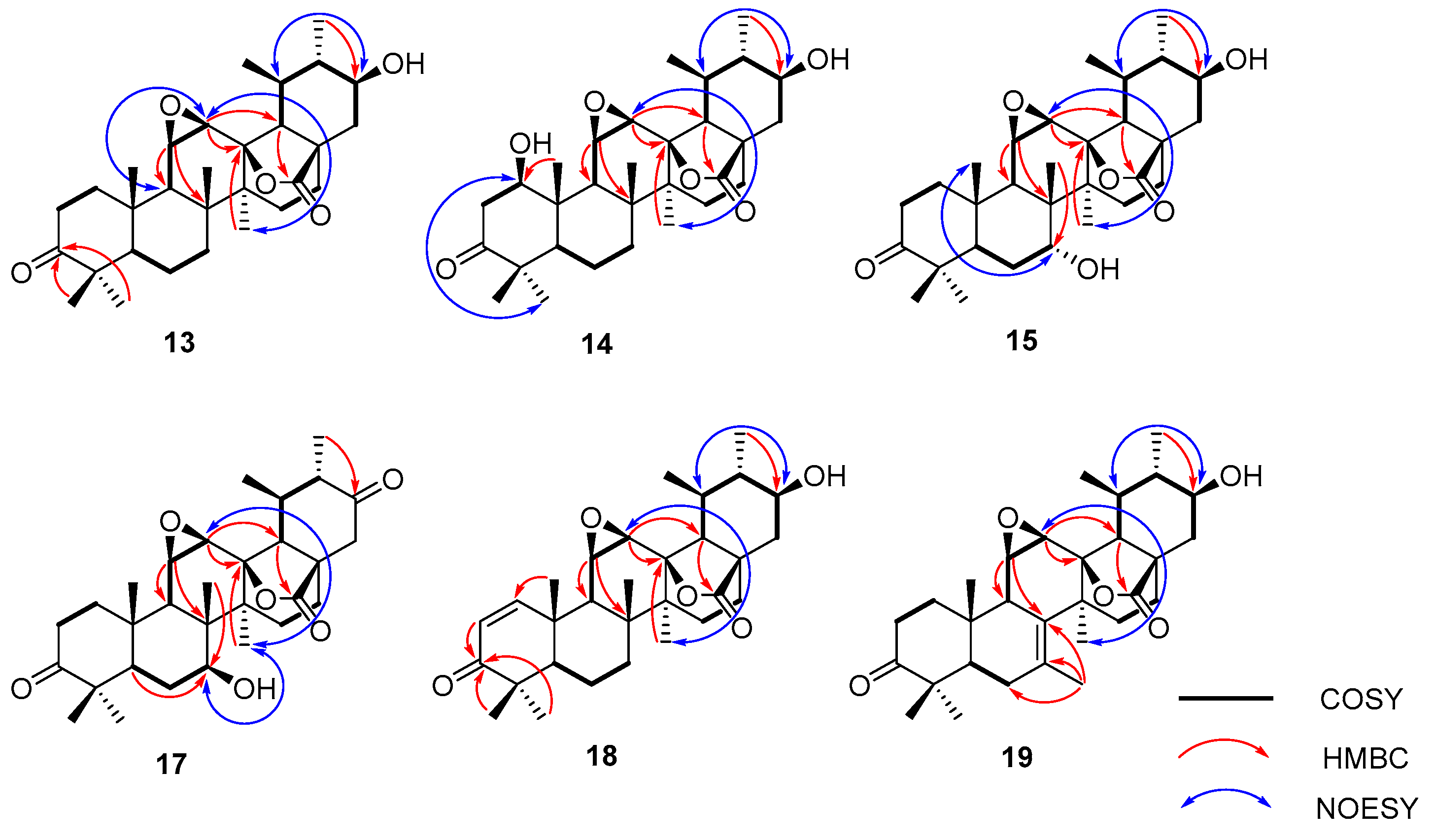
2.1. Biotransformation of Ursonic Acid

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 4 | 5 | 7 a | |||
|---|---|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | |
| 1 | 39.3 | 1.39, 1.83 m | 39.1 | 1.35, 1.86 m | 40.4 | 1.67, 2.22 m |
| 2 | 34.1 | 2.30, 2.48 m | 34.0 | 2.34, 2.48 m | 34.4 | 2.39, 2.53 m |
| 3 | 217.7 | 217.0 | 217.9 | |||
| 4 | 47.4 | 47.7 | 47.6 | |||
| 5 | 55.2 | 1.23 m | 52.5 | 1.37 m | 55.3 | 1.36 m |
| 6 | 19.5 | 1.24, 1.41 m | 26.6 | 1.55, 1.82 m | 19.6 | 1.32, 1.47 m |
| 7 | 32.3 | 1.29, 1.42 m | 73.4 | 3.89 dd (9.7, 6.0) | 33.1 | 1.31, 1.46 m |
| 8 | 39.5 | 45.2 | 42.3 | |||
| 9 | 46.7 | 1.53 m | 47.0 | 1.44 m | 51.3 | 1.74 d (9.1) |
| 10 | 36.7 | 36.8 | 37.7 | |||
| 11 | 23.6 | 1.53, 1.94 m | 23.7 | 1.93, 2.04 m | 76.5 | 3.84 d (8.6) |
| 12 | 127.2 | 5.35 d (3.8) | 127.5 | 5.42 dd (4.6, 2.7) | 125.7 | 5.52 m |
| 13 | 137.1 | 136.6 | 141.6 | |||
| 14 | 41.9 | 43.4 | 42.3 | |||
| 15 | 29.7 | 1.14 d (6.0), 1.18 m | 31.6 | 1.49, 1.92 m | 28.1 | 1.15, 1.77 m |
| 16 | 27.9 | 1.06, 1.80 m | 30.4 | 1.37, 1.54 m | 25.2 | 1.77, 1.88 m |
| 17 | 51.2 | 51.1 | 48.4 | |||
| 18 | 52.4 | 2.62 m | 51.1 | 2.08 m | 51.8 | 2.29 d (11.4) |
| 19 | 41.5 | 1.72 m | 41.7 | 1.74 m | 37.7 | 1.46 m |
| 20 | 51.1 | 2.08 m | 53.1 | 2.63 d (11.4) | 46.6 | 0.99 m |
| 21 | 209.8 | 210.4 | 71.1 | 3.44 m | ||
| 22 | 50.5 | 2.35, 2.57 m | 50.3 | 2.37 m, 2.56 d (12.9) | 44.5 | 1.58, 2.09 m |
| 23 | 26.6 | 1.02 s | 26.6 | 1.04 s | 26.7 | 1.09 s |
| 24 | 21.5 | 0.96 s | 21.5 | 0.98 s | 21.5 | 1.04 s |
| 25 | 15.3 | 0.99 s | 12.4 | 1.00 s | 15.7 | 1.13 s |
| 26 | 17.0 | 0.76 s | 9.7 | 0.81 s | 18.5 | 0.86 s |
| 27 | 23.8 | 0.99 s | 23.4 | 1.08 s | 22.7 | 1.15 s |
| 28 | 180.0 | 179.1 | 175.7 | |||
| 29 | 18.4 | 0.95 d (5.7) | 18.4 | 0.95 d (6.4) | 17.2 | 0.99 d (6.5) |
| 30 | 12.5 | 1.01 d (3.7) | 15.4 | 0.99 d (5.7) | 16.5 | 1.09 d (6.3) |
| -OCH3 | 54.7 | 3.29 s | ||||
| Position | 8 | 9 | 10 | |||
|---|---|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | |
| 1 | 39.0 | 1.38, 2.02 m | 38.5 | 1.34, 2.00 m | 39.7 | 1.70, 2.00 m |
| 2 | 33.9 | 2.36, 2.57 m | 33.7 | 2.37, 2.55 m | 89.6 | 4.39 dd (6.7, 1.8) |
| 3 | 216.8 | 216.1 | 160.6 | |||
| 4 | 47.6 | 47.2 | 37.1 | |||
| 5 | 54.6 | 1.27 m | 51.8 | 1.89 m | 53.0 | 1.12 m |
| 6 | 18.8 | 1.49, 1.62 m | 29.8 | 1.52, 1.64 m | 19.4 | 1.37, 1.48 m |
| 7 | 30.5 | 1.22, 1.38 m | 72.6 | 3.87 dd (9.9, 5.5) | 32.4 | 1.37,1.51 m |
| 8 | 42.0 | 46.8 | 39.4 | |||
| 9 | 52.4 | 1.98 m | 52.3 | 1.36 m | 46.1 | 1.54 m |
