

Introduction of Carbonyl Groups into Antibodies


Abstract
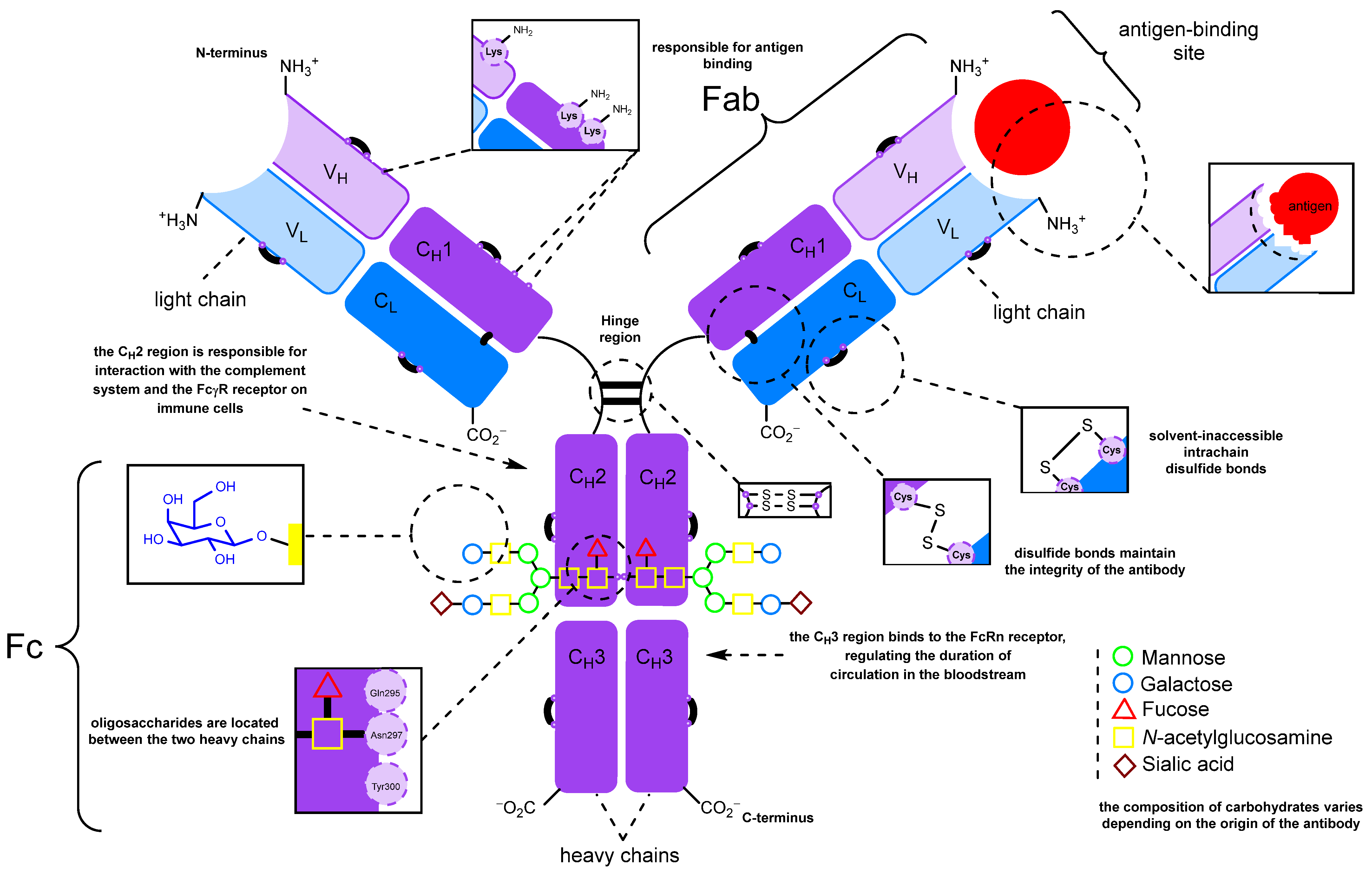
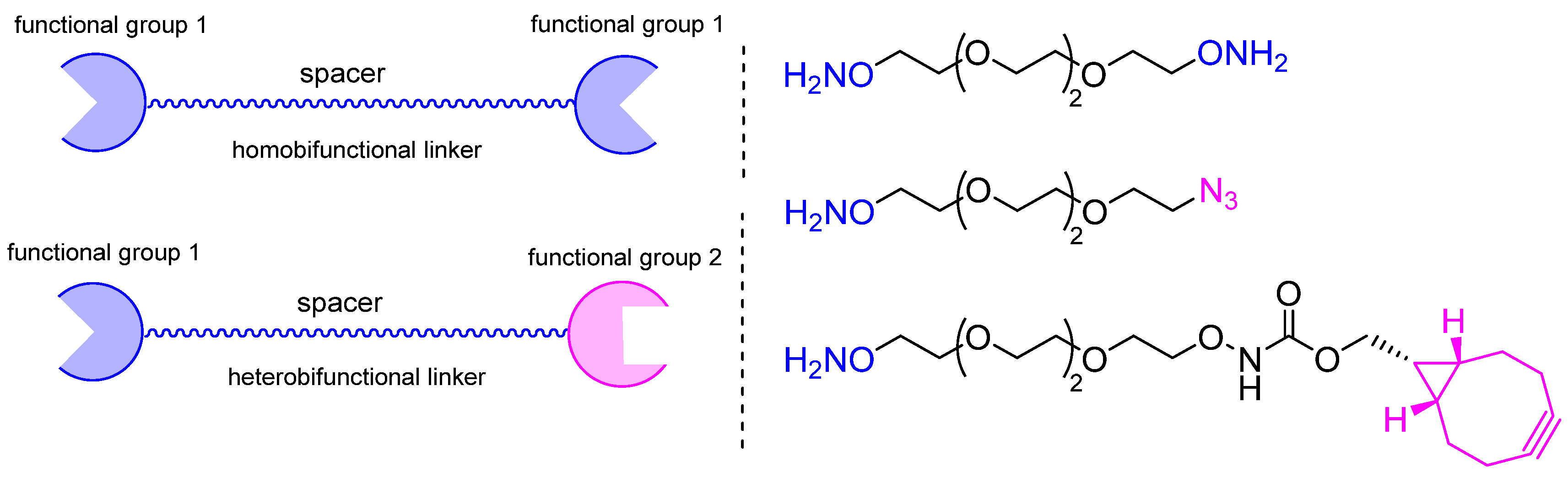
:1. Introduction
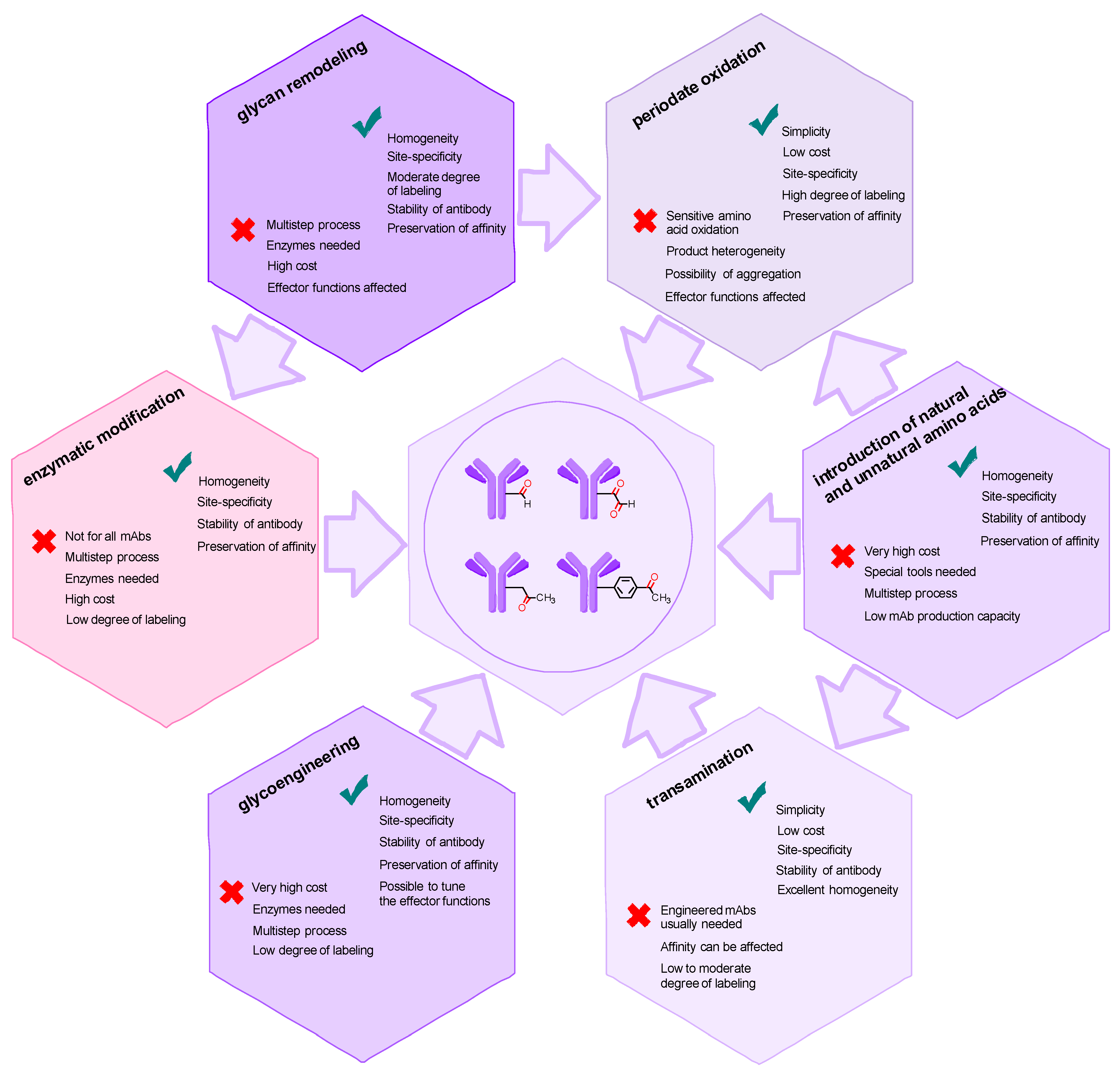
2. Methods for Introducing Carbonyl Groups into Antibodies
2.1. Introduction of the Carbonyl Group into Native Antibodies
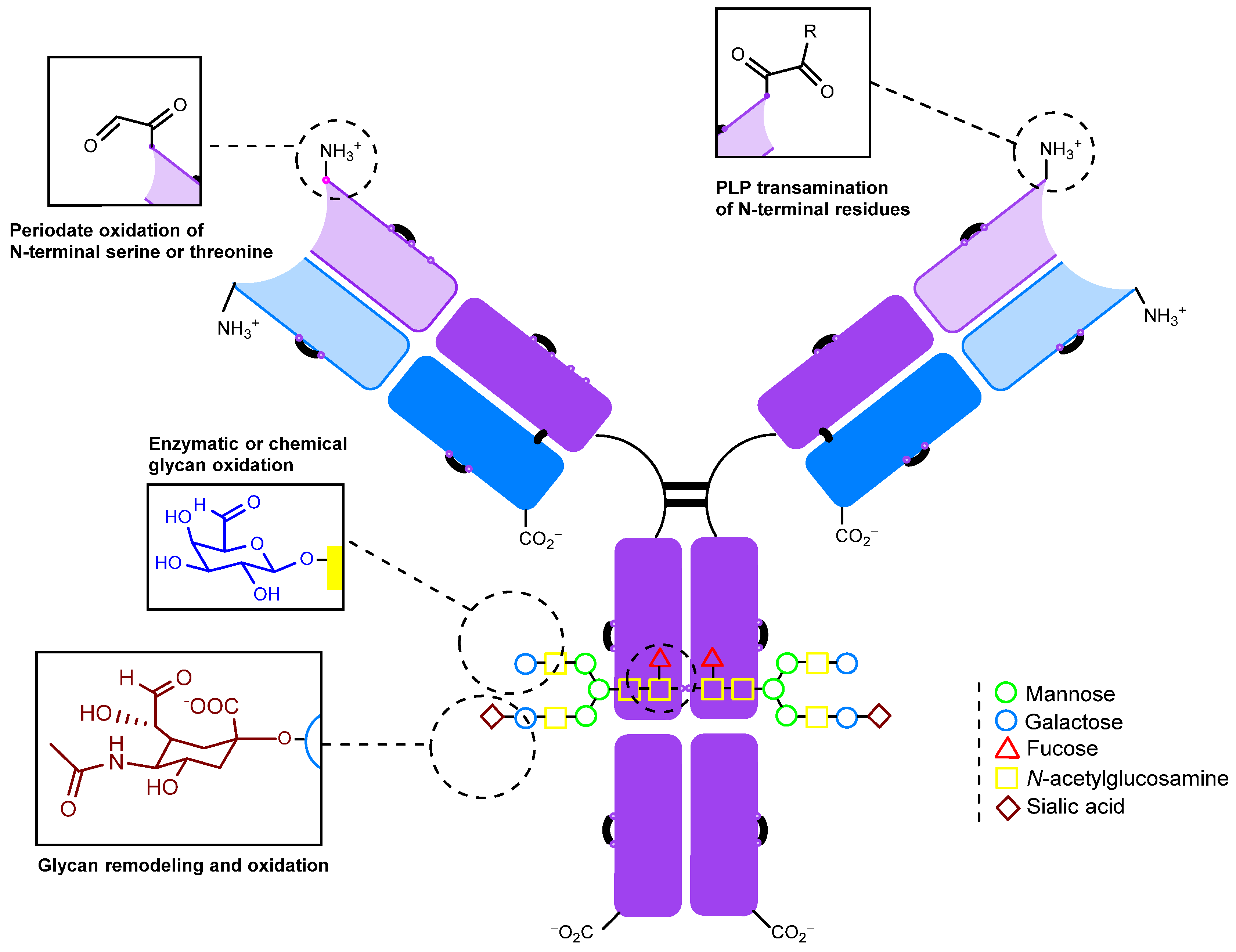
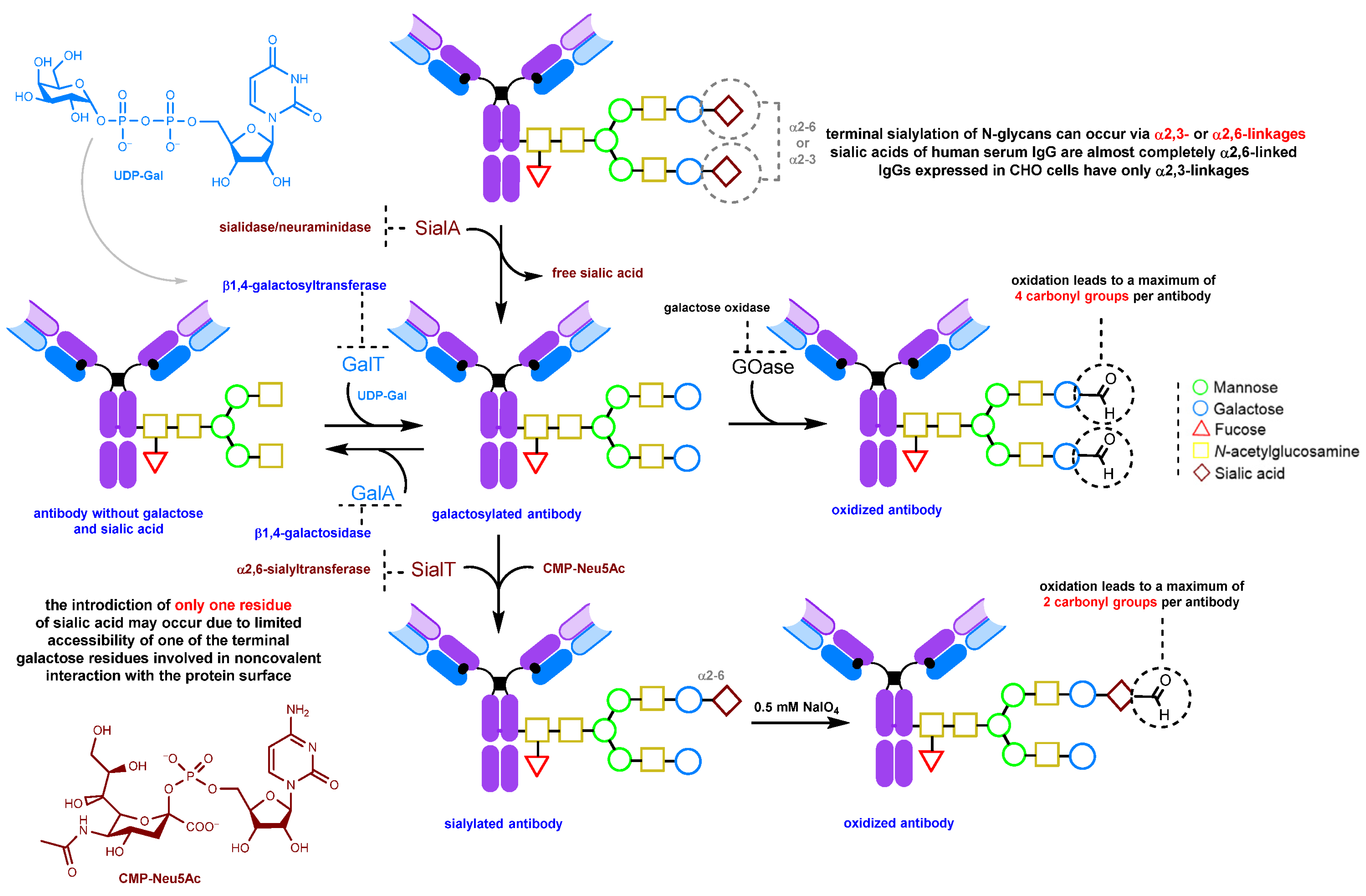
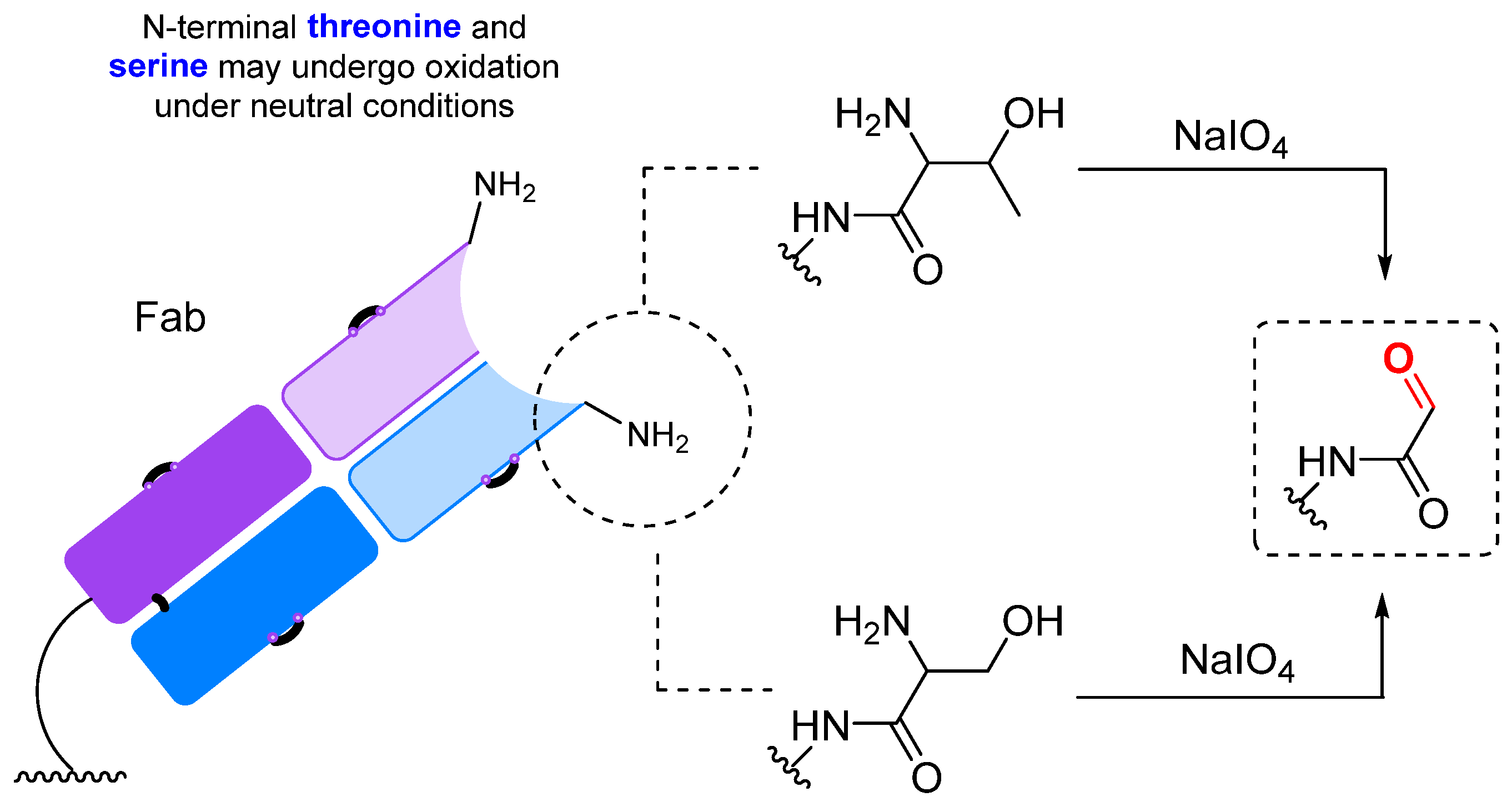
2.1.1. Periodate Oxidation of Glycans