| 10 | 36.1 | 36.0 | 35.9 | |||
| 11 | 133.3 | 5.92 dd (10.3, 1.6) | 132.1 | 5.85 d (10.4) | 23.3 | 1.91, 1.97 m |
| 12 | 128.8 | 5.51 dd (10.3, 3.2) | 129.5 | 5.53 dd (10.3, 3.1) | 126.1 | 5.27 t (3.6) |
| 13 | 89.5 | 89.4 | 137.7 | |||
| 14 | 41.6 | 42.9 | 42.0 | |||
| 15 | 25.6 | 1.17, 1.68 m | 29.4 | 1.18, 1.91 m | 28.0 | 1.11, 1.86 m |
| 16 | 23.7 | 1.50 m, 1.92 td (13.0, 5.7) | 23.8 | 1.48, 1.92 m | 24.1 | 1.67, 2.00 m |
| 17 | 45.6 | 45.5 | 48.1 | |||
| 18 | 59.9 | 1.64 m | 60.1 | 1.64 m | 52.7 | 2.19 dd (11.5, 1.7) |
| 19 | 36.0 | 1.82 m | 36.0 | 1.84 m | 39.1 | 1.34 m |
| 20 | 47.8 | 0.78 m | 47.8 | 0.80 m | 38.8 | 1.01 m |
| 21 | 71.8 | 3.39 m | 71.7 | 3.39 m | 30.7 | 1.34, 1.51 m |
| 22 | 40.3 | 1.40 m, 2.09 dd (12.8, 4.5) | 40.2 | 1.37 m, 2.09 dd (12.8, 4.5) | 36.8 | 1.67, 1.71 m |
| 23 | 26.0 | 1.03 s | 26.0 | 1.04 s | 28.6 | 1.04 s |
| 24 | 20.8 | 0.98 s | 20.8 | 0.98 s | 20.0 | 0.93 s |
| 25 | 17.8 | 0.99 s | 17.1 | 0.98 s | 15.4 | 0.95 s |
| 26 | 18.6 | 1.03 s | 13.7 | 1.07 s | 17.0 | 0.80 s |
| 27 | 16.0 | 1.08 s | 16.3 | 1.15 s | 23.5 | 1.08 s |
| 28 | 178.5 | 178.5 | 183.7 | |||
| 29 | 17.3 | 0.97 d (6.0) | 17.8 | 0.97 d (5.8) | 17.0 | 0.86 d (6.4) |
| 30 | 14.4 | 1.01 d (6.1) | 14.4 | 1.01 d (6.4) | 21.2 | 0.94 d (5.4) |
| -OCH3 | 54.2 | 3.47 s | ||||
| Position | 13 | 14 a | 15 b | |||
|---|---|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | |
| 1 | 39.6 | 1.62, 2.26 m | 78.8 | 4.25 dd (7.2, 6.6) | 39.1 | 1.67, 2.18 m |
| 2 | 33.9 | 2.48, 2.71 m | 45.8 | 2.99 dd (14.8, 6.6) 3.20 dd (14.8, 7.2) | 33.6 | 2.41, 2.62 m |
| 3 | 216.6 | 214.1 | 218.4 | |||
| 4 | 47.7 | 47.5 | 46.9 | |||
| 5 | 55.1 | 1.36 m | 51.1 | 1.40 dd (12.6, 3.1) | 46.3 | 2.02 m |
| 6 | 19.0 | 1.56, 1.72 m | 19.1 | 1.52, 1.71 m | 28.8 | 1.32, 2.05 m |
| 7 | 32.5 | 1.14, 1.39 m | 32.3 | 1.03, 1.36 m | 72.5 | 3.35 m |
| 8 | 40.1 | 40.7 | 42.5 | |||
| 9 | 49.2 | 1.62 m | 50.2 | 1.98 br.s | 44.9 | 2.14 m |
| 10 | 37.3 | 43.9 | 37.3 | |||
| 11 | 52.0 | 3.41 d (3.8) | 54.8 | 4.78 d (3.8) | 53.0 | 3.41 m |
| 12 | 51.3 | 3.16 d (3.8) | 52.1 | 3.28 d (3.8) | 51.4 | 3.13 d (3.8) |
| 13 | 88.8 | 89.2 | 90.2 | |||
| 14 | 41.9 | 42.2 | 42.6 | |||
| 15 | 25.5 | 1.20, 1.75 m | 26.1 | 1.16, 1.79 m | 24.4 | 1.60, 2.08 m |
| 16 | 23.4 | 1.58, 1.97 m | 23.9 | 1.55, 2.12 m | 22.9 | 1.38, 2.08 m |
| 17 | 46.3 | 46.6 | 46.2 | |||
| 18 | 60.8 | 1.99 m | 60.0 | 2.00 d (11.2) | 60.9 | 1.85 dd (11.6, 1.4) |
| 19 | 36.3 | 1.97 m | 36.7 | 2.09 m | 36.7 | 1.98 m |
| 20 | 47.5 | 0.97 m | 48.4 | 1.12 m | 47.2 | 0.81 m |
| 21 | 71.5 | 3.49 m | 70.7 | 3.76 m | 70.4 | 3.32 ddd (11.6, 10.0, 4.6) |