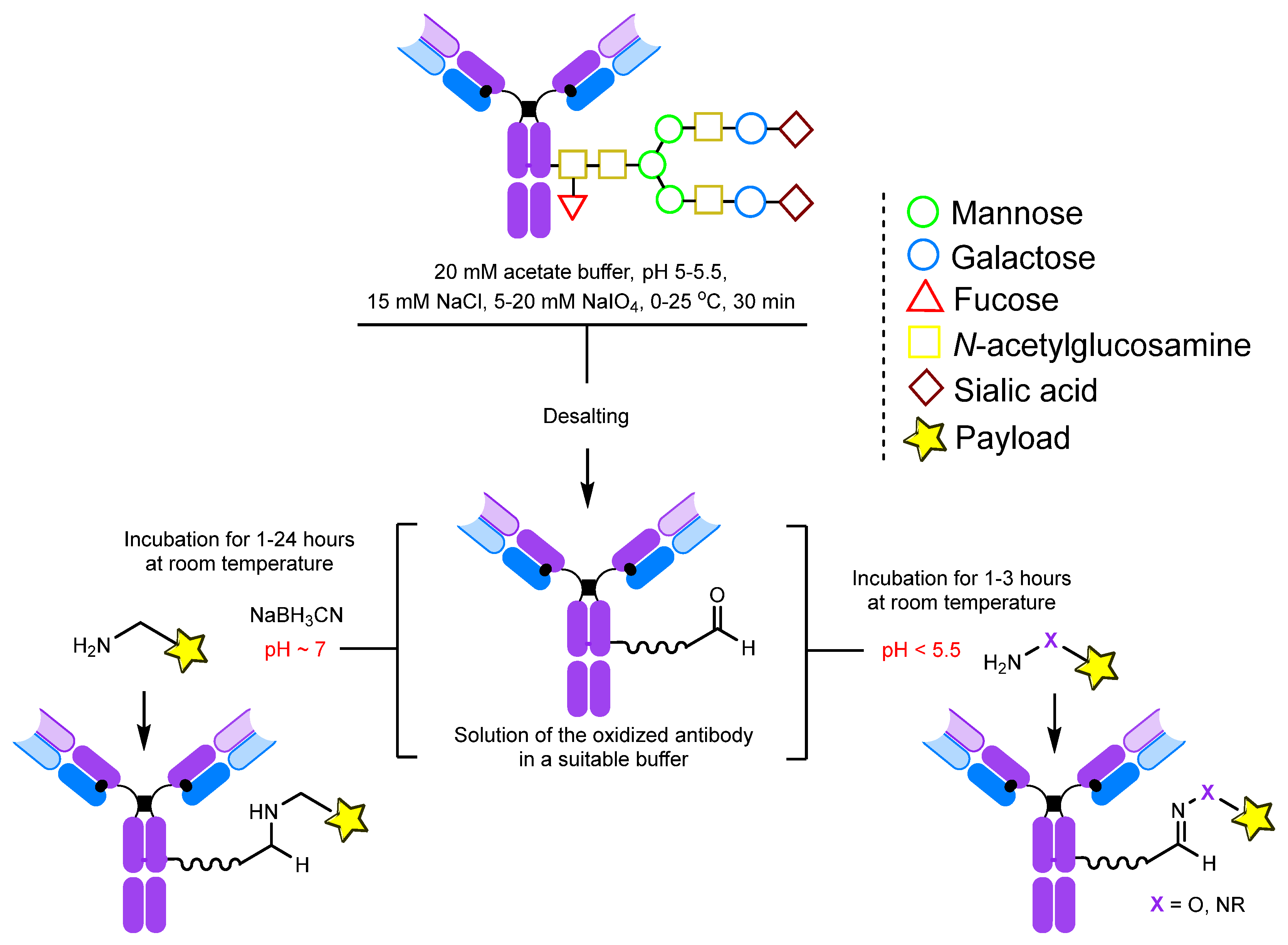
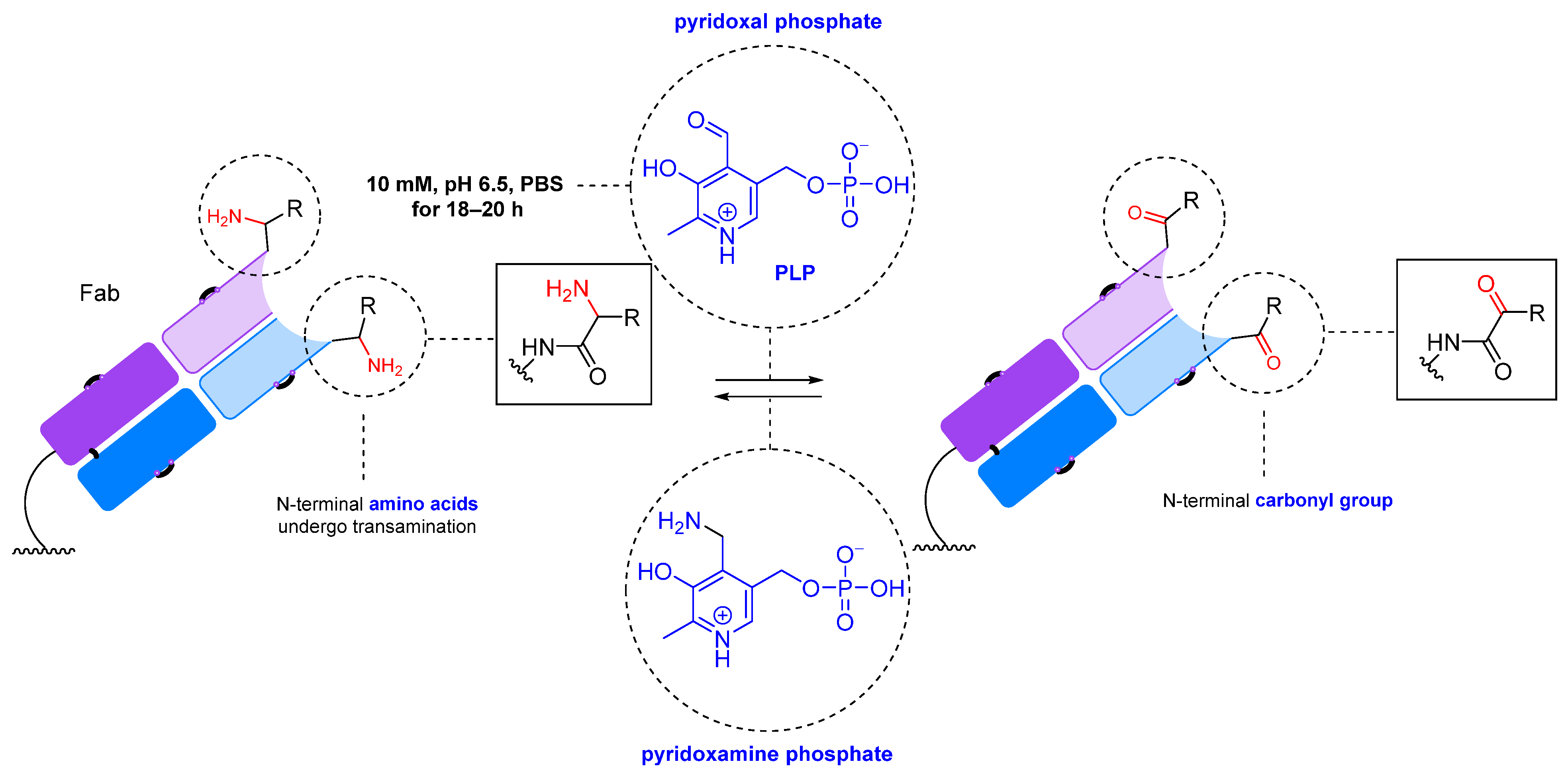
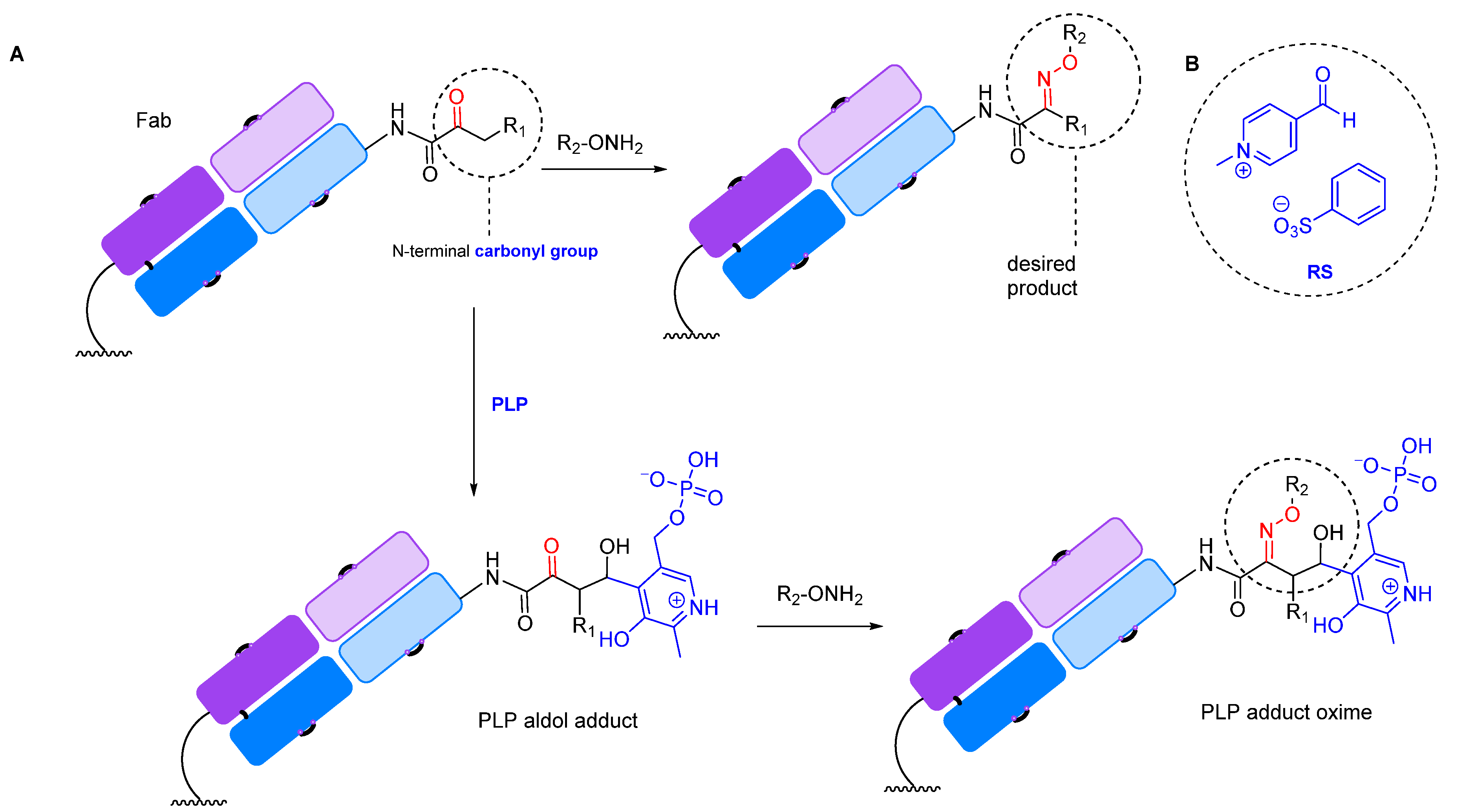
2.1.2. Transamination
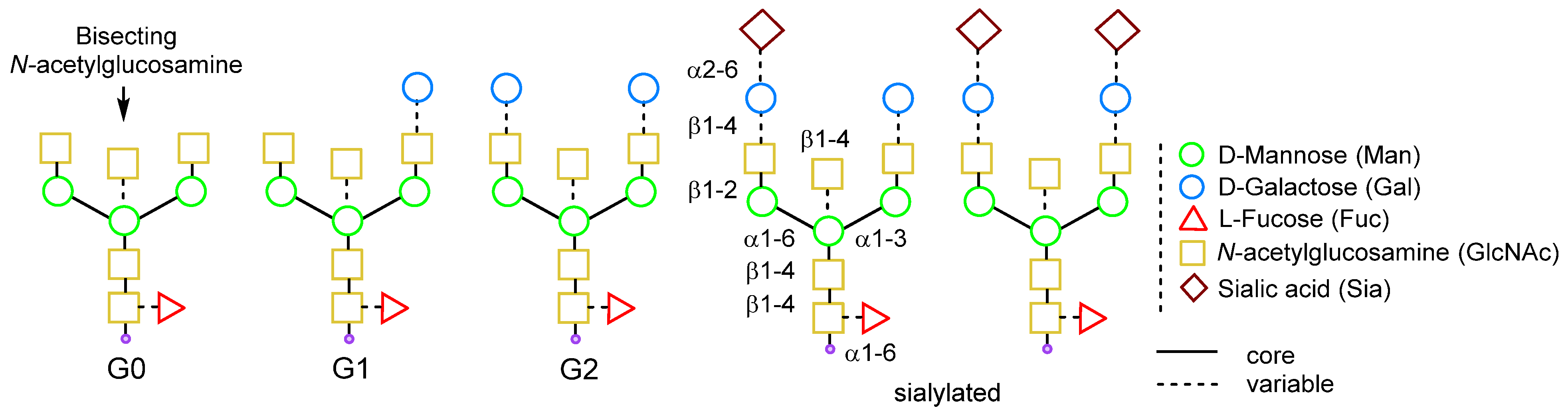
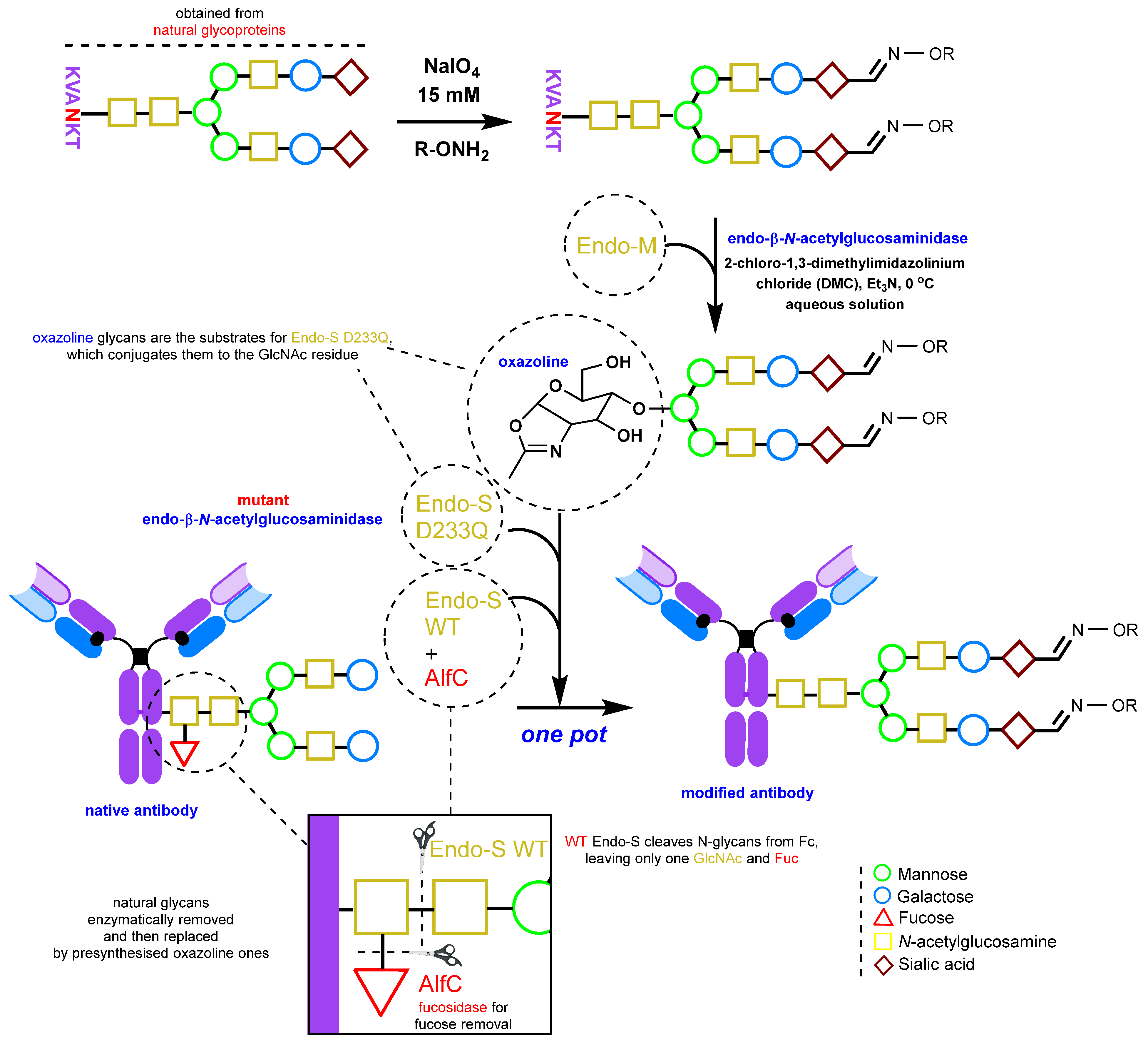
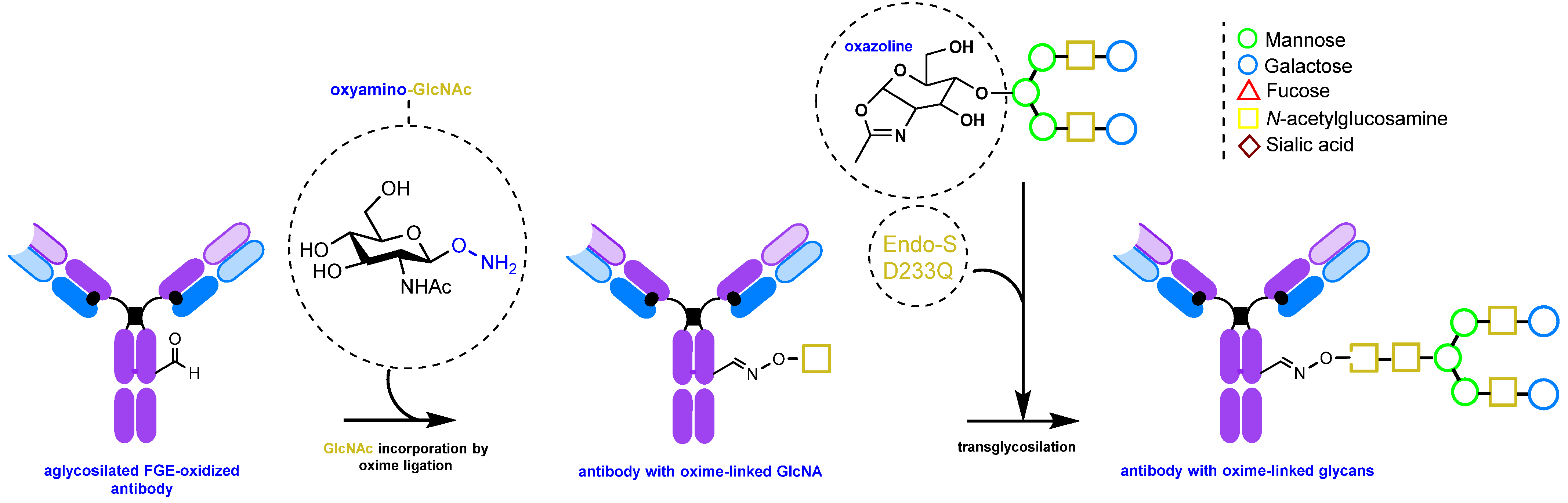
2.1.3. Glycan Remodeling
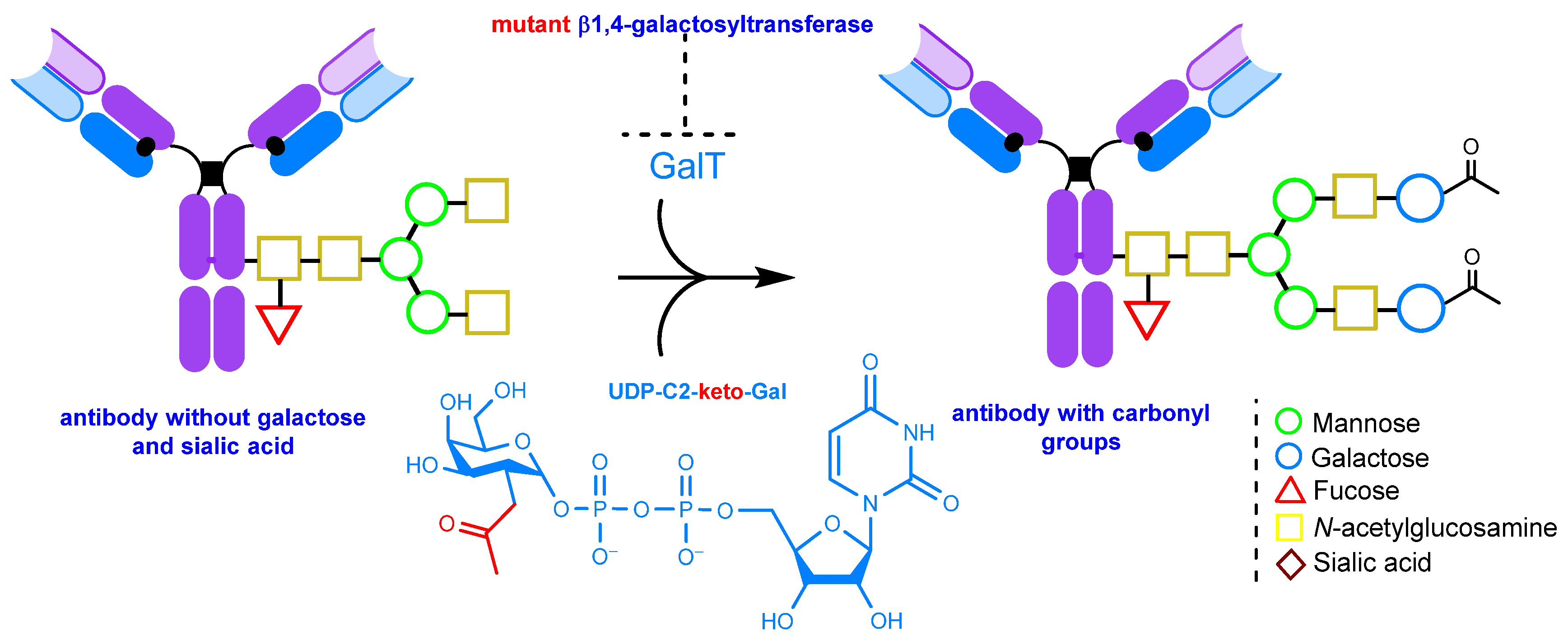
2.1.4. Glycoengineering Using Mutant Glycosyltransferases
2.2. Introduction of Carbonyl Groups with Genetic Engineering Tools