| 22 | 40.2 | 1.51, 2.21 m | 41.6 | 1.92, 2.56 m | 40.0 | 1.32, 1.98 m |
| 23 | 26.0 | 1.11 s | 26.8 | 1.16 s | 24.9 | 0.96 s |
| 24 | 20.8 | 1.09 s | 20.6 | 1.14 s | 20.0 | 0.95 s |
| 25 | 17.6 | 1.32 s | 14.1 | 1.55 s | 16.7 | 1.18 s |
| 26 | 19.3 | 1.31 s | 19.8 | 1.59 s | 20.2 | 1.10 s |
| 27 | 15.9 | 1.09 s | 16.0 | 1.19 s | 16.7 | 1.45 s |
| 28 | 177.7 | 178.5 | 179.6 | |||
| 29 | 18.6 | 1.20 d (5.0) | 18.6 | 1.13 d (6.2) | 17.7 | 1.11 d (5.7) |
| 30 | 14.6 | 1.13 d (6.3) | 15.0 | 1.33 d (6.4) | 13.5 | 0.99 d (6.4) |
| Position | 16 | 17 a | 18 | 19 a | ||||
|---|---|---|---|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | |
| 1 | 40.0 | 1.70, 2.19 m | 39.2 | 1.58, 2.23 m | 160.5 | 7.62 d (10.3) | 39.4 | 1.54, 2.27 m |
| 2 | 34.8 | 2.51, 2.67 m | 33.7 | 2.49, 2.69 m | 126.1 | 5.89 d (10.3) | 34.4 | 2.24 m 2.75 td (14.1, 5.5) |
| 3 | 219.2 | 215.6 | 207.3 | 216.1 | ||||
| 4 | 48.3 | 47.3 | 46.1 | 47.1 | ||||
| 5 | 53.3 | 1.60 m | 52.7 | 1.45 m | 54.5 | 1.76 m | 50.2 | 1.36 dd (12.5, 4.1) |
| 6 | 30.8 | 1.62, 1.77 m | 30.3 | 1.46, 1.70 m | 19.5 | 1.63, 1.76 m | 32.5 | 1.58, 2.61 m |
| 7 | 73.7 | 3.79 dd (10.7, 4.3) | 73.0 | 3.78 t (7.6) | 33.6 | 1.17, 1.50 m | 129.7 | |
| 8 | 46.7 | 45.4 | 42.3 | 128.5 | ||||
| 9 | 49.1 | 1.60 m | 48.1 | 1.46 m | 45.1 | 1.98 m | 49.5 | 2.37 m |
| 10 | 38.4 | 37.2 | 41.4 | 36.6 | ||||
| 11 | 53.9 | 3.44 m | 52.3 | 3.43 m | 52.8 | 3.75 d (3.8) | 52.6 | 3.30 dd (3.8, 1.5) |
| 12 | 53.0 | 3.19 d (3.9) | 51.4 | 3.24 d (3.8) | 52.8 | 3.28 d (3.8) | 52.5 | 3.09 d (3.8) |
| 13 | 91.3 | 88.7 | 90.9 | 87.8 | ||||
| 14 | 44.4 | 43.1 | 43.3 | 43.4 | ||||
| 15 | 30.4 | 1.29, 1.88 m | 29.2 | 1.70, 1.96 m | 26.7 | 1.28, 1.64 m | 35.5 | 1.70, 2.14 m |
| 16 | 24.6 | 1.41, 2.08 m | 23.9 | 1.58, 1.70 m | 24.4 | 1.43 m 2.14 td (13.3, 6.0) | 23.7 | 1.50, 1.88 m |
| 17 | 47.6 | 48.4 | 47.7 | 46.6 | ||||
| 18 | 62.4 | 1.99 d (11.7) | 60.6 | 2.49 m | 62.0 | 2.04 m | 59.5 | 1.91 m |
| 19 | 37.6 | 2.08 m | 38.3 | 2.28 m | 37.4 | 2.05 m | 35.3 | 1.88 m |
| 20 | 48.6 | 0.92 m | 51.0 | 2.04 m | 48.6 | 0.92 d | 47.5 | 0.90 m |
| 21 | 71.9 | 3.42 m | 208.4 | - | 71.8 | 3.42 m | 71.7 | 3.42 m |
| 22 | 41.1 | 1.41, 2.06 m | 47.0 | 2.58, 2.61 m | 41.1 | 1.44, 2.06 m | 39.8 | 1.44, 2.13 m |
| 23 | 26.6 | 1.10 s | 26.0 | 1.11 s | 27.6 | 1.15 s | 25.3 | 1.01 s |
| 24 | 21.2 | 1.07 s | 20.8 | 1.08 s | 21.8 | 1.11 s | 22.3 | 1.07 s |
| 25 | 18.1 | 1.25 s | 17.4 | 1.30 s | 21.5 | 1.41 s | 15.6 | 1.19 s |
| 26 | 14.9 | 1.28 s | 13.9 | 1.36 s | 20.4 | 1.32 s | 23.2 | 1.82 s |