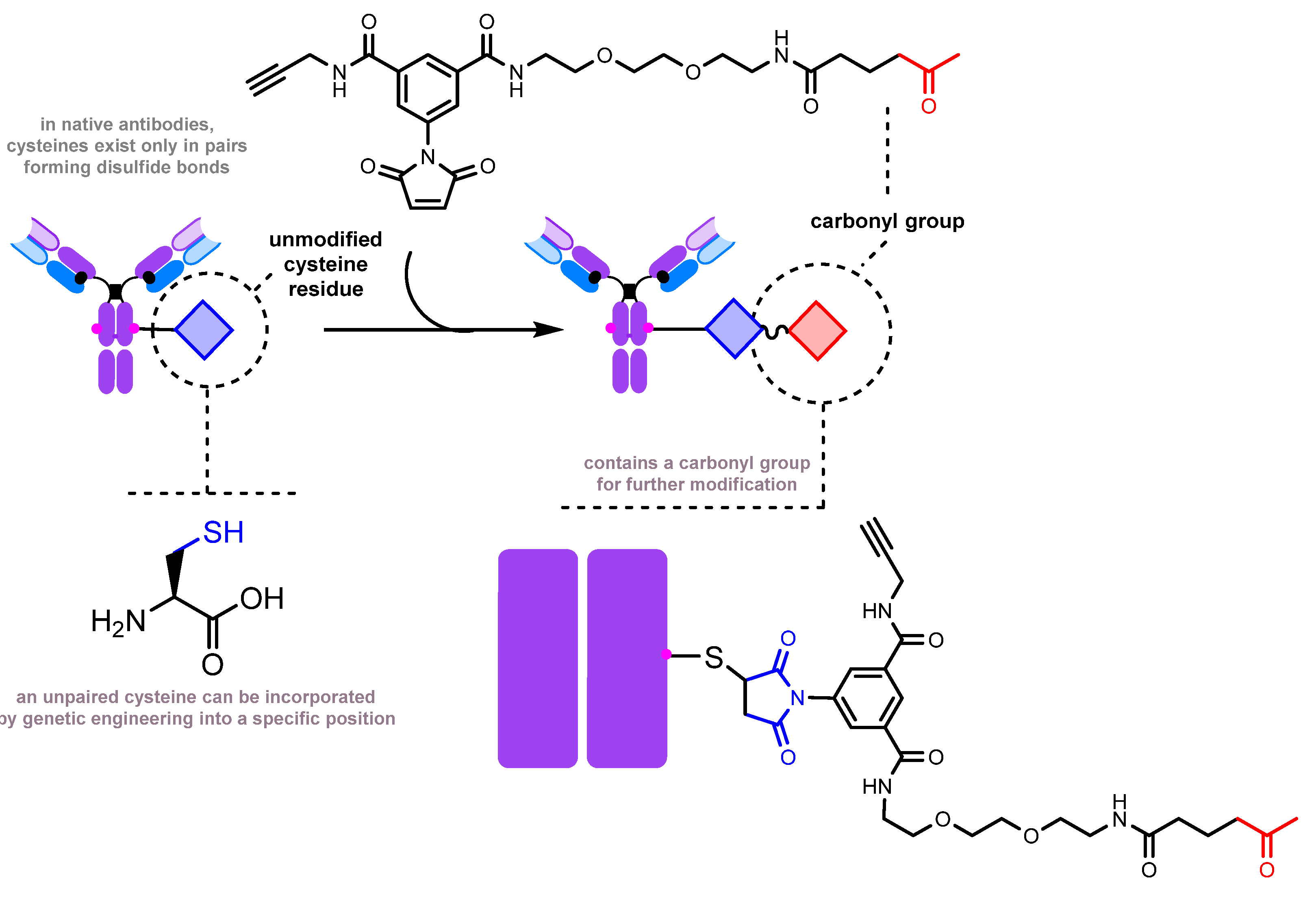
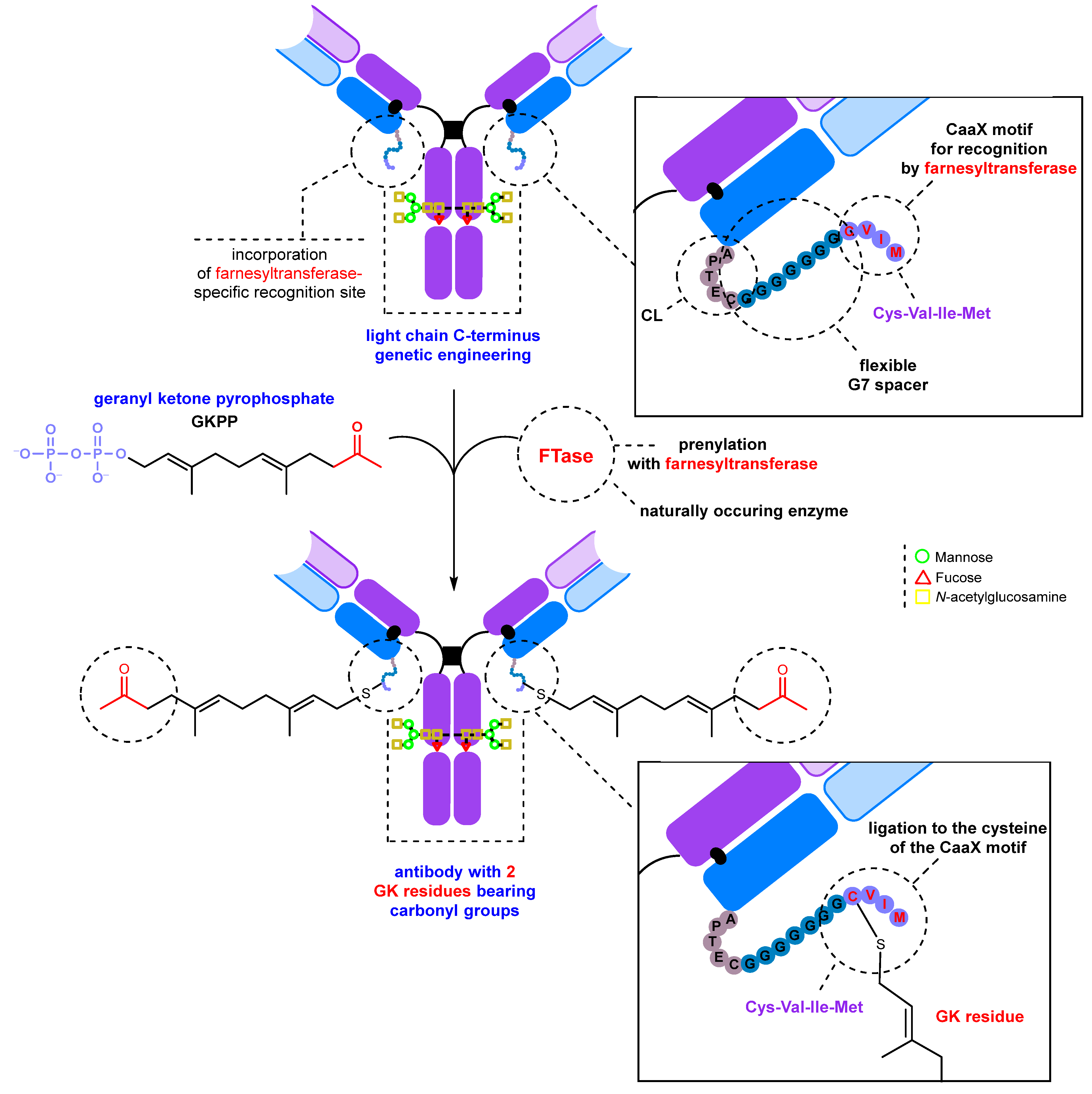
2.2.1. Introduction of a Natural Amino Acid as a Precursor of the Carbonyl Group
- Inserting an amino acid residue of interest outside the complementarity-determining region (CDR) near the N-terminus [204].
- Placing an amino acid residue of interest into the Fc region or Fab region, far from the antigen-binding site [203]. Subsequent to this step, enzymatic or chemical conjugation can be performed.
2.2.2. Introduction of a Protein Tag for Subsequent Carbonyl Group Insertion
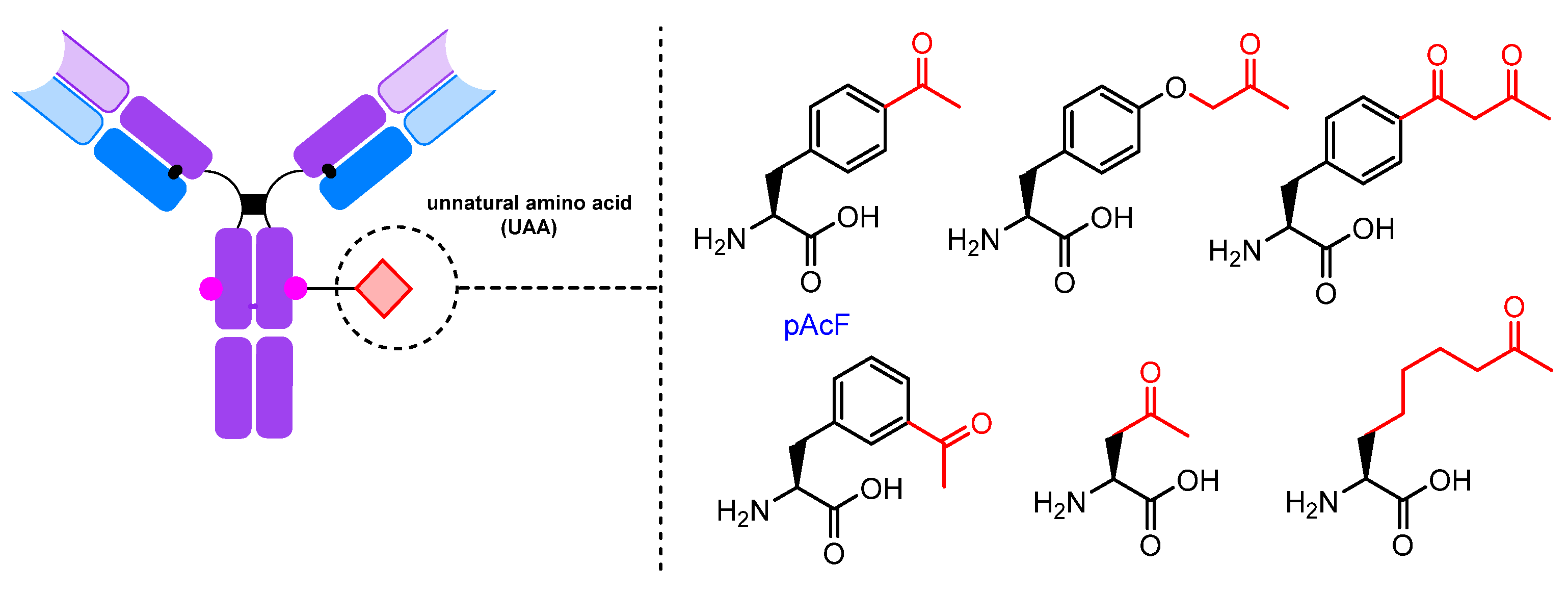
2.2.3. UAAs Containing Carbonyl Groups
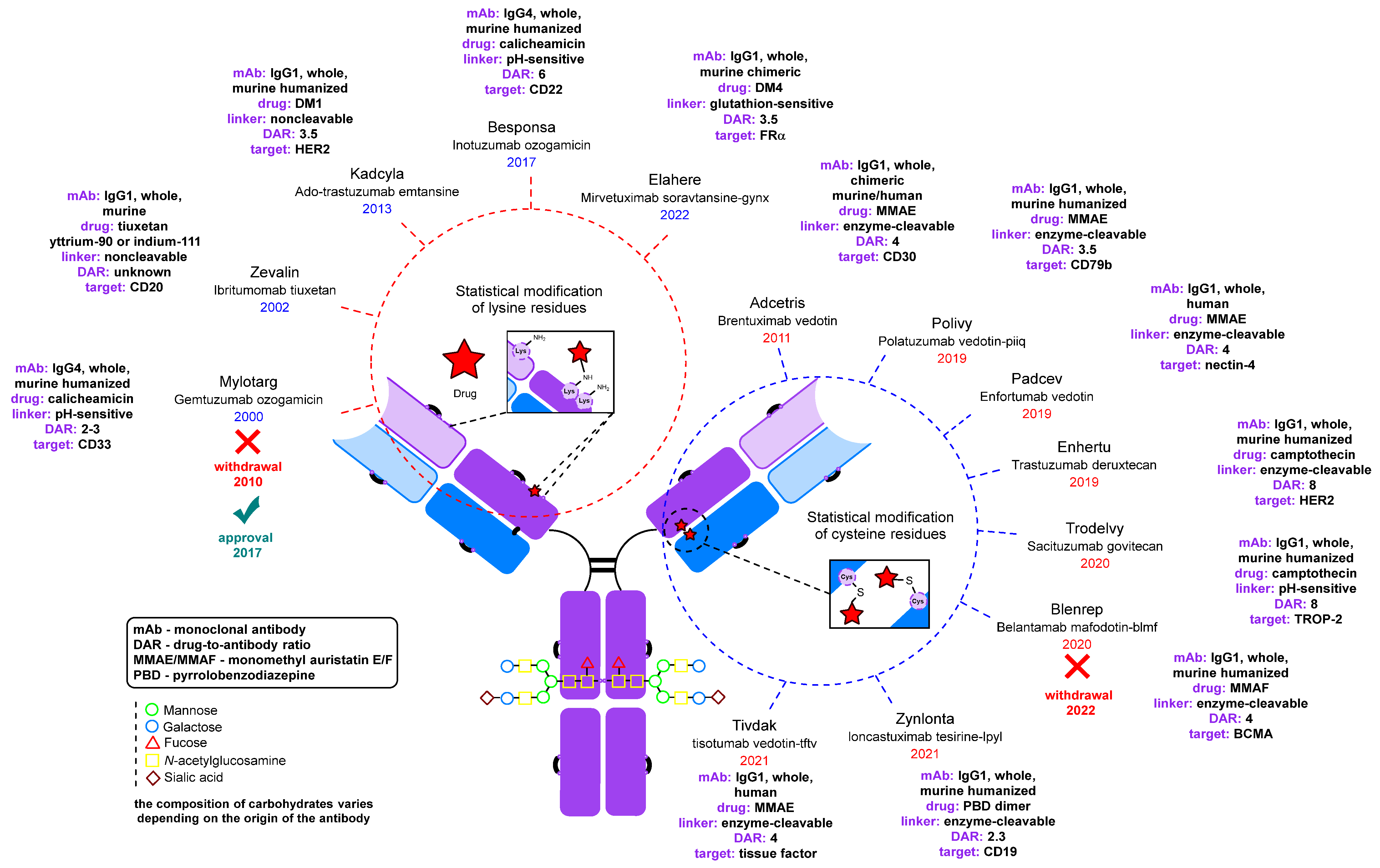
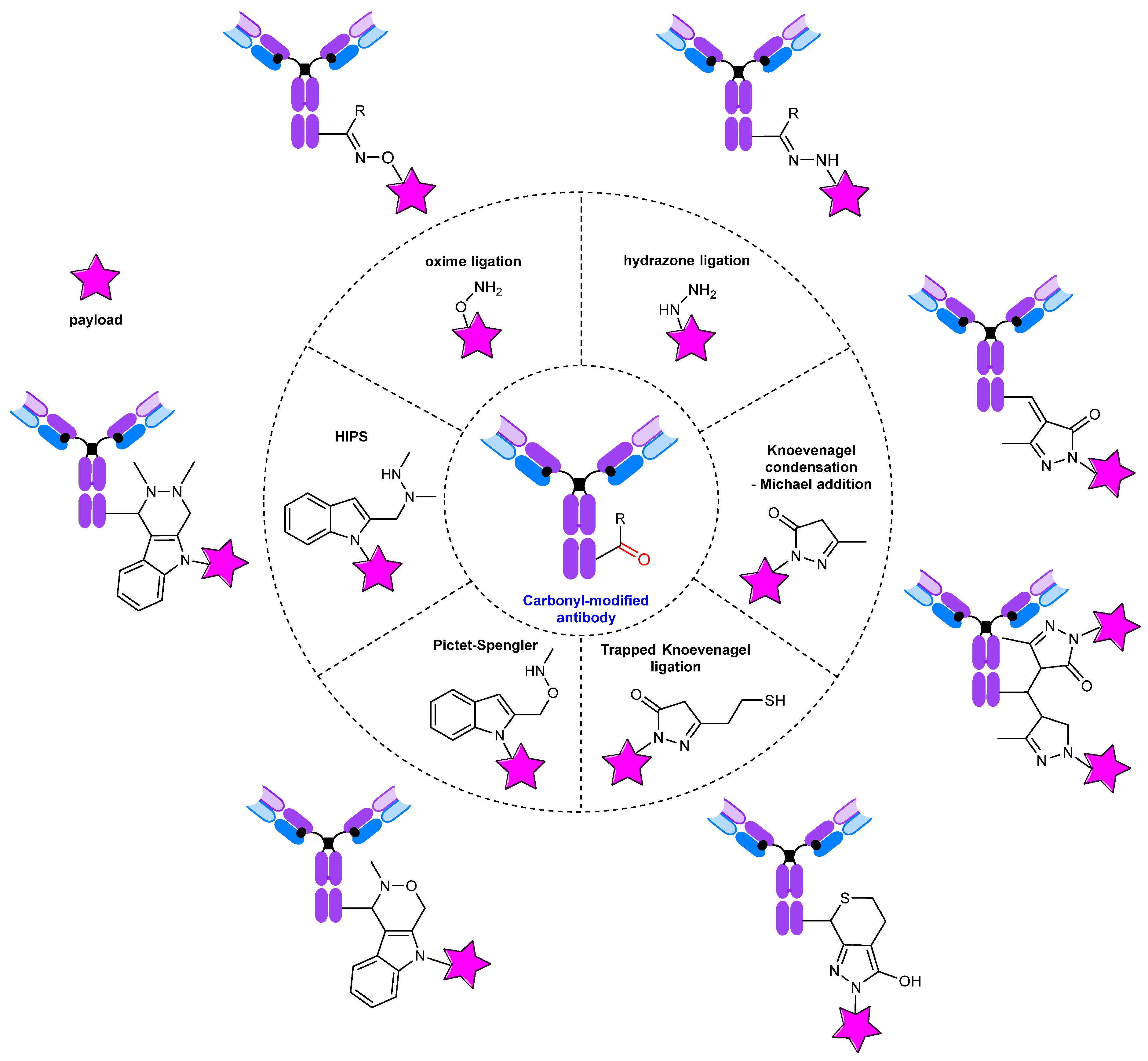
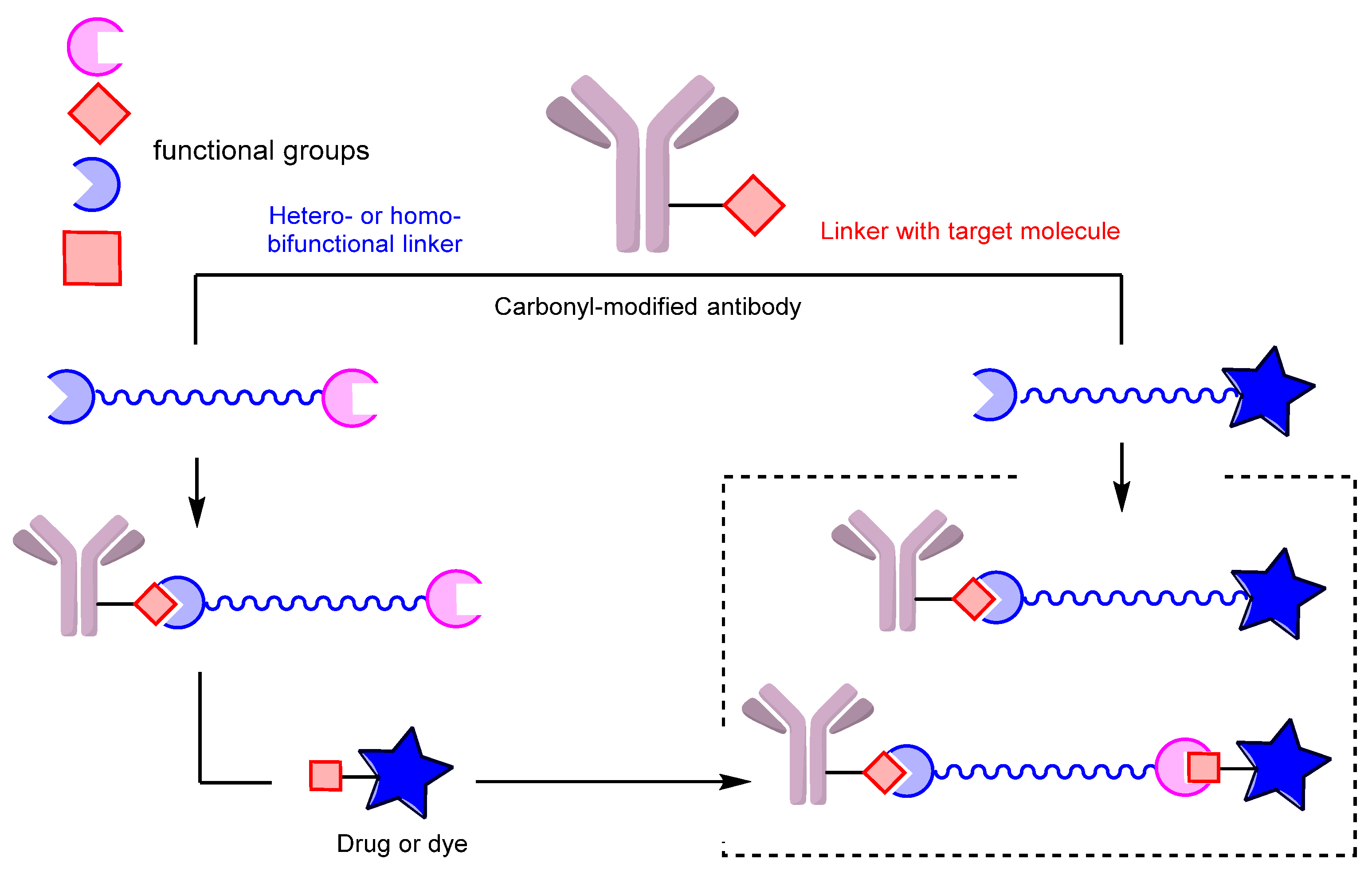
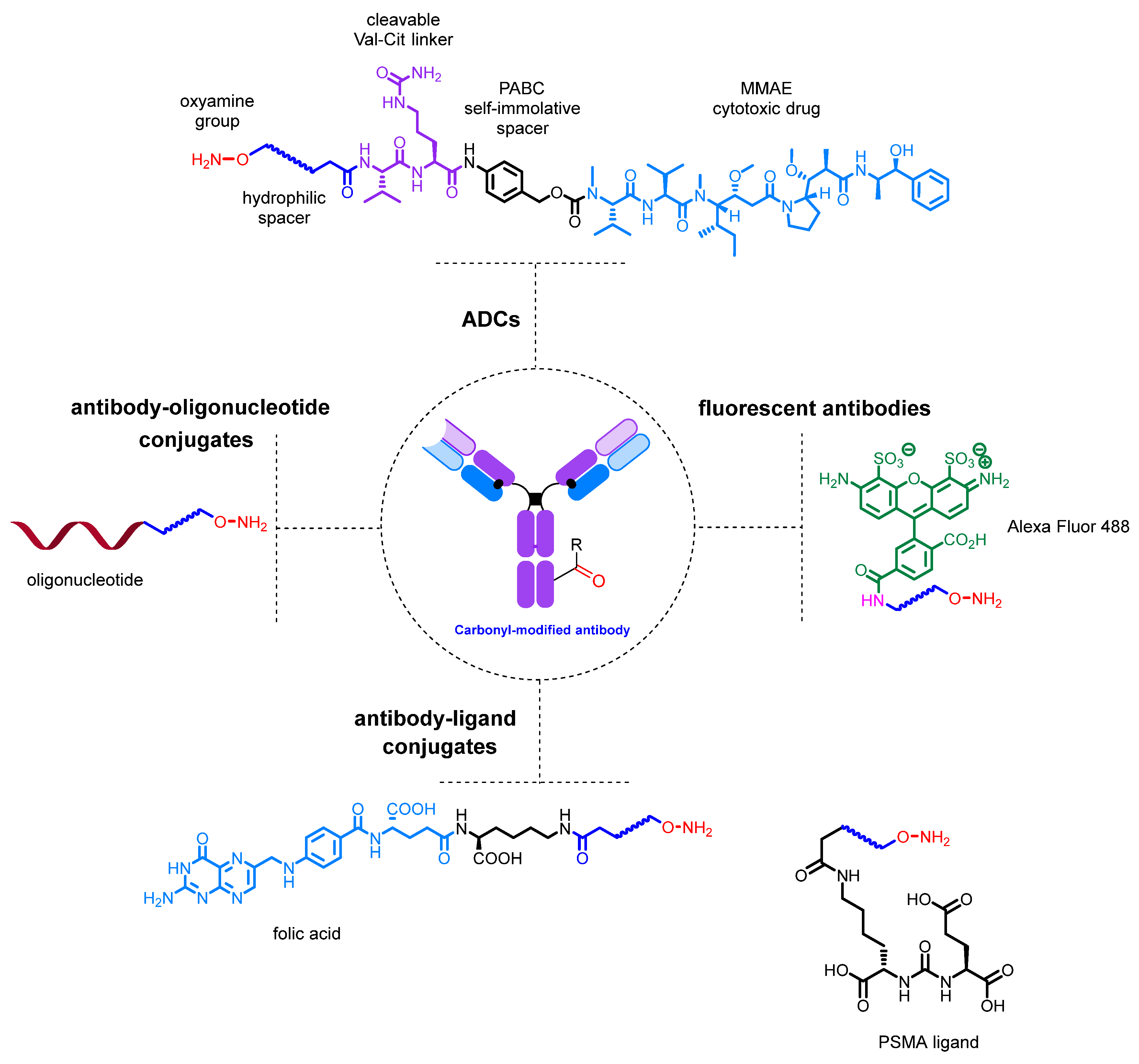
3. Conjugates: Modification of the Carbonyl Group
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Meares, C.F. Bioconjugate Chemistry. In Reference Module in Chemistry, Molecular Sciences and Chemical Engineering; Elsevier: Amsterdam, The Netherlands, 2014; p. B978012409547211087X. ISBN 978-0-12-409547-2. [Google Scholar] [CrossRef]
- Kalia, J.; Raines, R. Advances in Bioconjugation. Curr. Org. Chem. 2010, 14, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Narain, R. (Ed.) Chemistry of Bioconjugates: Synthesis, Characterization, and Biomedical Applications, 1st ed.; Wiley: Hoboken, NJ, USA, 2014; ISBN 978-1-118-35914-3. [Google Scholar] [CrossRef]
- Hermanson, G.T. Bioconjugate Techniques; Elsevier: Amsterdam, The Netherlands, 2013; ISBN 978-0-12-382239-0. [Google Scholar] [CrossRef]
- Algar, W.R.; Dawson, P.; Medintz, I.L. (Eds.) Chemoselective and Bioorthogonal Ligation Reactions; Wiley-VCH: Weinheim, Germany, 2017; Volume 2, ISBN 978-3-527-33436-0. [Google Scholar] [CrossRef]
- Prescher, J.A.; Bertozzi, C.R. Chemistry in Living Systems. Nat. Chem. Biol. 2005, 1, 13–21. [Google Scholar] [CrossRef]
- Best, M.D. Click Chemistry and Bioorthogonal Reactions: Unprecedented Selectivity in the Labeling of Biological Molecules. Biochemistry 2009, 48, 6571–6584. [Google Scholar] [CrossRef] [PubMed]
- Sletten, E.M.; Bertozzi, C.R. Bioorthogonal Chemistry: Fishing for Selectivity in a Sea of Functionality. Angew. Chem. Int. Ed. 2009, 48, 6974–6998. [Google Scholar] [CrossRef]
- Saxon, E.; Bertozzi, C.R. Cell Surface Engineering by a Modified Staudinger Reaction. Science 2000, 287, 2007–2010. [Google Scholar] [CrossRef] [PubMed]
- Prescher, J.A.; Dube, D.H.; Bertozzi, C.R. Chemical Remodelling of Cell Surfaces in Living Animals. Nature 2004, 430, 873–877. [Google Scholar] [CrossRef] [PubMed]
- Vocadlo, D.J.; Hang, H.C.; Kim, E.-J.; Hanover, J.A.; Bertozzi, C.R. A Chemical Approach for Identifying O -GlcNAc-Modified Proteins in Cells. Proc. Natl. Acad. Sci. USA 2003, 100, 9116–9121. [Google Scholar] [CrossRef]
- Hang, H.C.; Yu, C.; Pratt, M.R.; Bertozzi, C.R. Probing Glycosyltransferase Activities with the Staudinger Ligation. J. Am. Chem. Soc. 2004, 126, 6–7. [Google Scholar] [CrossRef]
- Zinn, S.; Vazquez-Lombardi, R.; Zimmermann, C.; Sapra, P.; Jermutus, L.; Christ, D. Advances in Antibody-Based Therapy in Oncology. Nat. Cancer. 2023, 4, 165–180. [Google Scholar] [CrossRef]
- Arlotta, K.J.; Owen, S.C. Antibody and Antibody Derivatives as Cancer Therapeutics. Wiley Interdiscipl. Rev. Nanomed. Nanobiotechnol. 2019, 11, e1556. [Google Scholar] [CrossRef]
- Jin, B.; Odongo, S.; Radwanska, M.; Magez, S. Nanobodies: A Review of Generation, Diagnostics and Therapeutics. Int. J. Mol. Sci. 2023, 24, 5994. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.; Wang, Z.; Hao, M.; Li, J. Bispecific Antibodies and Their Applications. J. Hematol. Oncol. 2015, 8, 130. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, S.S.; Khalili, S.; Baradaran, B.; Bidar, N.; Shahbazi, M.-A.; Mosafer, J.; Hashemzaei, M.; Mokhtarzadeh, A.; Hamblin, M.R. Bispecific Monoclonal Antibodies for Targeted Immunotherapy of Solid Tumors: Recent Advances and Clinical Trials. Int. J. Biol. Macromol. 2021, 167, 1030–1047. [Google Scholar] [CrossRef] [PubMed]
- Tetin, S.; Stroupe, S. Antibodies in Diagnostic Applications. Curr. Pharm. Biotechnol. 2004, 5, 9–16. [Google Scholar] [CrossRef]
- Zhang, X.; Soori, G.; Dobleman, T.J.; Xiao, G.G. The Application of Monoclonal Antibodies in Cancer Diagnosis. Expert Rev. Mol. Diagn. 2014, 14, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Dammes, N.; Peer, D. Monoclonal Antibody-Based Molecular Imaging Strategies and Theranostic Opportunities. Theranostics 2020, 10, 938–955. [Google Scholar] [CrossRef]
- Scott, E.C.; Baines, A.C.; Gong, Y.; Moore, R.; Pamuk, G.E.; Saber, H.; Subedee, A.; Thompson, M.D.; Xiao, W.; Pazdur, R.; et al. Trends in the Approval of Cancer Therapies by the FDA in the Twenty-First Century. Nat. Rev. Drug Discov. 2023, 22, 625–640. [Google Scholar] [CrossRef]
- Waldmann, T.A. Monoclonal Antibodies in Diagnosis and Therapy. Science 1991, 252, 1657–1662. [Google Scholar] [CrossRef]
- Fu, Z.; Li, S.; Han, S.; Shi, C.; Zhang, Y. Antibody Drug Conjugate: The “Biological Missile” for Targeted Cancer Therapy. Sign. Transduct. Target Ther. 2022, 7, 93. [Google Scholar] [CrossRef]
- Dumontet, C.; Reichert, J.M.; Senter, P.D.; Lambert, J.M.; Beck, A. Antibody–Drug Conjugates Come of Age in Oncology. Nat. Rev. Drug Discov. 2023, 22, 641–661. [Google Scholar] [CrossRef]
- Gogia, P.; Ashraf, H.; Bhasin, S.; Xu, Y. Antibody–Drug Conjugates: A Review of Approved Drugs and Their Clinical Level of Evidence. Cancers 2023, 15, 3886. [Google Scholar] [CrossRef]
- Ahmadi, S.E.; Shabannezhad, A.; Kahrizi, A.; Akbar, A.; Safdari, S.M.; Hoseinnezhad, T.; Zahedi, M.; Sadeghi, S.; Mojarrad, M.G.; Safa, M. Tissue Factor (Coagulation Factor III): A Potential Double-Edge Molecule to Be Targeted and Re-Targeted toward Cancer. Biomark. Res. 2023, 11, 60. [Google Scholar] [CrossRef]
- Breij, E.C.W.; De Goeij, B.E.C.G.; Verploegen, S.; Schuurhuis, D.H.; Amirkhosravi, A.; Francis, J.; Miller, V.B.; Houtkamp, M.; Bleeker, W.K.; Satijn, D.; et al. An Antibody–Drug Conjugate That Targets Tissue Factor Exhibits Potent Therapeutic Activity against a Broad Range of Solid Tumors. Cancer Res. 2014, 74, 1214–1226. [Google Scholar] [CrossRef]
- Arn, C.R.; Halla, K.J.; Gill, S. Tisotumab Vedotin Safety and Tolerability in Clinical Practice: Managing Adverse Events. J. Adv. Pract. Oncol. 2023, 14, 139–152. [Google Scholar] [CrossRef]
- Markham, A. Tisotumab Vedotin: First Approval. Drugs 2021, 81, 2141–2147. [Google Scholar] [CrossRef]
- Francisco, J.A.; Cerveny, C.G.; Meyer, D.L.; Mixan, B.J.; Klussman, K.; Chace, D.F.; Rejniak, S.X.; Gordon, K.A.; DeBlanc, R.; Toki, B.E.; et al. cAC10-vcMMAE, an Anti-CD30–Monomethyl Auristatin E Conjugate with Potent and Selective Antitumor Activity. Blood 2003, 102, 1458–1465. [Google Scholar] [CrossRef]
- Gualberto, A. Brentuximab Vedotin (SGN-35), an Antibody–Drug Conjugate for the Treatment of CD30-Positive Malignancies. Expert Opin. Investig. Drugs 2012, 21, 205–216. [Google Scholar] [CrossRef]
- Katz, J.; Janik, J.E.; Younes, A. Brentuximab Vedotin (SGN-35). Clin. Cancer Res. 2011, 17, 6428–6436. [Google Scholar] [CrossRef]
- DiJoseph, J.F.; Armellino, D.C.; Boghaert, E.R.; Khandke, K.; Dougher, M.M.; Sridharan, L.; Kunz, A.; Hamann, P.R.; Gorovits, B.; Udata, C.; et al. Antibody-Targeted Chemotherapy with CMC-544: A CD22-Targeted Immunoconjugate of Calicheamicin for the Treatment of B-Lymphoid Malignancies. Blood 2004, 103, 1807–1814. [Google Scholar] [CrossRef]
- Takeshita, A.; Yamakage, N.; Shinjo, K.; Ono, T.; Hirano, I.; Nakamura, S.; Shigeno, K.; Tobita, T.; Maekawa, M.; Kiyoi, H.; et al. CMC-544 (Inotuzumab Ozogamicin), an Anti-CD22 Immuno-Conjugate of Calicheamicin, Alters the Levels of Target Molecules of Malignant B-Cells. Leukemia 2009, 23, 1329–1336. [Google Scholar] [CrossRef]
- Li, X.; Zhou, M.; Qi, J.; Han, Y. Efficacy and Safety of Inotuzumab Ozogamicin (CMC-544) for the Treatment of Relapsed/Refractory Acute Lymphoblastic Leukemia and Non-Hodgkin Lymphoma: A Systematic Review and Meta-Analysis. Clin. Lymphoma Myeloma Leuk. 2021, 21, e227–e247. [Google Scholar] [CrossRef]
- Patel, K.C.; Hageman, K.; Cooper, M.R. Ado-Trastuzumab Emtansine for the Treatment of Human Epidermal Growth Factor Receptor 2-Positive Metastatic Breast Cancer. Am. J. Health Syst. Pharm. 2014, 71, 537–548. [Google Scholar] [CrossRef]
- Peddi, P.F.; Hurvitz, S.A. Ado-Trastuzumab Emtansine (T-DM1) in Human Epidermal Growth Factor Receptor 2 (HER2)-Positive Metastatic Breast Cancer: Latest Evidence and Clinical Potential. Ther. Adv. Med. Oncol. 2014, 6, 202–209. [Google Scholar] [CrossRef]
- Lewis Phillips, G.D.; Li, G.; Dugger, D.L.; Crocker, L.M.; Parsons, K.L.; Mai, E.; Blättler, W.A.; Lambert, J.M.; Chari, R.V.J.; Lutz, R.J.; et al. Targeting HER2-Positive Breast Cancer with Trastuzumab-DM1, an Antibody–Cytotoxic Drug Conjugate. Cancer Res. 2008, 68, 9280–9290. [Google Scholar] [CrossRef]
- Stadtmauer, E.A. Gemtuzumab Ozogamicin in the Treatment of Acute Myeloid Leukemia. Curr. Oncol. Rep. 2002, 4, 375–380. [Google Scholar] [CrossRef]
- Hosono, N.; Ookura, M.; Araie, H.; Morita, M.; Itoh, K.; Matsuda, Y.; Yamauchi, T. Clinical Outcomes of Gemtuzumab Ozogamicin for Relapsed Acute Myeloid Leukemia: Single-Institution Experience. Int. J. Hematol. 2021, 113, 362–369. [Google Scholar] [CrossRef]
- Yu, B.; Liu, D. Gemtuzumab Ozogamicin and Novel Antibody-Drug Conjugates in Clinical Trials for Acute Myeloid Leukemia. Biomark. Res. 2019, 7, 24. [Google Scholar] [CrossRef]
- Takeshita, A. Efficacy and Resistance of Gemtuzumab Ozogamicin for Acute Myeloid Leukemia. Int. J. Hematol. 2013, 97, 703–716. [Google Scholar] [CrossRef]
- Zaro, J.L. Mylotarg: Revisiting Its Clinical Potential Post-Withdrawal. In Antibody-Drug Conjugates; Wang, J., Shen, W.-C., Zaro, J.L., Eds.; AAPS Advances in the Pharmaceutical Sciences Series; Springer International Publishing: Cham, Switzerland, 2015; Volume 17, pp. 179–190. ISBN 978-3-319-13080-4. [Google Scholar] [CrossRef]
- Tennvall, J.; Fischer, M.; Bischof Delaloye, A.; Bombardieri, E.; Bodei, L.; Giammarile, F.; Lassmann, M.; Oyen, W.; Brans, B. EANM Procedure Guideline for Radio-Immunotherapy for B-Cell Lymphoma with 90Y-Radiolabelled Ibritumomab Tiuxetan (Zevalin). Eur. J. Nucl. Med. Mol. Imaging 2007, 34, 616–622. [Google Scholar] [CrossRef]
- Rizzieri, D. Zevalin® (Ibritumomab Tiuxetan): After More than a Decade of Treatment Experience, What Have We Learned? Crit. Rev. Oncol. Hematol. 2016, 105, 5–17. [Google Scholar] [CrossRef]
- Marcus, R. Use of 90Y-Ibritumomab Tiuxetan in Non-Hodgkin’s Lymphoma. Semin. Oncol. 2005, 32, 36–43. [Google Scholar] [CrossRef]
- Witzig, T.E.; Gordon, L.I.; Cabanillas, F.; Czuczman, M.S.; Emmanouilides, C.; Joyce, R.; Pohlman, B.L.; Bartlett, N.L.; Wiseman, G.A.; Padre, N.; et al. Randomized Controlled Trial of Yttrium-90–Labeled Ibritumomab Tiuxetan Radioimmunotherapy versus Rituximab Immunotherapy for Patients with Relapsed or Refractory Low-Grade, Follicular, or Transformed B-Cell Non-Hodgkin’s Lymphoma. J. Clin. Oncol. 2002, 20, 2453–2463. [Google Scholar] [CrossRef]
- Hagenbeek, A. Radioimmunotherapy for NHL: Experience of 90Y-Ibritumomab Tiuxetan in Clinical Practice. Leuk. Lymphoma. 2003, 44, S37–S47. [Google Scholar] [CrossRef]
- Tilly, H.; Morschhauser, F.; Sehn, L.H.; Friedberg, J.W.; Trněný, M.; Sharman, J.P.; Herbaux, C.; Burke, J.M.; Matasar, M.; Rai, S.; et al. Polatuzumab Vedotin in Previously Untreated Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2022, 386, 351–363. [Google Scholar] [CrossRef]
- Yip, V.; Lee, M.V.; Saad, O.M.; Ma, S.; Khojasteh, S.C.; Shen, B.-Q. Preclinical Characterization of the Distribution, Catabolism, and Elimination of a Polatuzumab Vedotin-Piiq (POLIVY®) Antibody–Drug Conjugate in Sprague Dawley Rats. J. Clin. Med. 2021, 10, 1323. [Google Scholar] [CrossRef]
- Cardillo, T.M.; Govindan, S.V.; Sharkey, R.M.; Trisal, P.; Arrojo, R.; Liu, D.; Rossi, E.A.; Chang, C.-H.; Goldenberg, D.M. Sacituzumab Govitecan (IMMU-132), an Anti-Trop-2/SN-38 Antibody–Drug Conjugate: Characterization and Efficacy in Pancreatic, Gastric, and Other Cancers. Bioconjug. Chem. 2015, 26, 919–931. [Google Scholar] [CrossRef]
- Weiss, J.; Glode, A.; Messersmith, W.A.; Diamond, J. Sacituzumab Govitecan: Breakthrough Targeted Therapy for Triple-Negative Breast Cancer. Expert Rev. Anticancer Ther. 2019, 19, 673–679. [Google Scholar] [CrossRef]
- Ocean, A.J.; Starodub, A.N.; Bardia, A.; Vahdat, L.T.; Isakoff, S.J.; Guarino, M.; Messersmith, W.A.; Picozzi, V.J.; Mayer, I.A.; Wegener, W.A.; et al. Sacituzumab Govitecan (IMMU-132), an anti-Trop-2-SN-38 Antibody-drug Conjugate for the Treatment of Diverse Epithelial Cancers: Safety and Pharmacokinetics. Cancer 2017, 123, 3843–3854. [Google Scholar] [CrossRef]