| 27 | 16.4 | 1.20 s | 16.4 | 1.14 s | 16.4 | 1.14 s | 24.3 | 1.18 s |
| 28 | 181.0 | 176.3 | 180.7 | 177.7 | ||||
| 29 | 18.9 | 1.19 d (6.3) | 19.6 | 1.29 d (6.3) | 18.9 | 1.21 d (5.6) | 18.4 | 1.11 d (5.8) |
| 30 | 14.7 | 1.08 d (6.6) | 11.2 | 1.09 d (6.5) | 14.8 | 1.09 d (6.4) | 14.6 | 1.05 d (6.4) |
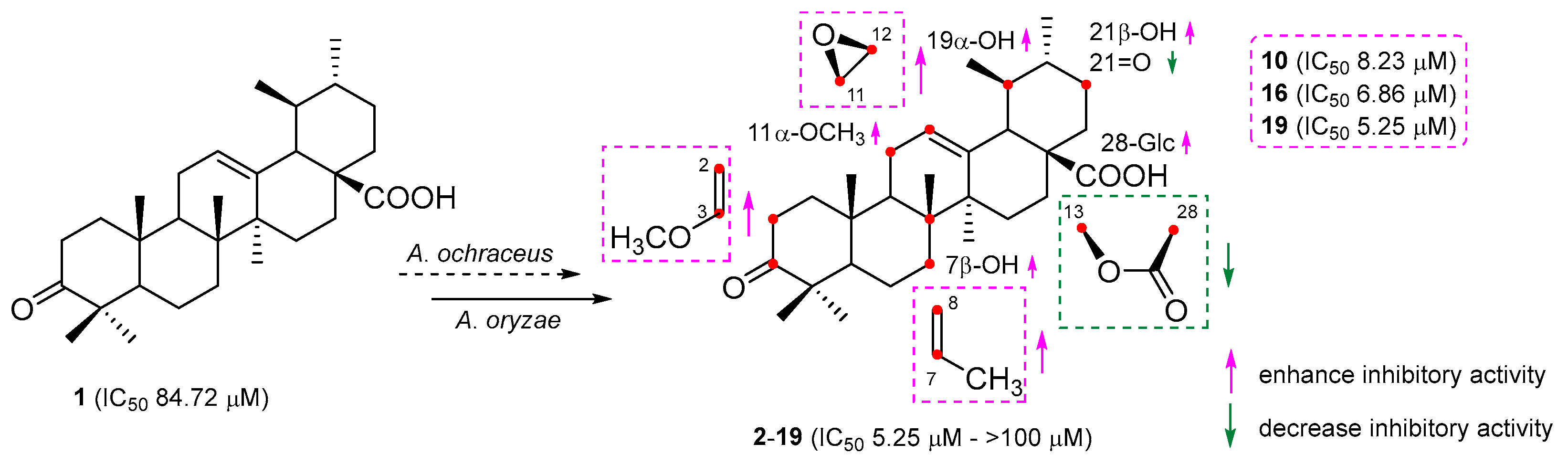
2.2. Anti-Neuroinflammatory Activities
| Compounds | IC50 (μM) | Cell Viability (%) | Compounds | IC50 (μM) | Cell Viability (%) |
|---|---|---|---|---|---|
| L-NMMA a | 28.25 ± 2.97 | 100.51 ± 4.36 | 10 | 8.23 ± 2.61 | 104.41 ± 4.05 |
| Ursonic acid (1) | 84.72 ± 3.22 | 98.84 ± 3.61 | 11 | 42.48 ± 3.70 | 101.29 ± 3.42 |
| 2 | 38.17 ± 4.09 | 101.25 ± 2.45 | 12 | 31.05 ± 3.98 | 98.23 ± 4.37 |
| 3 | 20.93 ± 2.13 | 100.33 ± 3.92 | 13 | 15.19 ± 3.07 | 102.64 ± 3.59 |
| 4 | >100 | 103.62 ± 3.18 | 14 | 22.57 ± 3.44 | 104.05 ± 4.92 |
| 5 | 52.81 ± 3.34 | 100.56 ± 4.11 | 15 | 36.64 ± 2.33 | 101.78 ± 3.18 |
| 6 | 50.24 ± 3.16 | 104.79 ± 4.23 | 16 | 6.86 ± 3.52 | 97.82 ± 4.75 |
| 7 | 11.58 ± 2.01 | 99.17 ± 3.72 | 17 | 40.79 ± 3.26 | 100.37 ± 3.54 |
| 8 | >100 | 102.48 ± 3.54 | 18 | 17.42 ± 2.72 | 103.24 ± 3.26 |
| 9 | >100 | 103.95 ± 3.86 | 19 | 5.25 ± 3.19 | 102.83 ± 3.03 |
3. Discussion
4. Materials and Methods
4.1. General
4.2. Microorganism and Substance
4.3. Biotransformation Procedures
4.4. Extraction and Isolation
4.5. Compound Characterization
4.6. Anti-Neuroinflammatory Activities
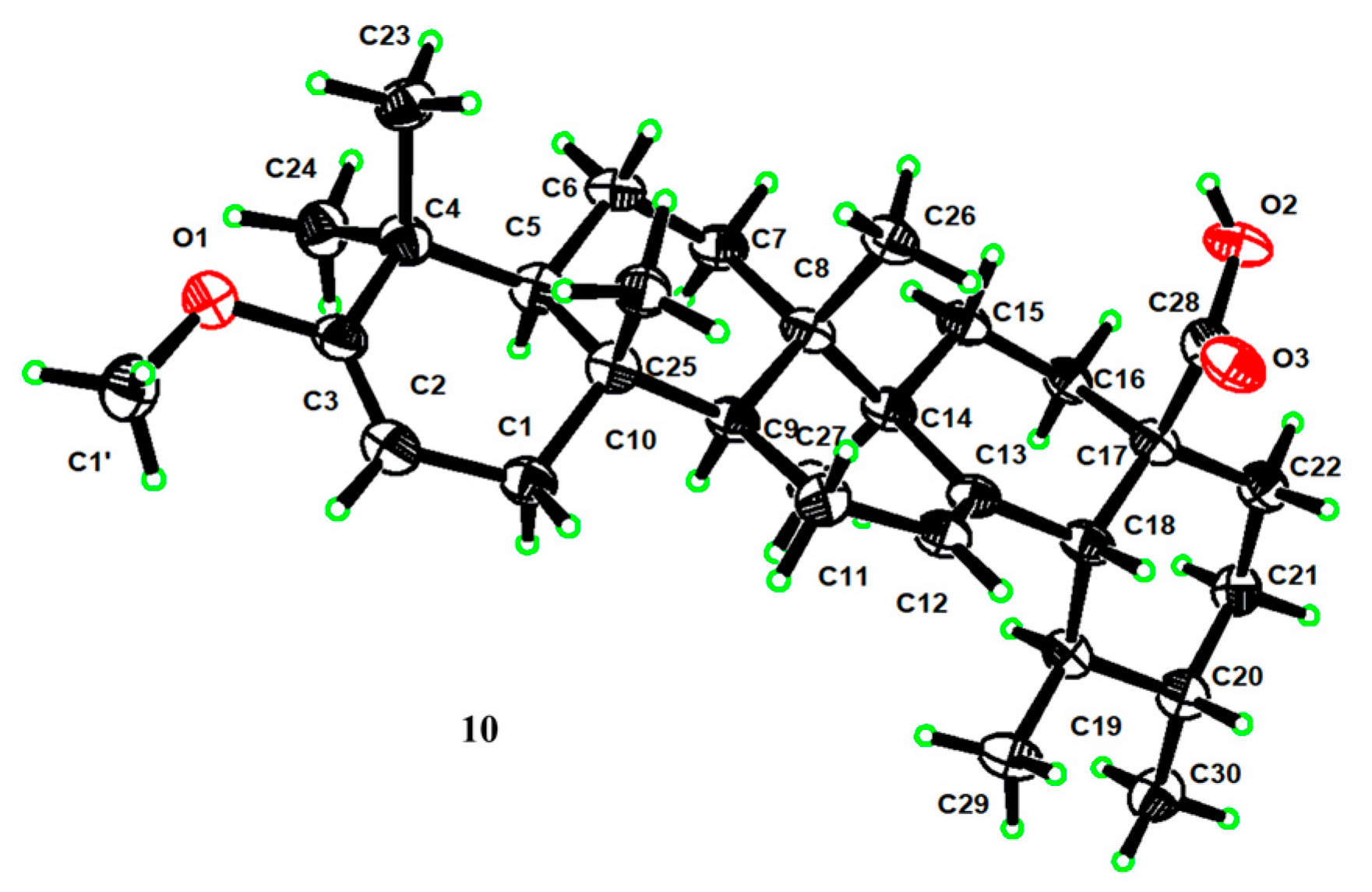
4.7. X-ray Crystallographic Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Romano, R.; Bucci, C. Antisense therapy: A potential breakthrough in the treatment of neurodegenerative diseases. Neural Regen. Res. 2024, 19, 1027–1035. [Google Scholar] [CrossRef] [PubMed]
- Arjunan, A.; Sah, D.K.; Woo, M.; Song, J. Identification of the molecular mechanism of insulin-like growth factor-1 (IGF-1): A promising therapeutic target for neurodegenerative diseases associated with metabolic syndrome. Cell Biosci. 2023, 13, 16. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.B.; Lin, H.Y.; Hong, X.; Ji, D.L.; Wu, F. Poloxamer 188-mediated anti-inflammatory effect rescues cognitive deficits in paraquat and maneb-induced mouse model of Parkinson’s disease. Toxicology 2020, 436, 152437. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Duan, J.A.; Qian, D.; Tang, Y.P.; Wu, D.W.; Su, S.L.; Wang, H.Q.; Zhao, Y.N. Content variations of triterpenic acid, nucleoside, nucleobase, and sugar in jujube (Ziziphus jujuba) fruit during ripening. Food Chem. 2015, 167, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Mahjoub, F.; Akhavan, R.K.; Yousef, M.; Mohebbi, M.; Salari, R. Pistacia atlantica Desf. A review of its traditional uses, phytochemicals and pharmacology. J. Med. Life 2018, 11, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Son, J.; Lee, S.Y. Therapeutic potential of ursonic acid: Comparison with ursolic acid. Biomolecules 2020, 10, 1505. [Google Scholar] [CrossRef] [PubMed]
- Capela, R.; Moreira, R.; Lopes, F. An overview of drug resistance in Protozoal diseases. Int. J. Mol. Sci. 2019, 20, 5748. [Google Scholar] [CrossRef]
- Hevener, E.; Verstak, T.A.; Lutat, K.E.; Riggsbee, D.L.; Mooney, J.W. Recent developments in topoisomerase targeted cancer chemotherapy. Acta Pharm. Sin. B 2018, 8, 844–861. [Google Scholar] [CrossRef]
- Son, J.; Lee, S.Y. Ursonic acid exerts inhibitory effects on matrix metalloproteinases via ERK signaling pathway. Chem.-Biol. Interact. 2020, 315, 108910. [Google Scholar] [CrossRef]
- Furtado, N.A.J.C.; Pirson, K.; Edelberg, H.; Miranda, L.M.; Loira-Pastoriza, C.; Preat, V.; Larondelle, Y.; Andre, C.M. Pentacyclic triterpene bioavailability: An overview of in vitro and in vivo studies. Molecules 2017, 22, 400. [Google Scholar] [CrossRef]
- Borkova, L.; Frydrych, I.; Jakubcova, N.; Adamek, R.; Liskova, B.; Gurska, S.; Medvedíkova, M.; Hajduch, M. Synthesis and biological evaluation of triterpenoid thiazoles derived from betulonic acid, dihydrobetulonic acid, and ursonic acid. Eur. J. Med. Chem. 2020, 185, 111806. [Google Scholar] [CrossRef] [PubMed]
- Mlala, S.; Oyedeji, A.O.; Gondwe, M.; Oyedeji, O.O. Ursolic acid and its derivatives as bioactive agents. Molecules 2019, 24, 2751. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.P.; Zhang, B.J.; Cui, X.P.; Yang, Y.; Jiang, Z.Y.; Zhou, Z.H.; Zhong, Y.Y.; Mai, Y.Y.; Ouyang, Z.; Chen, H.S.; et al. Synthesis and biological evaluation of novel ursolic acid analogues as potential α-glucosidase inhibitors. Sci. Rep. 2017, 7, 45578. [Google Scholar] [CrossRef] [PubMed]
- Diao, M.X.; Li, C.; Li, J.X.; Lu, J.; Xie, N.Z. Probing the biotransformation process of sclareol by resting cells of Hyphozyma roseonigra. Food Chem. 2022, 70, 10563–10570. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.H.; Xin, Y.; Qiu, Z.D.; Zhang, Q.; He, T.Z.; Qiu, Y.; Wang, W.N. Cordyceps sinensis-mediated biotransformation of notoginsenoside R1 into 25-OH-20(S/R)-R2 with elevated cardioprotective effect against DOX-induced cell injury. RSC Adv. 2022, 12, 12938–12946. [Google Scholar] [CrossRef]