- Bardia, A.; Messersmith, W.A.; Kio, E.A.; Berlin, J.D.; Vahdat, L.; Masters, G.A.; Moroose, R.; Santin, A.D.; Kalinsky, K.; Picozzi, V.; et al. Sacituzumab Govitecan, a Trop-2-Directed Antibody-Drug Conjugate, for Patients with Epithelial Cancer: Final Safety and Efficacy Results from the Phase I/II IMMU-132-01 Basket Trial. Ann. Oncol. 2021, 32, 746–756. [Google Scholar] [CrossRef]
- Challita-Eid, P.M.; Satpayev, D.; Yang, P.; An, Z.; Morrison, K.; Shostak, Y.; Raitano, A.; Nadell, R.; Liu, W.; Lortie, D.R.; et al. Enfortumab Vedotin Antibody–Drug Conjugate Targeting Nectin-4 Is a Highly Potent Therapeutic Agent in Multiple Preclinical Cancer Models. Cancer Res. 2016, 76, 3003–3013. [Google Scholar] [CrossRef]
- McGregor, B.A.; Sonpavde, G. Enfortumab Vedotin, a Fully Human Monoclonal Antibody against Nectin 4 Conjugated to Monomethyl Auristatin E for Metastatic Urothelial Carcinoma. Expert Opin. Investig. Drugs 2019, 28, 821–826. [Google Scholar] [CrossRef]
- Maas, M.; Stühler, V.; Walz, S.; Stenzl, A.; Bedke, J. Enfortumab Vedotin–next Game-Changer in Urothelial Cancer. Expert Opin. Biol. Ther. 2021, 21, 801–809. [Google Scholar] [CrossRef]
- Spreen, L. Mirvetuximab Soravtansine-Gynx (ElahereTM). Oncol. Times 2023, 45, 6. [Google Scholar] [CrossRef]
- Parvez, S.; Prakash, K.; Soni, D. A Review on Chemistry, Mechanism, Clinical Trials Studies of ELAHERE (Mirvetuximab Soravtansine-Gynx): A Novel FDA Approved Drug to Treat Ovarian Cancer. World J. Pharm. Pharm. Sci. 2023, 12, 891–898. [Google Scholar] [CrossRef]
- Heo, Y.-A. Mirvetuximab Soravtansine: First Approval. Drugs 2023, 83, 265–273. [Google Scholar] [CrossRef]
- Nwabufo, C.K. Mirvetuximab Soravtansine in Ovarian Cancer Therapy: Expert Opinion on Pharmacological Considerations. Cancer Chemother. Pharmacol. 2023; online ahead of print. [Google Scholar] [CrossRef]
- Yam, C.; Rauch, G.M.; Rahman, T.; Karuturi, M.; Ravenberg, E.; White, J.; Clayborn, A.; McCarthy, P.; Abouharb, S.; Lim, B.; et al. A Phase II Study of Mirvetuximab Soravtansine in Triple-Negative Breast Cancer. Investig. New Drugs 2021, 39, 509–515. [Google Scholar] [CrossRef]
- Mosele, F.; Deluche, E.; Lusque, A.; Le Bescond, L.; Filleron, T.; Pradat, Y.; Ducoulombier, A.; Pistilli, B.; Bachelot, T.; Viret, F.; et al. Trastuzumab Deruxtecan in Metastatic Breast Cancer with Variable HER2 Expression: The Phase 2 DAISY Trial. Nat. Med. 2023, 29, 2110–2120. [Google Scholar] [CrossRef]
- Andrikopoulou, A.; Zografos, E.; Liontos, M.; Koutsoukos, K.; Dimopoulos, M.-A.; Zagouri, F. Trastuzumab Deruxtecan (DS-8201a): The Latest Research and Advances in Breast Cancer. Clin. Breast Cancer 2021, 21, e212–e219. [Google Scholar] [CrossRef]
- Ogitani, Y.; Hagihara, K.; Oitate, M.; Naito, H.; Agatsuma, T. Bystander Killing Effect of DS -8201a, a Novel Anti-human Epidermal Growth Factor Receptor 2 Antibody–Drug Conjugate, in Tumors with Human Epidermal Growth Factor Receptor 2 Heterogeneity. Cancer Sci. 2016, 107, 1039–1046. [Google Scholar] [CrossRef]
- Markham, A. Belantamab Mafodotin: First Approval. Drugs 2020, 80, 1607–1613. [Google Scholar] [CrossRef]
- Trudel, S.; Lendvai, N.; Popat, R.; Voorhees, P.M.; Reeves, B.; Libby, E.N.; Richardson, P.G.; Hoos, A.; Gupta, I.; Bragulat, V.; et al. Antibody–Drug Conjugate, GSK2857916, in Relapsed/Refractory Multiple Myeloma: An Update on Safety and Efficacy from Dose Expansion Phase I Study. Blood Cancer J. 2019, 9, 37. [Google Scholar] [CrossRef]
- Calabretta, E.; Hamadani, M.; Carlo-Stella, C. The Antibody-Drug Conjugate Loncastuximab Tesirine for the Treatment of Diffuse Large B-Cell Lymphoma. Blood 2022, 140, 303–308. [Google Scholar] [CrossRef]
- Lee, A. Loncastuximab Tesirine: First Approval. Drugs 2021, 81, 1229–1233. [Google Scholar] [CrossRef]
- Xu, B. Loncastuximab Tesirine: An Effective Therapy for Relapsed or Refractory Diffuse Large B-Cell Lymphoma. Eur. J. Clin. Pharmacol. 2022, 78, 707–719. [Google Scholar] [CrossRef]
- Zammarchi, F.; Corbett, S.; Adams, L.; Tyrer, P.C.; Kiakos, K.; Janghra, N.; Marafioti, T.; Britten, C.E.; Havenith, C.E.G.; Chivers, S.; et al. ADCT-402, a PBD Dimer–Containing Antibody Drug Conjugate Targeting CD19-Expressing Malignancies. Blood 2018, 131, 1094–1105. [Google Scholar] [CrossRef]
- Senter, P.D.; Sievers, E.L. The Discovery and Development of Brentuximab Vedotin for Use in Relapsed Hodgkin Lymphoma and Systemic Anaplastic Large Cell Lymphoma. Nat. Biotechnol. 2012, 30, 631–637. [Google Scholar] [CrossRef]
- Sasso, J.M.; Tenchov, R.; Bird, R.; Iyer, K.A.; Ralhan, K.; Rodriguez, Y.; Zhou, Q.A. The Evolving Landscape of Antibody–Drug Conjugates: In Depth Analysis of Recent Research Progress. Bioconjug. Chem. 2023, 34, 1951–2000. [Google Scholar] [CrossRef]
- Song, C.H.; Jeong, M.; In, H.; Kim, J.H.; Lin, C.-W.; Han, K.H. Trends in the Development of Antibody-Drug Conjugates for Cancer Therapy. Antibodies 2023, 12, 72. [Google Scholar] [CrossRef]
- Kaplon, H.; Crescioli, S.; Chenoweth, A.; Visweswaraiah, J.; Reichert, J.M. Antibodies to Watch in 2023. mAbs 2023, 15, 2153410. [Google Scholar] [CrossRef]
- Murphy, K.M.; Weaver, C.; Berg, L.J.; Janeway, C. Janeway’s Immunobiology, 10th ed.; International Student Edition; W.W. Norton & Company: New York, NY, USA; London, UK, 2022; ISBN 978-0-393-88491-3. [Google Scholar]
- Chua, M.M.; Fan, S.T.; Karush, F. Attachment of Immunoglobulin to Liposomal Membrane via Protein Carbohydrate. Biochim. Biophys. Acta 1984, 800, 291–300. [Google Scholar] [CrossRef]
- Nezlin, R.S.; Sykulev, Y.K. Structural Studies of Immunoglobulins Spin-Labeled at the Carbohydrate Moiety. Mol. Immunol. 1982, 19, 347–356. [Google Scholar] [CrossRef]
- Biswas, S.; Mandal, G.; Anadon, C.M.; Chaurio, R.A.; Lopez-Bailon, L.U.; Nagy, M.Z.; Mine, J.A.; Hänggi, K.; Sprenger, K.B.; Innamarato, P.; et al. Targeting Intracellular Oncoproteins with Dimeric IgA Promotes Expulsion from the Cytoplasm and Immune-Mediated Control of Epithelial Cancers. Immunity 2023, 56, 2570–2583. [Google Scholar] [CrossRef]
- Almagro, J.C.; Daniels-Wells, T.R.; Perez-Tapia, S.M.; Penichet, M.L. Progress and Challenges in the Design and Clinical Development of Antibodies for Cancer Therapy. Front. Immunol. 2018, 8, 1751. [Google Scholar] [CrossRef]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG Subclasses and Allotypes: From Structure to Effector Functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef]
- Li, T.; Wang, X.; Niu, M.; Wang, M.; Zhou, J.; Wu, K.; Yi, M. Bispecific Antibody Targeting TGF-β and PD-L1 for Synergistic Cancer Immunotherapy. Front. Immunol. 2023, 14, 1196970. [Google Scholar] [CrossRef]
- Beishenaliev, A.; Loke, Y.L.; Goh, S.J.; Geo, H.N.; Mugila, M.; Misran, M.; Chung, L.Y.; Kiew, L.V.; Roffler, S.; Teo, Y.Y. Bispecific Antibodies for Targeted Delivery of Anti-Cancer Therapeutic Agents: A Review. J. Control. Release 2023, 359, 268–286. [Google Scholar] [CrossRef]
- Ma, J.; Mo, Y.; Tang, M.; Shen, J.; Qi, Y.; Zhao, W.; Huang, Y.; Xu, Y.; Qian, C. Bispecific Antibodies: From Research to Clinical Application. Front. Immunol. 2021, 12, 626616. [Google Scholar] [CrossRef]
- Shim, H. Bispecific Antibodies and Antibody–Drug Conjugates for Cancer Therapy: Technological Considerations. Biomolecules 2020, 10, 360. [Google Scholar] [CrossRef]
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P.W.H.I. Bispecific Antibodies: A Mechanistic Review of the Pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef]
- Szijj, P.; Chudasama, V. The Renaissance of Chemically Generated Bispecific Antibodies. Nat. Rev. Chem. 2021, 5, 78–92. [Google Scholar] [CrossRef]
- Wei, J.; Yang, Y.; Wang, G.; Liu, M. Current Landscape and Future Directions of Bispecific Antibodies in Cancer Immunotherapy. Front. Immunol. 2022, 13, 1035276. [Google Scholar] [CrossRef]
- Dimasi, N.; Fleming, R.; Zhong, H.; Bezabeh, B.; Kinneer, K.; Christie, R.J.; Fazenbaker, C.; Wu, H.; Gao, C. Efficient Preparation of Site-Specific Antibody–Drug Conjugates Using Cysteine Insertion. Mol. Pharm. 2017, 14, 1501–1516. [Google Scholar] [CrossRef]
- Andreev, J.; Thambi, N.; Perez Bay, A.E.; Delfino, F.; Martin, J.; Kelly, M.P.; Kirshner, J.R.; Rafique, A.; Kunz, A.; Nittoli, T.; et al. Bispecific Antibodies and Antibody–Drug Conjugates (ADCs) Bridging HER2 and Prolactin Receptor Improve Efficacy of HER2 ADCs. Mol. Cancer Ther. 2017, 16, 681–693. [Google Scholar] [CrossRef]
- Cal, P.M.S.D.; Bernardes, G.J.L.; Gois, P.M.P. Cysteine-Selective Reactions for Antibody Conjugation. Angew. Chem. Int. Ed. 2014, 53, 10585–10587. [Google Scholar] [CrossRef]
- Harding, F.A.; Stickler, M.M.; Razo, J.; DuBridge, R. The Immunogenicity of Humanized and Fully Human Antibodies: Residual Immunogenicity Resides in the CDR Regions. mAbs 2010, 2, 256–265. [Google Scholar] [CrossRef]
- Doevendans, E.; Schellekens, H. Immunogenicity of Innovative and Biosimilar Monoclonal Antibodies. Antibodies 2019, 8, 21. [Google Scholar] [CrossRef]
- Safdari, Y.; Farajnia, S.; Asgharzadeh, M.; Khalili, M. Antibody Humanization Methods—A Review and Update. Biotechnol. Genet. Eng. Rev. 2013, 29, 175–186. [Google Scholar] [CrossRef]
- Panowski, S.; Bhakta, S.; Raab, H.; Polakis, P.; Junutula, J.R. Site-Specific Antibody Drug Conjugates for Cancer Therapy. mAbs 2014, 6, 34–45. [Google Scholar] [CrossRef]
- Adlersberg, J.B. The Immunoglobulin Hinge (Interdomain) Region. Ric. Clin. Lab. 1976, 6, 191–205. [Google Scholar] [CrossRef]
- Liu, H.; May, K. Disulfide Bond Structures of IgG Molecules: Structural Variations, Chemical Modifications and Possible Impacts to Stability and Biological Function. mAbs 2012, 4, 17–23. [Google Scholar] [CrossRef]
- Deveuve, Q.; Gouilleux-Gruart, V.; Thibault, G.; Lajoie, L. La Région Charnière Des Anticorps Thérapeutiques: L’importance Capitale d’une Courte Séquence. Med. Sci. 2019, 35, 1098–1105. [Google Scholar] [CrossRef]
- Hagihara, Y.; Saerens, D. Engineering Disulfide Bonds within an Antibody. Biochim. Biophys. Acta 2014, 1844, 2016–2023. [Google Scholar] [CrossRef]
- Mthembu, S.N.; Sharma, A.; Albericio, F.; De La Torre, B.G. Breaking a Couple: Disulfide Reducing Agents. ChemBioChem 2020, 21, 1947–1954. [Google Scholar] [CrossRef]
- Junutula, J.R.; Raab, H.; Clark, S.; Bhakta, S.; Leipold, D.D.; Weir, S.; Chen, Y.; Simpson, M.; Tsai, S.P.; Dennis, M.S.; et al. Site-Specific Conjugation of a Cytotoxic Drug to an Antibody Improves the Therapeutic Index. Nat. Biotechnol. 2008, 26, 925–932. [Google Scholar] [CrossRef]
- You, J.; Zhang, J.; Wang, J.; Jin, M. Cysteine-Based Coupling: Challenges and Solutions. Bioconjug. Chem. 2021, 32, 1525–1534. [Google Scholar] [CrossRef]
- Gunnoo, S.B.; Madder, A. Chemical Protein Modification through Cysteine. ChemBioChem 2016, 17, 529–553. [Google Scholar] [CrossRef]
- Akkapeddi, P.; Azizi, S.-A.; Freedy, A.M.; Cal, P.M.S.D.; Gois, P.M.P.; Bernardes, G.J.L. Construction of Homogeneous Antibody–Drug Conjugates Using Site-Selective Protein Chemistry. Chem. Sci. 2016, 7, 2954–2963. [Google Scholar] [CrossRef]
- Spears, R.J.; Fascione, M.A. Site-Selective Incorporation and Ligation of Protein Aldehydes. Org. Biomol. Chem. 2016, 14, 7622–7638. [Google Scholar] [CrossRef]
- Kölmel, D.K.; Kool, E.T. Oximes and Hydrazones in Bioconjugation: Mechanism and Catalysis. Chem. Rev. 2017, 117, 10358–10376. [Google Scholar] [CrossRef]
- O’Shannessy, D.J.; Quarles, R.H. Labeling of the Oligosaccharide Moieties of Immunoglobulins. J. Immunol. Meth. 1987, 99, 153–161. [Google Scholar] [CrossRef]
- Nakane, P.K.; Kawaoi, A. Peroxidase-Labeled Antibody a New Method of Conjugation. J. Histochem Cytochem. 1974, 22, 1084–1091. [Google Scholar] [CrossRef]
- Wang, L.; Brock, A.; Herberich, B.; Schultz, P.G. Expanding the Genetic Code of Escherichia coli. Science 2001, 292, 498–500. [Google Scholar] [CrossRef]
- Liu, S.; Liu, X. IgG N-Glycans. In Advances in Clinical Chemistry; Elsevier: Amsterdam, The Netherlands, 2021; Volume 105, pp. 1–47. ISBN 978-0-12-824627-6. [Google Scholar] [CrossRef]
- Zhong, X.; D’Antona, A.M.; Scarcelli, J.J.; Rouse, J.C. New Opportunities in Glycan Engineering for Therapeutic Proteins. Antibodies 2022, 11, 5. [Google Scholar] [CrossRef]
- Rodwell, J.D.; Alvarez, V.L.; Lee, C.; Lopes, A.D.; Goers, J.W.; King, H.D.; Powsner, H.J.; McKearn, T.J. Site-Specific Covalent Modification of Monoclonal Antibodies: In Vitro and in Vivo Evaluations. Proc. Natl. Acad. Sci. USA 1986, 83, 2632–2636. [Google Scholar] [CrossRef]
- Kaur, H. Characterization of Glycosylation in Monoclonal Antibodies and Its Importance in Therapeutic Antibody Development. Crit. Rev. Biotechnol. 2021, 41, 300–315. [Google Scholar] [CrossRef]
- Zauner, G.; Selman, M.H.J.; Bondt, A.; Rombouts, Y.; Blank, D.; Deelder, A.M.; Wuhrer, M. Glycoproteomic Analysis of Antibodies. Mol. Cell Proteomics. 2013, 12, 856–865. [Google Scholar] [CrossRef]
- Wang, L.-X.; Tong, X.; Li, C.; Giddens, J.P.; Li, T. Glycoengineering of Antibodies for Modulating Functions. Annu. Rev. Biochem. 2019, 88, 433–459. [Google Scholar] [CrossRef]
- Mimura, Y.; Katoh, T.; Saldova, R.; O’Flaherty, R.; Izumi, T.; Mimura-Kimura, Y.; Utsunomiya, T.; Mizukami, Y.; Yamamoto, K.; Matsumoto, T.; et al. Glycosylation Engineering of Therapeutic IgG Antibodies: Challenges for the Safety, Functionality and Efficacy. Protein Cell 2018, 9, 47–62. [Google Scholar] [CrossRef]
- Boune, S.; Hu, P.; Epstein, A.L.; Khawli, L.A. Principles of N-Linked Glycosylation Variations of IgG-Based Therapeutics: Pharmacokinetic and Functional Considerations. Antibodies 2020, 9, 22. [Google Scholar] [CrossRef]
- Sjögren, J.; Lood, R.; Nägeli, A. On Enzymatic Remodeling of IgG Glycosylation; Unique Tools with Broad Applications. Glycobiology 2020, 30, 254–267. [Google Scholar] [CrossRef]
- Golay, J.; Andrea, A.E.; Cattaneo, I. Role of Fc Core Fucosylation in the Effector Function of IgG1 Antibodies. Front. Immunol. 2022, 13, 929895. [Google Scholar] [CrossRef]
- Zhang, L.; Luo, S.; Zhang, B. Glycan Analysis of Therapeutic Glycoproteins. mAbs 2016, 8, 205–215. [Google Scholar] [CrossRef]
- Huhn, C.; Selman, M.H.J.; Ruhaak, L.R.; Deelder, A.M.; Wuhrer, M. IgG Glycosylation Analysis. Proteomics 2009, 9, 882–913. [Google Scholar] [CrossRef]
- Maverakis, E.; Kim, K.; Shimoda, M.; Gershwin, M.E.; Patel, F.; Wilken, R.; Raychaudhuri, S.; Ruhaak, L.R.; Lebrilla, C.B. Glycans in the Immune System and The Altered Glycan Theory of Autoimmunity: A Critical Review. J. Autoimmun. 2015, 57, 1–13. [Google Scholar] [CrossRef]
- Quast, I.; Peschke, B.; Lünemann, J.D. Regulation of Antibody Effector Functions through IgG Fc N-Glycosylation. Cell. Mol. Life Sci. 2017, 74, 837–847. [Google Scholar] [CrossRef]
- Van De Bovenkamp, F.S.; Hafkenscheid, L.; Rispens, T.; Rombouts, Y. The Emerging Importance of IgG Fab Glycosylation in Immunity. J. Immunol. 2016, 196, 1435–1441. [Google Scholar] [CrossRef]
- Vattepu, R.; Sneed, S.L.; Anthony, R.M. Sialylation as an Important Regulator of Antibody Function. Front. Immunol. 2022, 13, 818736. [Google Scholar] [CrossRef]
- O’Shannessy, D.J.; Dobersen, M.J.; Quarles, R.H. A Novel Procedure for Labeling Immunoglobulins by Conjugation to Oligosaccharide Moieties. Immunol. Lett. 1984, 8, 273–277. [Google Scholar] [CrossRef]
- Fan, Y.; Kildegaard, H.F.; Andersen, M.R. Engineer Medium and Feed for Modulating N-Glycosylation of Recombinant Protein Production in CHO Cell Culture. In Heterologous Protein Production in CHO Cells; Meleady, P., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2017; Volume 1603, pp. 209–226. ISBN 978-1-4939-6971-5. [Google Scholar] [CrossRef]
- Sheeley, D.M.; Merrill, B.M.; Taylor, L.C.E. Characterization of Monoclonal Antibody Glycosylation: Comparison of Expression Systems and Identification of Terminal α-Linked Galactose. Anal. Biochem. 1997, 247, 102–110. [Google Scholar] [CrossRef]
- Grainger, R.K.; James, D.C. CHO Cell Line Specific Prediction and Control of Recombinant Monoclonal Antibody N-glycosylation. Biotechnol. Bioeng. 2013, 110, 2970–2983. [Google Scholar] [CrossRef]
- Routier, F.H.; Davies, M.J.; Bergemann, K.; Hounsell, E.F. The Glycosylation Pattern of a Humanized IgGl Antibody (D1.3) Expressed in CHO Cells. Glycoconj. J. 1997, 14, 201–207. [Google Scholar] [CrossRef]
- Kim, W.-D.; Tokunaga, M.; Ozaki, H.; Ishibashi, T.; Honda, K.; Kajiura, H.; Fujiyama, K.; Asano, R.; Kumagai, I.; Omasa, T.; et al. Glycosylation Pattern of Humanized IgG-like Bispecific Antibody Produced by Recombinant CHO Cells. Appl. Microbiol. Biotechnol. 2010, 85, 535–542. [Google Scholar] [CrossRef]
- Rothfus, J.A.; Smith, E.L. Glycopeptides. IV. The Periodate Oxidation of Glycopeptides from Human Gamma-Globulin. J. Biol. Chem. 1963, 238, 1402–1410. [Google Scholar] [CrossRef]
- Malaprade Reaction: (Malaprade Oxidation). In Comprehensive Organic Name Reactions and Reagents; Wiley: Hoboken, NJ, USA, 2010; pp. 1807–1810. ISBN 978-0-471-70450-8. [CrossRef]
- Nicolet, B.H.; Shinn, L.A. The Action of Periodic Acid on α-Amino Alcohols. J. Am. Chem. Soc. 1939, 61, 1615. [Google Scholar] [CrossRef]
- Zuberbühler, K.; Casi, G.; Bernardes, G.J.L.; Neri, D. Fucose-Specific Conjugation of Hydrazide Derivatives to a Vascular-Targeting Monoclonal Antibody in IgG Format. Chem. Commun. 2012, 48, 7100. [Google Scholar] [CrossRef]
- Wolfe, C.A.C.; Hage, D.S. Studies on the Rate and Control of Antibody Oxidation by Periodate. Anal. Biochem. 1995, 231, 123–130. [Google Scholar] [CrossRef]
- Sapozhnikova, K.A.; Gulyak, E.L.; Misyurin, V.A.; Simonova, M.A.; Ryabukhina, E.V.; Alexeeva, A.V.; Tikhonova, N.A.; Lyzhko, N.A.; Popova, G.P.; Misyurin, A.V.; et al. Branched Linkers for Site-Specific Fluorescent Labeling of Antibodies. Molecules 2023, 28, 425. [Google Scholar] [CrossRef]
- Fischer-Durand, N.; Salmain, M.; Vessières, A.; Jaouen, G. A New Bioorthogonal Cross-Linker with Alkyne and Hydrazide End Groups for Chemoselective Ligation. Application to Antibody Labelling. Tetrahedron 2012, 68, 9638–9644. [Google Scholar] [CrossRef]
- Abraham, R.; Moller, D.; Gabel, D.; Senter, P.; Hellström, I.; Hellström, K.E. The Influence of Periodate Oxidation on Monoclonal Antibody Avidity and Immunoreactivity. J. Immunol. Meth. 1991, 144, 77–86. [Google Scholar] [CrossRef]
- Akira, M.; Kohkichi, S.; Tadashi, Y. Modification of Immunoglobulin G Using Specific Reactivity of Sugar Moiety. Immunochemistry 1978, 15, 523–528. [Google Scholar] [CrossRef]
- Kurth, M.; Pelegrin, A.; Rose, K.; Offord, R.E.; Pochon, S.; Mach, J.P.; Buchegger, F. Site-Specific Conjugation of a Radioiodinated Phenethylamine Derivative to a Monoclonal Antibody Results in Increased Radioactivity Localization in Tumor. J. Med. Chem. 1993, 36, 1255–1261. [Google Scholar] [CrossRef]
- Andersen, B.R.; Abele, D.C.; Vannier, W.E. Effects of Mild Periodate Oxidation on Antibodies. J. Immunol. 1966, 97, 913–924. [Google Scholar] [CrossRef]
- Zhou, Q.; Stefano, J.E.; Manning, C.; Kyazike, J.; Chen, B.; Gianolio, D.A.; Park, A.; Busch, M.; Bird, J.; Zheng, X.; et al. Site-Specific Antibody–Drug Conjugation through Glycoengineering. Bioconjug. Chem. 2014, 25, 510–520. [Google Scholar] [CrossRef]
- Inglis, A.S.; Rivett, D.E.; McMahon, D.T.W. The Identification of Tryptophan Residues in Proteins as Oxidised Derivatives during Amino Acid Sequence Determinations. FEBS Lett. 1979, 104, 115–118. [Google Scholar] [CrossRef]
- Clamp, J.; Hough, L. The Periodate Oxidation of Amino Acids with Reference to Studies on Glycoproteins. Biochem. J. 1965, 94, 17–24. [Google Scholar] [CrossRef]
- Robinson, G.M.; Smyth, M.R. Simultaneous Determination of Products and Intermediates of L-Dopa Oxidation Using Capillary Electrophoresis With Diode-Array Detection. Analyst 1997, 122, 797–802. [Google Scholar] [CrossRef]
- Hsuan, J.J. The Cross-Linking of Tyrosine Residues in Apo-Ovotransferrin by Treatment with Periodate Anions. Biochem. J. 1987, 247, 467–473. [Google Scholar] [CrossRef]
- Stadtman, E.R. Role of Oxidized Amino Acids in Protein Breakdown and Stability. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1995; Volume 258, pp. 379–393. ISBN 978-0-12-182159-3. [Google Scholar] [CrossRef]
- Burzio, L.A.; Waite, J.H. Reactivity of Peptidyl-tyrosine to Hydroxylation and Cross-linking. Protein Sci. 2001, 10, 735–740. [Google Scholar] [CrossRef]
- Yamasaki, R.B.; Osuga, D.T.; Feeney, R.E. Periodate Oxidation of Methionine in Proteins. Anal. Biochem. 1982, 126, 183–189. [Google Scholar] [CrossRef]
- Willan, K.J.; Golding, B.; Givol, D.; Dwek, R.A. Specific Spin Labelling of the Fc Region of Immunoglobulins. FEBS Lett. 1977, 80, 133–136. [Google Scholar] [CrossRef]
- Qasba, P.K. Glycans of Antibodies as a Specific Site for Drug Conjugation Using Glycosyltransferases. Bioconjug. Chem. 2015, 26, 2170–2175. [Google Scholar] [CrossRef]
- Hageman, T.; Wei, H.; Kuehne, P.; Fu, J.; Ludwig, R.; Tao, L.; Leone, A.; Zocher, M.; Das, T.K. Impact of Tryptophan Oxidation in Complementarity-Determining Regions of Two Monoclonal Antibodies on Structure-Function Characterized by Hydrogen-Deuterium Exchange Mass Spectrometry and Surface Plasmon Resonance. Pharm. Res. 2019, 36, 24. [Google Scholar] [CrossRef]
- McDonnell, K.A.; Low, S.C.; Hoehn, T.; Donnelly, R.; Palmieri, H.; Fraley, C.; Sakorafas, P.; Mezo, A.R. Synthesis and Structure−Activity Relationships of Dimeric Peptide Antagonists of the Human Immunoglobulin G−Human Neonatal Fc Receptor (IgG−FcRn) Interaction. J. Med. Chem. 2010, 53, 1587–1596. [Google Scholar] [CrossRef]
- Umaña, P.; Jean–Mairet, J.; Moudry, R.; Amstutz, H.; Bailey, J.E. Engineered Glycoforms of an Antineuroblastoma IgG1 with Optimized Antibody-Dependent Cellular Cytotoxic Activity. Nat. Biotechnol. 1999, 17, 176–180. [Google Scholar] [CrossRef]
- Fleminger, G.; Solomon, B.; Wolf, T.; Hadas, E. Single Step Oxidative Binding of Antibodies to Hydrazide-Modified Eupergit C. Appl. Biochem. Biotechnol. 1990, 26, 231–238. [Google Scholar] [CrossRef]
- O’shannessy, D.J. Hydrazido-Derivatized Supports in Affinity Chromatography. J. Chromatogr. A 1990, 510, 13–21. [Google Scholar] [CrossRef]
- Fleminger, G.; Hadas, E.; Wolf, T.; Solomon, B. Oriented Immobilization of Periodateoxidized Monoclonal Antibodies on Amino and Hydrazide Derivatives of Eupergit C. Appl. Biochem. Biotechnol. 1990, 23, 123–137. [Google Scholar] [CrossRef]
- Hoffman, W.L.; O’Shannessy, D.J. Site-Specific Immobilization of Antibodies by Their Oligosaccharide Moieties to New Hydrazide Derivatized Solid Supports. J. Immunol. Meth. 1988, 112, 113–120. [Google Scholar] [CrossRef]
- Scheck, R.A.; Francis, M.B. Regioselective Labeling of Antibodies through N-Terminal Transamination. ACS Chem. Biol. 2007, 2, 247–251. [Google Scholar] [CrossRef]
- Netirojjanakul, C.; Witus, L.S.; Behrens, C.R.; Weng, C.-H.; Iavarone, A.T.; Francis, M.B. Synthetically Modified Fc Domains as Building Blocks for Immunotherapy Applications. Chem. Sci. 2013, 4, 266–272. [Google Scholar] [CrossRef]
- Witus, L.S.; Francis, M. Site-Specific Protein Bioconjugation via a Pyridoxal 5′-Phosphate-Mediated N-Terminal Transamination Reaction. Curr. Prot. Chem. Biol. 2010, 2, 125–134. [Google Scholar] [CrossRef]
- Witus, L.S.; Netirojjanakul, C.; Palla, K.S.; Muehl, E.M.; Weng, C.-H.; Iavarone, A.T.; Francis, M.B. Site-Specific Protein Transamination Using N -Methylpyridinium-4-Carboxaldehyde. J. Am. Chem. Soc. 2013, 135, 17223–17229. [Google Scholar] [CrossRef]
- Zhang, Z.; Shah, B.; Richardson, J. Impact of Fc N-Glycan Sialylation on IgG Structure. mAbs 2019, 11, 1381–1390. [Google Scholar] [CrossRef]
- Schauer, R. Chemistry, Metabolism, and Biological Functions of Sialic Acids. In Advances in Carbohydrate Chemistry and Biochemistry; Elsevier: Amsterdam, The Netherlands, 1982; Volume 40, pp. 131–234. ISBN 978-0-12-007240-8. [Google Scholar] [CrossRef]
- Cabezas, J.A. Some Questions and Suggestions on the Type References of the Official Nomenclature (IUB) for Sialidase(s) and Endosialidase. Biochem. J. 1991, 278, 311–312. [Google Scholar] [CrossRef]
- Haxho, F.; Neufeld, R.J.; Szewczuk, M.R. Neuraminidase-1: A Novel Therapeutic Target in Multistage Tumorigenesis. Oncotarget 2016, 7, 40860–40881. [Google Scholar] [CrossRef]
- Shields, R.L.; Lai, J.; Keck, R.; O’Connell, L.Y.; Hong, K.; Meng, Y.G.; Weikert, S.H.A.; Presta, L.G. Lack of Fucose on Human IgG1 N-Linked Oligosaccharide Improves Binding to Human FcγRIII and Antibody-Dependent Cellular Toxicity. J. Biol. Chem. 2002, 277, 26733–26740. [Google Scholar] [CrossRef]