- Luo, J.; Mobley, R.; Woodfne, S.; Drijfhout, F.; Horrocks, P.; Ren, X.D.; Li, W.W. Biotransformation of artemisinin to a novel derivative via ring rearrangement by Aspergillus niger. Microb. Biotechnol. 2022, 106, 2433–2444. [Google Scholar] [CrossRef]
- Hudlicky, T.; Reed, J.W. Applications of biotransformations and biocatalysis to complexity generation in organic synthesis. Chem. Soc. Rev. 2009, 38, 2981–2982. [Google Scholar] [CrossRef]
- Gerothanassis, I.P. Ligand-observed in-tube NMR in natural products research: A review on enzymatic biotransformations, protein-ligand interactions, and in-cell NMR spectroscopy. Arab. J. Chem. 2023, 16, 104536. [Google Scholar] [CrossRef]
- Tsagogiannis, E.; Vandera, E.; Primikyri, A.; Asimakoula, S.; Tzakos, A.G.; Gerothanassis, I.P.; Koukkou, A.-I. Characterization of protocatechuate 4,5-dioxygenase from Pseudarthrobacter phenanthrenivorans Sphe3 and in situ reaction monitoring in the NMR tube. Int. J. Mol. Sci. 2021, 22, 9647. [Google Scholar] [CrossRef]
- Siddiqui, M.; Atia, T.W.; Choudhary, M.I.; Atta, U.R. Biotransformation studies on bioactive compounds: 25 years of interesting research at the ICCBS. Chem. Synth. 2023, 3, 25. [Google Scholar] [CrossRef]
- Lu, Y.J.; Tang, Y.F.; Wu, Y.N.; Zhang, X.Y.; Yi, Y.; Wang, W.L.; Wang, A.D.; Yang, M.; Fan, B.Y.; Chen, G.T. Microbial transformation of betulonic acid by Circinella muscae CGMCC 3.2695 and anti-neuroinflammatory activity of the products. Phytochemistry 2022, 204, 113431. [Google Scholar] [CrossRef] [PubMed]
- Song, K.N.; Lu, Y.J.; Chu, C.J.; Wu, Y.N.; Huang, H.L.; Fan, B.Y.; Chen, G.T. Biotransformation of betulonic acid by the fungus Rhizopus arrhizus CGMCC 3.868 and antineuroinflammatory activity of the biotransformation products. J. Nat. Prod. 2021, 84, 2664–2674. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Chen, Y.H.; Cheng, Z.; Li, H.J.; Bian, H.H.; Yang, X.C.; Lv, J.; Liu, W.; Su, L.; Sun, P. Anti-inflammatory oleanane-type triterpenoids produced by Nonomuraea sp. MYH522 through microbial transformation. J. Agric. Food Chem. 2023, 71, 3777–3789. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.H.; Zhang, C.; Wang, W.W.; Yu, B.Y.; Zhang, J. Site-selective biotransformation of ursane triterpenes by Streptomyces griseus ATCC 13273. RSC Adv. 2017, 7, 20754–20759. [Google Scholar] [CrossRef]
- Yadav, V.R.; Prasad, S.; Sung, B.; Kannappan, R.; Aggarwal, B.B. Targeting inflammatory pathways by triterpenoids for prevention and treatment of cancer. Toxins 2010, 2, 2428–2466. [Google Scholar] [CrossRef]