- Zhu, Z.; Ramakrishnan, B.; Li, J.; Wang, Y.; Feng, Y.; Prabakaran, P.; Colantonio, S.; Dyba, M.A.; Qasba, P.K.; Dimitrov, D.S. Site-Specific Antibody-Drug Conjugation through an Engineered Glycotransferase and a Chemically Reactive Sugar. mAbs 2014, 6, 1190–1200. [Google Scholar] [CrossRef]
- Tang, F.; Wang, L.-X.; Huang, W. Chemoenzymatic Synthesis of Glycoengineered IgG Antibodies and Glycosite-Specific Antibody–Drug Conjugates. Nat. Protoc. 2017, 12, 1702–1721. [Google Scholar] [CrossRef]
- Li, T.; DiLillo, D.J.; Bournazos, S.; Giddens, J.P.; Ravetch, J.V.; Wang, L.-X. Modulating IgG Effector Function by Fc Glycan Engineering. Proc. Natl. Acad. Sci. USA 2017, 114, 3485–3490. [Google Scholar] [CrossRef]
- Li, T.; Tong, X.; Yang, Q.; Giddens, J.P.; Wang, L.-X. Glycosynthase Mutants of Endoglycosidase S2 Show Potent Transglycosylation Activity and Remarkably Relaxed Substrate Specificity for Antibody Glycosylation Remodeling. J. Biol. Chem. 2016, 291, 16508–16518. [Google Scholar] [CrossRef]
- Fairbanks, A.J. Synthetic and Semi-Synthetic Approaches to Unprotected N -Glycan Oxazolines. Beilstein J. Org. Chem. 2018, 14, 416–429. [Google Scholar] [CrossRef]
- Tong, X.; Li, T.; Orwenyo, J.; Toonstra, C.; Wang, L.-X. One-Pot Enzymatic Glycan Remodeling of a Therapeutic Monoclonal Antibody by Endoglycosidase S (Endo-S) from Streptococcus Pyogenes. Bioorg. Med. Chem. 2018, 26, 1347–1355. [Google Scholar] [CrossRef]
- Giddens, J.P.; Lomino, J.V.; DiLillo, D.J.; Ravetch, J.V.; Wang, L.-X. Site-Selective Chemoenzymatic Glycoengineering of Fab and Fc Glycans of a Therapeutic Antibody. Proc. Natl. Acad. Sci. USA 2018, 115, 12023–12027. [Google Scholar] [CrossRef]
- Faridoon, F.; Shi, W.; Qin, K.; Tang, Y.; Li, M.; Guan, D.; Tian, X.; Jiang, B.; Dong, J.; Tang, F.; et al. New Linker Structures Applied in Glycosite-Specific Antibody Drug Conjugates. Org. Chem. Front. 2019, 6, 3144–3149. [Google Scholar] [CrossRef]
- Kadowaki, S.; Yamamoto, K.; Fujisaki, M.; Izumi, K.; Tochikura, T.; Yokoyama, T. Purification and Characterization of a Novel Fungal Endo-β-N-Acetylglucosaminidase Acting on Complex Oligosaccharides of Glycoproteins. Agric. Biol. Chem. 1990, 54, 97–106. [Google Scholar] [CrossRef]
- Yamamoto, K.J.; Kadowaki, S.; Watanabe, J.; Kumagai, H. Transglycosylation Activity of Mucor Hiemalis Endo-β-N-Acetylglucosaminidase Which Transfers Complex Oligosaccharides to the N-acetylglucosamine Moieties of Peptides. Biochem. Biophys. Res. Commun. 1994, 203, 244–252. [Google Scholar] [CrossRef]
- Haneda, K.; Inazu, T.; Yamamoto, K.; Kumagai, H.; Nakahara, Y.; Kobata, A. Transglycosylation of Intact Sialo Complex-Type Oligosaccharides to the N-acetylglucosamine Moieties of Glycopeptides by Mucor Hiemalis Endo-β-N-Acetylglucosaminidase. Carbohydr. Res. 1996, 292, 61–70. [Google Scholar] [CrossRef]
- Mizuno, M.; Muramoto, I.; Kawakami, T.; Seike, M.; Aimoto, S.; Haneda, K.; Inazu, T. A Synthesis of a Glycopeptide Analogue of Eel Calcitonin. Tetrahedron Lett. 1998, 39, 55–58. [Google Scholar] [CrossRef]
- Yamamoto, K. Chemo-Enzymatic Synthesis of Bioactive Glycopeptide Using Microbial Endoglycosidase. J. Biosci. Bioeng. 2001, 92, 493–501. [Google Scholar] [CrossRef]
- Manabe, S. Attempts to Synthesize Homogeneous Glycan-Conjugated Antibody-Drug Conjugates. Transl. Regul. Sci. 2020, 2, 84–89. [Google Scholar] [CrossRef]
- Le, N.P.L.; Bowden, T.A.; Struwe, W.B.; Crispin, M. Immune Recruitment or Suppression by Glycan Engineering of Endogenous and Therapeutic Antibodies. Biochim. Biophys. Acta 2016, 1860, 1655–1668. [Google Scholar] [CrossRef]
- Griffin, M.E.; Hsieh-Wilson, L.C. Glycan Engineering for Cell and Developmental Biology. Cell Chem. Biol. 2016, 23, 108–121. [Google Scholar] [CrossRef]
- Wang, L.-X.; Lomino, J.V. Emerging Technologies for Making Glycan-Defined Glycoproteins. ACS Chem. Biol. 2012, 7, 110–122. [Google Scholar] [CrossRef]
- Basu, K.; Green, E.M.; Cheng, Y.; Craik, C.S. Why Recombinant Antibodies—Benefits and Applications. Curr. Opin. Biotechnol. 2019, 60, 153–158. [Google Scholar] [CrossRef]
- De St. Groth, S.F.; Scheidegger, D. Production of Monoclonal Antibodies: Strategy and Tactics. J. Immunol. Meth. 1980, 35, 1–21. [Google Scholar] [CrossRef]
- Alejandra, W.-P.; Miriam Irene, J.-P.; Fabio Antonio, G.-S.; Patricia, R.-G.R.; Elizabeth, T.-A.; Aleman-Aguilar, J.P.; Rebeca, G.-V. Production of Monoclonal Antibodies for Therapeutic Purposes: A Review. Int. Immunopharmacol. 2023, 120, 110376. [Google Scholar] [CrossRef]
- Bjerrum, K.B.; Aagaard, J.B.; Soucy, J.A.; Kabiljagic, A.A.; Skjoedt, K.; Graversen, J.H.; Henriksen, M.L.; Hansen, S.W.K. Facile Generation of Monoclonal Antibodies Suitable for Conjugation. J. Immunol. Meth. 2020, 483, 112807. [Google Scholar] [CrossRef]
- Kumar, R.; Parray, H.A.; Shrivastava, T.; Sinha, S.; Luthra, K. Phage Display Antibody Libraries: A Robust Approach for Generation of Recombinant Human Monoclonal Antibodies. Int. J. Biol. Macromol. 2019, 135, 907–918. [Google Scholar] [CrossRef]
- Hentrich, C.; Ylera, F.; Frisch, C.; Ten Haaf, A.; Knappik, A. Monoclonal Antibody Generation by Phage Display. In Handbook of Immunoassay Technologies; Elsevier: Amsterdam, The Netherlands, 2018; pp. 47–80. ISBN 978-0-12-811762-0. [Google Scholar]
- Lembke, W.; Locher, M. Monoclonal Antibodies: Discovery and Protein Engineering. In Regulatory Toxicology; Reichl, F.-X., Schwenk, M., Eds.; Springer International Publishing: Cham, Switzerland, 2021; pp. 83–98. ISBN 978-3-030-57498-7. [Google Scholar] [CrossRef]
- Klein, C.; Schaefer, W.; Regula, J.T.; Dumontet, C.; Brinkmann, U.; Bacac, M.; Umaña, P. Engineering Therapeutic Bispecific Antibodies Using CrossMab Technology. Methods 2019, 154, 21–31. [Google Scholar] [CrossRef]
- Li, H.; Er Saw, P.; Song, E. Challenges and Strategies for Next-Generation Bispecific Antibody-Based Antitumor Therapeutics. Cell Mol. Immunol. 2020, 17, 451–461. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, Y.; Park, J.; Liu, X.; Hu, Y.; Wang, T.; McFarland, K.; Betenbaugh, M.J. Design and Production of Bispecific Antibodies. Antibodies 2019, 8, 43. [Google Scholar] [CrossRef]
- Muyldermans, S. A Guide to: Generation and Design of Nanobodies. FEBS J. 2021, 288, 2084–2102. [Google Scholar] [CrossRef]
- De Marco, A. Recombinant Expression of Nanobodies and Nanobody-Derived Immunoreagents. Protein Expr. Purif. 2020, 172, 105645. [Google Scholar] [CrossRef]
- Todorovska, A.; Roovers, R.C.; Dolezal, O.; Kortt, A.A.; Hoogenboom, H.R.; Hudson, P.J. Design and Application of Diabodies, Triabodies and Tetrabodies for Cancer Targeting. J. Immunol. Meth. 2001, 248, 47–66. [Google Scholar] [CrossRef]
- Vaks, L.; Benhar, I. Production of Stabilized scFv Antibody Fragments in the E. coli Bacterial Cytoplasm. In Human Monoclonal Antibodies; Steinitz, M., Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2014; Volume 1060, pp. 171–184. ISBN 978-1-62703-585-9. [Google Scholar] [CrossRef]
- Felices, M.; Lenvik, T.R.; Davis, Z.B.; Miller, J.S.; Vallera, D.A. Generation of BiKEs and TriKEs to Improve NK Cell-Mediated Targeting of Tumor Cells. In Natural Killer Cells; Somanchi, S.S., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2016; Volume 1441, pp. 333–346. ISBN 978-1-4939-3682-3. [Google Scholar] [CrossRef]
- Lyons, A.; King, D.J.; Owens, R.J.; Yarranton, G.T.; Millican, A.; Whittle, N.R.; Adair, J.R. Site-Specific Attachment to Recombinant Antibodies via Introduced Surface Cysteine Residues. Protein Eng. Des. Sel. 1990, 3, 703–708. [Google Scholar] [CrossRef]
- Tumey, L.N.; Li, F.; Rago, B.; Han, X.; Loganzo, F.; Musto, S.; Graziani, E.I.; Puthenveetil, S.; Casavant, J.; Marquette, K.; et al. Site Selection: A Case Study in the Identification of Optimal Cysteine Engineered Antibody Drug Conjugates. AAPS J. 2017, 19, 1123–1135. [Google Scholar] [CrossRef]
- Hofer, T.; Skeffington, L.R.; Chapman, C.M.; Rader, C. Molecularly Defined Antibody Conjugation through a Selenocysteine Interface. Biochemistry 2009, 48, 12047–12057. [Google Scholar] [CrossRef]
- Thompson, P.; Bezabeh, B.; Fleming, R.; Pruitt, M.; Mao, S.; Strout, P.; Chen, C.; Cho, S.; Zhong, H.; Wu, H.; et al. Hydrolytically Stable Site-Specific Conjugation at the N-Terminus of an Engineered Antibody. Bioconjug. Chem. 2015, 26, 2085–2096. [Google Scholar] [CrossRef]
- Li, X.; Nelson, C.G.; Nair, R.R.; Hazlehurst, L.; Moroni, T.; Martinez-Acedo, P.; Nanna, A.R.; Hymel, D.; Burke, T.R.; Rader, C. Stable and Potent Selenomab-Drug Conjugates. Cell Chem. Biol. 2017, 24, 433–442.e6. [Google Scholar] [CrossRef]
- Deeks, E.D. Polatuzumab Vedotin: First Global Approval. Drugs 2019, 79, 1467–1475. [Google Scholar] [CrossRef]
- Lehar, S.M.; Pillow, T.; Xu, M.; Staben, L.; Kajihara, K.K.; Vandlen, R.; DePalatis, L.; Raab, H.; Hazenbos, W.L.; Hiroshi Morisaki, J.; et al. Novel Antibody–Antibiotic Conjugate Eliminates Intracellular S. aureus. Nature 2015, 527, 323–328. [Google Scholar] [CrossRef]
- Zhou, C.; Lehar, S.; Gutierrez, J.; Rosenberger, C.M.; Ljumanovic, N.; Dinoso, J.; Koppada, N.; Hong, K.; Baruch, A.; Carrasco-Triguero, M.; et al. Pharmacokinetics and Pharmacodynamics of DSTA4637A: A Novel THIOMABTM Antibody Antibiotic Conjugate against Staphylococcus aureus in Mice. mAbs 2016, 8, 1612–1619. [Google Scholar] [CrossRef]
- Deng, R.; Zhou, C.; Li, D.; Cai, H.; Sukumaran, S.; Carrasco-Triguero, M.; Saad, O.; Nazzal, D.; Lowe, C.; Ramanujan, S.; et al. Preclinical and Translational Pharmacokinetics of a Novel THIOMABTM Antibody-Antibiotic Conjugate against Staphylococcus aureus. mAbs 2019, 11, 1162–1174. [Google Scholar] [CrossRef]
- Lim, J.; Lewin-Koh, N.; Chu, T.; Rymut, S.M.; Berhanu, A.; Carrasco-Triguero, M.; Rosenberger, C.C.; Hazenbos, W.L.; Miller, L.G.; Fowler, V.G.; et al. 167. A Phase 1b, Randomized, Double-Blind, Placebo-Controlled, Multiple-Ascending Dose Study to Investigate the Safety, Tolerability, and Pharmacokinetics of DSTA4637S in Patients with Staphylococcus Aureus Bacteremia Receiving Standard-of-Care Antibiotics. Open Forum Infect. Dis. 2020, 7, S213. [Google Scholar] [CrossRef]
- Kumar, A.; Kinneer, K.; Masterson, L.; Ezeadi, E.; Howard, P.; Wu, H.; Gao, C.; Dimasi, N. Synthesis of a Heterotrifunctional Linker for the Site-Specific Preparation of Antibody-Drug Conjugates with Two Distinct Warheads. Bioorg. Med. Chem. Lett. 2018, 28, 3617–3621. [Google Scholar] [CrossRef]
- Department of Medicinal Chemistry, University of Utah; Nervig, C.S.; Owen, S.C.; Department of Molecular Pharmaceutics, University of Utah; Department of Biomedical Engineering, University of Utah. Advances in the Development of Dual-Drug Antibody Drug Conjugates. J. Antib. Drug Conjug. 2023. [Google Scholar] [CrossRef]
- Peng, Q.; Zang, B.; Zhao, W.; Li, D.; Ren, J.; Ji, F.; Jia, L. Efficient Continuous-Flow Aldehyde Tag Conversion Using Immobilized Formylglycine Generating Enzyme. Catal. Sci. Technol. 2020, 10, 484–492. [Google Scholar] [CrossRef]
- Drake, P.M.; Albers, A.E.; Baker, J.; Banas, S.; Barfield, R.M.; Bhat, A.S.; De Hart, G.W.; Garofalo, A.W.; Holder, P.; Jones, L.C.; et al. Aldehyde Tag Coupled with HIPS Chemistry Enables the Production of ADCs Conjugated Site-Specifically to Different Antibody Regions with Distinct In Vivo Efficacy and PK Outcomes. Bioconjug. Chem. 2014, 25, 1331–1341. [Google Scholar] [CrossRef]
- Pomplun, S.; Mohamed, M.Y.H.; Oelschlaegel, T.; Wellner, C.; Bergmann, F. Efficient Pictet–Spengler Bioconjugation with N-Substituted Pyrrolyl Alanine Derivatives. Angew. Chem. Int. Ed. 2019, 58, 3542–3547. [Google Scholar] [CrossRef]
- Dickgiesser, S.; Rasche, N.; Nasu, D.; Middel, S.; Hörner, S.; Avrutina, O.; Diederichsen, U.; Kolmar, H. Self-Assembled Hybrid Aptamer-Fc Conjugates for Targeted Delivery: A Modular Chemoenzymatic Approach. ACS Chem. Biol. 2015, 10, 2158–2165. [Google Scholar] [CrossRef]
- Smith, E.L.; Giddens, J.P.; Iavarone, A.T.; Godula, K.; Wang, L.-X.; Bertozzi, C.R. Chemoenzymatic Fc Glycosylation via Engineered Aldehyde Tags. Bioconjug. Chem. 2014, 25, 788–795. [Google Scholar] [CrossRef]
- Dahlgren, G.; Hodsdon, J.M. The Kinetics of the Periodate Oxidation of 2-Aminoethanol. J. Phys. Chem. 1964, 68, 416–418. [Google Scholar] [CrossRef]