- Coleman, J.W. Nitric oxide in immunity and inflammation. Int. Immunopharmacol. 2001, 1, 1397–1406. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Xu, S.H.; Ma, B.L.; Wang, W.W.; Yu, B.Y.; Zhang, J. New derivatives of ursolic acid through the biotransformation by Bacillus megaterium CGMCC 1.1741 as inhibitors on nitric oxide production. Bioorg. Med. Chem. Lett. 2017, 27, 2575–2578. [Google Scholar] [CrossRef]
- Tu, W.C.; Luo, R.H.; Yuan, E.; Sakah, K.J.; Yang, Q.Y.; Xiao, W.L.; Zheng, Y.T.; Liu, M.F. Triterpene constituents from the fruits of Cyclocarya paliurus and their anti-HIV-1IIIB activity. Nat. Prod. Res. 2023, 37, 1787–1796. [Google Scholar] [CrossRef]
- Chu, C.J.; Song, K.N.; Zhang, Y.Z.; Yang, M.; Fan, B.Y.; Huang, H.J.; Chen, G.T. Biotransformation of ursolic acid by Circinella muscae and their anti-neuroinflammatory activities of metabolites. Nat. Prod. Res. 2022, 36, 2777–2782. [Google Scholar] [CrossRef]
- Leal, A.S.; Wang, R.; Salvador, J.A.R.; Jing, Y. Synthesis of novel ursolic acid heterocyclic derivatives with improved abilities of antiproliferation and induction of p53, p21waf1 and NOXA in pancreatic cancer cells. Bioorg. Med. Chem. 2012, 20, 5774–5786. [Google Scholar] [CrossRef]
- Cheng, D.L.; Cao, X.P. Pomolic acid derivatives from the root of Sanguisorba officinalis. Phytochemistry 1992, 31, 1317–1320. [Google Scholar] [CrossRef]
- Ikuta, A.; Morikawa, A. Triterpenes from Stauntonia hexaphylla callus tissues. J. Nat. Prod. 1992, 55, 1230–1233. [Google Scholar] [CrossRef]
- Shah, S.A.A.; Tan, H.L.; Sultan, S.; Faridz, M.A.B.M.; Shah, M.A.B.M.; Nurfazilah, S.; Hussain, M. Microbial-catalyzed biotransformation of multifunctional triterpenoids derived from phytonutrients. Int. J. Mol. Sci. 2014, 15, 12027–12060. [Google Scholar] [CrossRef] [PubMed]
- Edyta, K.S.; Tomasz, J. Microbial transformations of 7-methoxyflavanone. Molecules 2012, 17, 14810–14820. [Google Scholar]
- Letizia, C.M.; Benedetta, G.; Immacolata, S.; Valerio, D.; Diego, R.; Andrea, P.; Roberto, L.; Pinheiro, S.O.R.; Francesco, M. Development of a high-yielding bioprocess for 11α-hydroxylation of canrenone under conditions of oxygen-enriched air supply. Steroids 2016, 116, 1–4. [Google Scholar]
- Rong, S.; Tang, X.; Guan, S.; Zhang, B.; Li, Q.; Cai, B.; Huang, J. Effects of impeller geometry on the 11α-hydroxylation of canrenone in Rushton turbine-stirred tanks. J. Microbiol. Biotechnol. 2021, 31, 890–901. [Google Scholar] [CrossRef] [PubMed]
- Tomasz, T.; Agnieszka, B.; Jaroslaw, P.; Ewa, H. Transformation of xanthohumol by Aspergillus ochraceus. J. Basic Microb. 2014, 54, 66–71. [Google Scholar]