- Ta, H.T.; Peter, K.; Hagemeyer, C.E. Enzymatic Antibody Tagging: Toward a Universal Biocompatible Targeting Tool. Tr. Cardiovasc. Med. 2012, 22, 105–111. [Google Scholar] [CrossRef]
- Min, B.; Jin, J.; Kim, H.; Her, N.-G.; Park, C.; Kim, D.; Yang, J.; Hwang, J.; Kim, E.; Choi, M.; et al. cIRCR201-dPBD, a Novel Pyrrolobenzodiazepine Dimer-Containing Site-Specific Antibody–Drug Conjugate Targeting c-Met Overexpression Tumors. ACS Omega 2020, 5, 25798–25809. [Google Scholar] [CrossRef]
- Shin, S.H.; Park, Y.; Park, S.S.; Ju, E.J.; Park, J.; Ko, E.J.; Bae, D.J.; Kim, S.; Chung, C.; Song, H.Y.; et al. An Elaborate New Linker System Significantly Enhances the Efficacy of an HER2-Antibody-Drug Conjugate against Refractory HER2-Positive Cancers. Adv. Sci. 2021, 8, 2102414. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Y.; Uslu, S.; Venkatachalapathy, S.; Rashidian, M.; Schaefer, J.V.; Plückthun, A.; Distefano, M.D. Enzymatic Construction of DARPin-Based Targeted Delivery Systems Using Protein Farnesyltransferase and a Capture and Release Strategy. Int. J. Mol. Sci. 2022, 23, 11537. [Google Scholar] [CrossRef]
- Mortensen, M.R.; Skovsgaard, M.B.; Märcher, A.; Andersen, V.L.; Palmfeldt, J.; Nielsen, T.B.; Tørring, T.; Laursen, N.S.; Andersen, K.R.; Kjems, J.; et al. Introduction of an Aldehyde Handle on Nanobodies by Affinity-Guided Labeling. Bioconjug. Chem. 2020, 31, 1295–1300. [Google Scholar] [CrossRef]
- Wals, K.; Ovaa, H. Unnatural Amino Acid Incorporation in E. coli: Current and Future Applications in the Design of Therapeutic Proteins. Front. Chem. 2014, 2, 15. [Google Scholar] [CrossRef]
- Hallam, T.J.; Smider, V.V. Unnatural Amino Acids in Novel Antibody Conjugates. Future Med. Chem. 2014, 6, 1309–1324. [Google Scholar] [CrossRef]
- Young, D.D.; Schultz, P.G. Playing with the Molecules of Life. ACS Chem. Biol. 2018, 13, 854–870. [Google Scholar] [CrossRef]
- Evans, E.G.B.; Millhauser, G.L. Genetic Incorporation of the Unnatural Amino Acid P-Acetyl Phenylalanine into Proteins for Site-Directed Spin Labeling. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2015; Volume 563, pp. 503–527. ISBN 978-0-12-802834-6. [Google Scholar] [CrossRef]
- Tharp, J.M.; Vargas-Rodriguez, O.; Schepartz, A.; Söll, D. Genetic Encoding of Three Distinct Noncanonical Amino Acids Using Reprogrammed Initiator and Nonsense Codons. ACS Chem. Biol. 2021, 16, 766–774. [Google Scholar] [CrossRef]
- Palei, S.; Mootz, H.D. Cyclic Peptides Made by Linking Synthetic and Genetically Encoded Fragments. ChemBioChem 2016, 17, 378–382. [Google Scholar] [CrossRef]
- Tian, F.; Lu, Y.; Manibusan, A.; Sellers, A.; Tran, H.; Sun, Y.; Phuong, T.; Barnett, R.; Hehli, B.; Song, F.; et al. A General Approach to Site-Specific Antibody Drug Conjugates. Proc. Natl. Acad. Sci. USA 2014, 111, 1766–1771. [Google Scholar] [CrossRef]
- Costa, S.A.; Mozhdehi, D.; Dzuricky, M.J.; Isaacs, F.J.; Brustad, E.M.; Chilkoti, A. Active Targeting of Cancer Cells by Nanobody Decorated Polypeptide Micelle with Bio-Orthogonally Conjugated Drug. Nano Lett. 2019, 19, 247–254. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, Z.; Wang, Z.; Luo, F.; Guan, M.; Xu, M.; Li, Y.; Zhang, Y.; Wang, Z.; Wang, W. A Simple and Efficient Method to Generate Dual Site-Specific Conjugation ADCs with Cysteine Residue and an Unnatural Amino Acid. Bioconjug. Chem. 2021, 32, 1094–1104. [Google Scholar] [CrossRef]
- Cornish, V.W.; Hahn, K.M.; Schultz, P.G. Site-Specific Protein Modification Using a Ketone Handle. J. Am. Chem. Soc. 1996, 118, 8150–8151. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, Z.; Brock, A.; Schultz, P.G. Addition of the Keto Functional Group to the Genetic Code of Escherichia coli. Proc. Natl. Acad. Sci. USA 2003, 100, 56–61. [Google Scholar] [CrossRef]
- Zhang, Z.; Smith, B.A.C.; Wang, L.; Brock, A.; Cho, C.; Schultz, P.G. A New Strategy for the Site-Specific Modification of Proteins in Vivo. Biochemistry 2003, 42, 6735–6746. [Google Scholar] [CrossRef]
- Zeng, H.; Xie, J.; Schultz, P.G. Genetic Introduction of a Diketone-Containing Amino Acid into Proteins. Bioorg. Med. Chem. Lett. 2006, 16, 5356–5359. [Google Scholar] [CrossRef]
- Huang, Y.; Wan, W.; Russell, W.K.; Pai, P.-J.; Wang, Z.; Russell, D.H.; Liu, W. Genetic Incorporation of an Aliphatic Keto-Containing Amino Acid into Proteins for Their Site-Specific Modifications. Bioorg. Med. Chem. Lett. 2010, 20, 878–880. [Google Scholar] [CrossRef]
- Kazane, S.A.; Sok, D.; Cho, E.H.; Uson, M.L.; Kuhn, P.; Schultz, P.G.; Smider, V.V. Site-Specific DNA-Antibody Conjugates for Specific and Sensitive Immuno-PCR. Proc. Natl. Acad. Sci. USA 2012, 109, 3731–3736. [Google Scholar] [CrossRef]
- Kazane, S.A.; Axup, J.Y.; Kim, C.H.; Ciobanu, M.; Wold, E.D.; Barluenga, S.; Hutchins, B.A.; Schultz, P.G.; Winssinger, N.; Smider, V.V. Self-Assembled Antibody Multimers through Peptide Nucleic Acid Conjugation. J. Am. Chem. Soc. 2013, 135, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Wold, E.D.; McBride, R.; Axup, J.Y.; Kazane, S.A.; Smider, V.V. Antibody Microarrays Utilizing Site-Specific Antibody–Oligonucleotide Conjugates. Bioconjugate Chem. 2015, 26, 807–811. [Google Scholar] [CrossRef]
- Axup, J.Y.; Bajjuri, K.M.; Ritland, M.; Hutchins, B.M.; Kim, C.H.; Kazane, S.A.; Halder, R.; Forsyth, J.S.; Santidrian, A.F.; Stafin, K.; et al. Synthesis of Site-Specific Antibody-Drug Conjugates Using Unnatural Amino Acids. Proc. Natl. Acad. Sci. USA 2012, 109, 16101–16106. [Google Scholar] [CrossRef]
- Kim, C.H.; Axup, J.Y.; Lawson, B.R.; Yun, H.; Tardif, V.; Choi, S.H.; Zhou, Q.; Dubrovska, A.; Biroc, S.L.; Marsden, R.; et al. Bispecific Small Molecule–Antibody Conjugate Targeting Prostate Cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 17796–17801. [Google Scholar] [CrossRef]
- Kularatne, S.A.; Deshmukh, V.; Ma, J.; Tardif, V.; Lim, R.K.V.; Pugh, H.M.; Sun, Y.; Manibusan, A.; Sellers, A.J.; Barnett, R.S.; et al. A CXCR4-Targeted Site-Specific Antibody–Drug Conjugate. Angew. Chem. Int. Ed. 2014, 53, 11863–11867. [Google Scholar] [CrossRef]
- Kim, C.H.; Axup, J.Y.; Dubrovska, A.; Kazane, S.A.; Hutchins, B.A.; Wold, E.D.; Smider, V.V.; Schultz, P.G. Synthesis of Bispecific Antibodies Using Genetically Encoded Unnatural Amino Acids. J. Am. Chem. Soc. 2012, 134, 9918–9921. [Google Scholar] [CrossRef]
- Hutchins, B.M.; Kazane, S.A.; Staflin, K.; Forsyth, J.S.; Felding-Habermann, B.; Schultz, P.G.; Smider, V.V. Site-Specific Coupling and Sterically Controlled Formation of Multimeric Antibody Fab Fragments with Unnatural Amino Acids. J. Mol. Biol. 2011, 406, 595–603. [Google Scholar] [CrossRef]
- Agarwal, P.; Van Der Weijden, J.; Sletten, E.M.; Rabuka, D.; Bertozzi, C.R. A Pictet-Spengler Ligation for Protein Chemical Modification. Proc. Natl. Acad. Sci. USA 2013, 110, 46–51. [Google Scholar] [CrossRef]
- Kudirka, R.A.; Barfield, R.M.; McFarland, J.M.; Drake, P.M.; Carlson, A.; Bañas, S.; Zmolek, W.; Garofalo, A.W.; Rabuka, D. Site-Specific Tandem Knoevenagel Condensation–Michael Addition To Generate Antibody–Drug Conjugates. ACS Med. Chem. Lett. 2016, 7, 994–998. [Google Scholar] [CrossRef]
- Kudirka, R.; Barfield, R.M.; McFarland, J.; Albers, A.E.; de Hart, G.W.; Drake, P.M.; Holder, P.G.; Banas, S.; Jones, L.C.; Garofalo, A.W.; et al. Generating Site-Specifically Modified Proteins via a Versatile and Stable Nucleophilic Carbon Ligation. Chem. Biol. 2015, 22, 293–298. [Google Scholar] [CrossRef]
- Melnyk, O.; Fehrentz, J.-A.; Martinez, J.; Gras-Masse, H. Functionalization of Peptides and Proteins by Aldehyde or Keto Groups. Pept. Sci. 2000, 55, 165–186. [Google Scholar] [CrossRef]
- El-Mahdi, O.; Melnyk, O. α-Oxo Aldehyde or Glyoxylyl Group Chemistry in Peptide Bioconjugation. Bioconjug. Chem. 2013, 24, 735–765. [Google Scholar] [CrossRef] [PubMed]
- Agten, S.M.; Dawson, P.E.; Hackeng, T.M. Oxime Conjugation in Protein Chemistry: From Carbonyl Incorporation to Nucleophilic Catalysis. J. Pept. Sci. 2016, 22, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Kalia, J.; Raines, R.T. Hydrolytic Stability of Hydrazones and Oximes. Angew. Chem. Int. Ed. 2008, 47, 7523–7526. [Google Scholar] [CrossRef]
- Dong, X.; Obermeyer, A.C.; Olsen, B.D. Three-Dimensional Ordered Antibody Arrays Through Self-Assembly of Antibody–Polymer Conjugates. Angew. Chem. Int. Ed. 2017, 56, 1273–1277. [Google Scholar] [CrossRef] [PubMed]
- Lyu, Z.; Kang, L.; Buuh, Z.Y.; Jiang, D.; McGuth, J.C.; Du, J.; Wissler, H.L.; Cai, W.; Wang, R.E. A Switchable Site-Specific Antibody Conjugate. ACS Chem. Biol. 2018, 13, 958–964. [Google Scholar] [CrossRef] [PubMed]
- Sapozhnikova, K.A.; Misyurin, V.A.; Ryazantsev, D.Y.; Kokin, E.A.; Finashutina, Y.P.; Alexeeva, A.V.; Ivanov, I.A.; Kocharovskaya, M.V.; Tikhonova, N.A.; Popova, G.P.; et al. Sensitive Immunofluorescent Detection of the PRAME Antigen Using a Practical Antibody Conjugation Approach. Int. J. Mol. Sci. 2021, 22, 12845. [Google Scholar] [CrossRef] [PubMed]
- Snyder, J.T.; Malinao, M.-C.; Dugal-Tessier, J.; Atkinson, J.E.; Anand, B.S.; Okada, A.; Mendelsohn, B.A. Metabolism of an Oxime-Linked Antibody Drug Conjugate, AGS62P1, and Characterization of Its Identified Metabolite. Mol. Pharm. 2018, 15, 2384–2390. [Google Scholar] [CrossRef]
- Sapozhnikova, K.A.; Gulyak, E.L.; Brylev, V.A.; Misyurin, V.A.; Oreshkov, S.D.; Alexeeva, A.V.; Ryazantsev, D.Y.; Simonova, M.A.; Ryabukhina, E.V.; Popova, G.P.; et al. Aminooxy Click Modification of a Periodate-Oxidized Immunoglobulin G: A General Approach to Antibody–Drug Conjugates with Dye-Mediated Expeditious Stoichiometry Control. Int. J. Mol. Sci. 2023, 24, 5134. [Google Scholar] [CrossRef]
- Kularatne, S.A.; Deshmukh, V.; Gymnopoulos, M.; Biroc, S.L.; Xia, J.; Srinagesh, S.; Sun, Y.; Zou, N.; Shimazu, M.; Pinkstaff, J.; et al. Recruiting Cytotoxic T Cells to Folate-Receptor-Positive Cancer Cells. Angew. Chem. Int. Ed. 2013, 52, 12101–12104. [Google Scholar] [CrossRef]
- Lim, R.K.V.; Yu, S.; Cheng, B.; Li, S.; Kim, N.-J.; Cao, Y.; Chi, V.; Kim, J.Y.; Chatterjee, A.K.; Schultz, P.G.; et al. Targeted Delivery of LXR Agonist Using a Site-Specific Antibody–Drug Conjugate. Bioconjug. Chem. 2015, 26, 2216–2222. [Google Scholar] [CrossRef]
- Dovgan, I.; Koniev, O.; Kolodych, S.; Wagner, A. Antibody–Oligonucleotide Conjugates as Therapeutic, Imaging, and Detection Agents. Bioconjug. Chem. 2019, 30, 2483–2501. [Google Scholar] [CrossRef]
- Zhang, J.; Ji, D.; Shen, W.; Xiao, Q.; Gu, Y.; O’Shaughnessy, J.; Hu, X. Phase I Trial of a Novel Anti-HER2 Antibody–Drug Conjugate, ARX788, for the Treatment of HER2-Positive Metastatic Breast Cancer. Clin. Cancer Res. 2022, 28, 4212–4221. [Google Scholar] [CrossRef]
- Skidmore, L.; Mills, D.; Kim, J.Y.; Shastri, P.; Knudsen, N.A.; Steen, J.; Nelson, J.; Buechler, Y.; Tian, F.; Zhang, S. Abstract 3997: Preclinical Characterization of ARX517, a next-Generation Anti-PSMA Antibody Drug Conjugate for the Treatment of Metastatic Castration-Resistant Prostate Cancer. Cancer Res. 2023, 83, 3997. [Google Scholar] [CrossRef]
- Zammarchi, F.; Havenith, K.E.; Chivers, S.; Hogg, P.; Bertelli, F.; Tyrer, P.; Janghra, N.; Reinert, H.W.; Hartley, J.A.; Van Berkel, P.H. Preclinical Development of ADCT-601, a Novel Pyrrolobenzodiazepine Dimer-Based Antibody–Drug Conjugate Targeting AXL-Expressing Cancers. Mol. Cancer Ther. 2022, 21, 582–593. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gulyak, E.L.; Alferova, V.A.; Korshun, V.A.; Sapozhnikova, K.A. Introduction of Carbonyl Groups into Antibodies. Molecules 2023, 28, 7890. https://doi.org/10.3390/molecules28237890
Gulyak EL, Alferova VA, Korshun VA, Sapozhnikova KA. Introduction of Carbonyl Groups into Antibodies. Molecules. 2023; 28(23):7890. https://doi.org/10.3390/molecules28237890
Chicago/Turabian StyleGulyak, Evgeny L., Vera A. Alferova, Vladimir A. Korshun, and Ksenia A. Sapozhnikova. 2023. "Introduction of Carbonyl Groups into Antibodies" Molecules 28, no. 23: 7890. https://doi.org/10.3390/molecules28237890
APA StyleGulyak, E. L., Alferova, V. A., Korshun, V. A., & Sapozhnikova, K. A. (2023). Introduction of Carbonyl Groups into Antibodies. Molecules, 28(23), 7890. https://doi.org/10.3390/molecules28237890