- Converti, A.; Gandolfi, R.; Zilli, M.; Molinari, F.; Binaghi, L.; Perego, P.; Borghi, M. Synthesis of ethyl phenylacetate by lyophilized mycelium of Aspergillus oryzae. Appl. Microbiol. Biotechnol. 2005, 67, 637–640. [Google Scholar] [CrossRef]
- Dong, Y.S.; Teng, H.; Qi, S.S.; Liu, L.; Wang, H.; Zhao, Y.K.; Xiu, Z.L. Pathways and kinetics analysis of biotransformation of Dioscorea zingiberensis by Aspergillus oryzae. Biochem. Eng. J. 2010, 52, 123–130. [Google Scholar] [CrossRef]
- Sikander, A.; Wajeeha, N. Biotransformation of L-tyrosine to dopamine by a calcium alginate immobilized mutant strain of Aspergillus oryzae. Appl. Biochem. Biotechnol. 2016, 179, 1435–1444. [Google Scholar]
- Wang, H.; Liu, L.; Guo, Y.X.; Dong, Y.S.; Zhang, D.J.; Xiu, Z.L. Biotransformation of piceid in Polygonum cuspidatum to resveratrol by Aspergillus oryzae. Appl. Microbiol. Biotechnol. 2007, 75, 763–768. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Ni, Y.H.; Jiang, B.C.; Song, Y.; Xu, B.H.; Fan, B.Y.; Huang, H.L.; Chen, G.T. Anti-aging derivatives of cycloastragenol produced by biotransformation. Nat. Prod. Res. 2021, 35, 2685–2690. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Y.-N.; Su, D.; Yang, J.; Yi, Y.; Wang, A.-D.; Yang, M.; Li, J.-L.; Fan, B.-Y.; Chen, G.-T.; Wang, W.-L.; et al. Biotransformation of Ursonic Acid by Aspergillus ochraceus and Aspergillus oryzae to Discover Anti-Neuroinflammatory Derivatives. Molecules 2023, 28, 7943. https://doi.org/10.3390/molecules28247943
Wu Y-N, Su D, Yang J, Yi Y, Wang A-D, Yang M, Li J-L, Fan B-Y, Chen G-T, Wang W-L, et al. Biotransformation of Ursonic Acid by Aspergillus ochraceus and Aspergillus oryzae to Discover Anti-Neuroinflammatory Derivatives. Molecules. 2023; 28(24):7943. https://doi.org/10.3390/molecules28247943
Chicago/Turabian StyleWu, Yan-Ni, Dan Su, Jia Yang, Ying Yi, An-Dong Wang, Min Yang, Jian-Lin Li, Bo-Yi Fan, Guang-Tong Chen, Wen-Li Wang, and et al. 2023. "Biotransformation of Ursonic Acid by Aspergillus ochraceus and Aspergillus oryzae to Discover Anti-Neuroinflammatory Derivatives" Molecules 28, no. 24: 7943. https://doi.org/10.3390/molecules28247943
APA StyleWu, Y.-N., Su, D., Yang, J., Yi, Y., Wang, A.-D., Yang, M., Li, J.-L., Fan, B.-Y., Chen, G.-T., Wang, W.-L., & Ling, B. (2023). Biotransformation of Ursonic Acid by Aspergillus ochraceus and Aspergillus oryzae to Discover Anti-Neuroinflammatory Derivatives. Molecules, 28(24), 7943. https://doi.org/10.3390/molecules28247943