
Gasdermin D-Mediated Pyroptosis in Diabetic Cardiomyopathy: Molecular Mechanisms and Pharmacological Implications


Abstract
:
1. Introduction
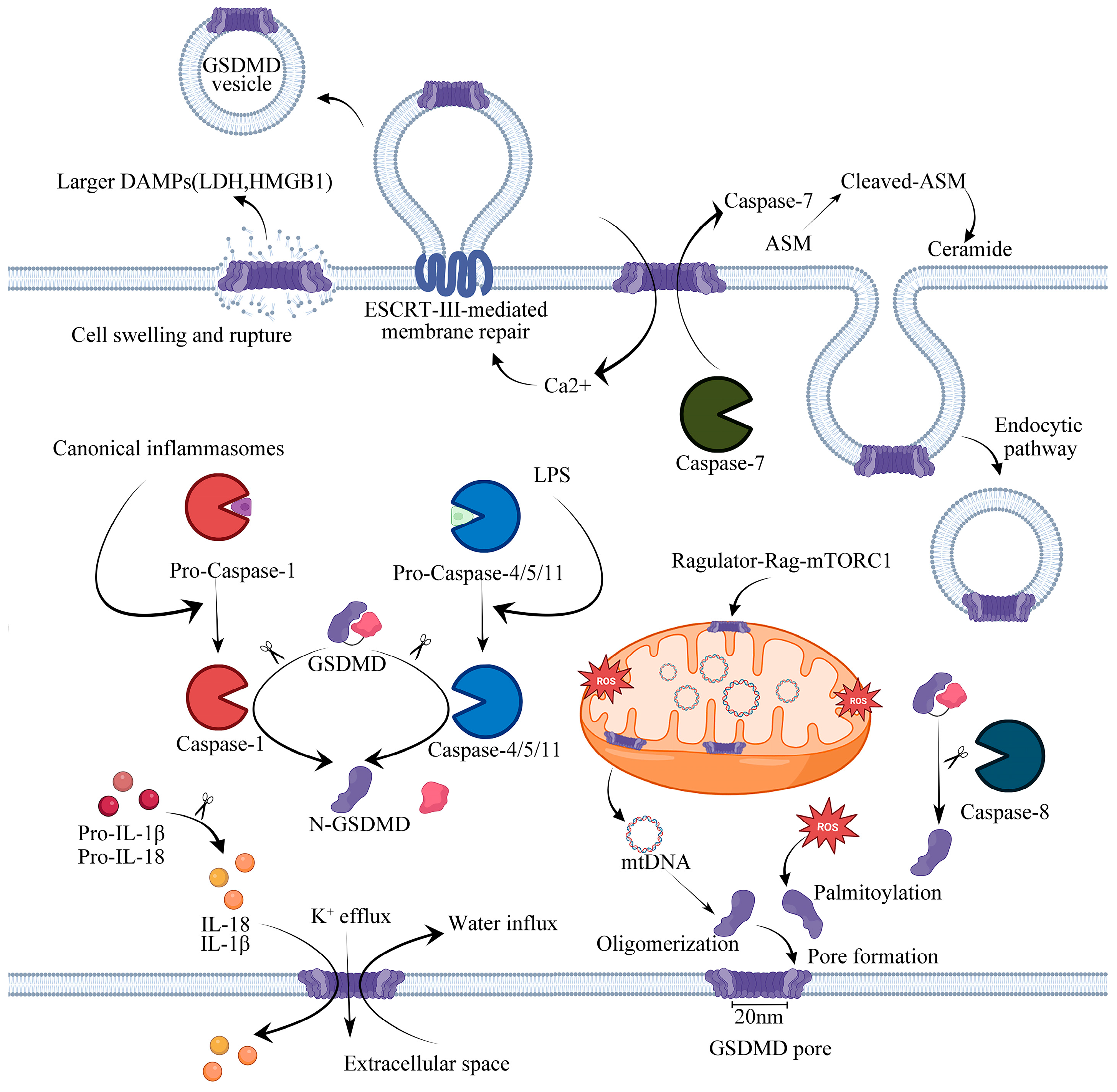
2. Pyroptosis
3. GSDMD and the Mechanism of Pore Formation
4. Inflammasome-Mediated GSDMD Activation Pathways
4.1. Canonical Pathway
4.2. Noncanonical Pathway
4.3. Other Dependent Pathway
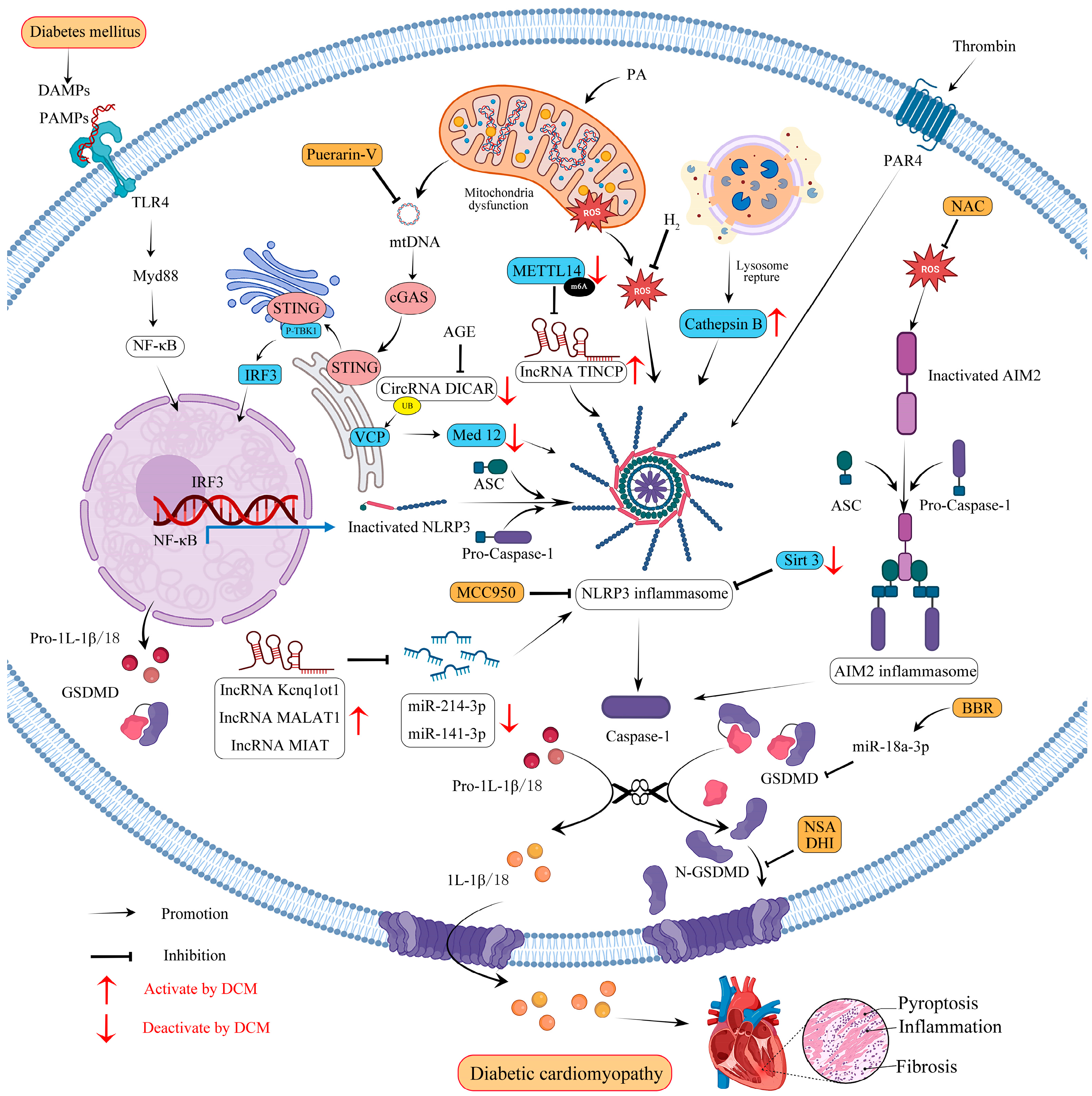
5. Molecular Mechanisms of GSDMD-Mediated Pyroptosis in Diabetic Cardiomyopathy
5.1. NLRP3 and ROS Crosstalk
5.2. AIM2 and ROS Crosstalk
5.3. mtDNA
5.4. CircRNAs
5.5. lncRNAs
6. Therapeutic Potential of GSDMD in DCM
6.1. Clinical Drugs
6.2. GSDMD Inhibitors
6.3. Herbal Medicine and Monomers
7. Summary and Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Cornier, M.-A.; Dabelea, D.; Hernandez, T.L.; Lindstrom, R.C.; Steig, A.J.; Stob, N.R.; Van Pelt, R.E.; Wang, H.; Eckel, R.H. The metabolic syndrome. Endocr. Rev. 2008, 29, 777–822. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Hill, M.A.; Sowers, J.R. Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity. Circ. Res. 2018, 122, 624–638. [Google Scholar] [CrossRef] [PubMed]
- Dillmann, W.H. Diabetic Cardiomyopathy. Circ. Res. 2019, 124, 1160–1162. [Google Scholar] [CrossRef] [PubMed]
- Frustaci, A.; Kajstura, J.; Chimenti, C.; Jakoniuk, I.; Leri, A.; Maseri, A.; Nadal-Ginard, B.; Anversa, P. Myocardial cell death in human diabetes. Circ. Res. 2000, 87, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Zhao, Y.; Liang, H.; Du, W.; Wang, L. Preliminary evidence for the presence of multiple forms of cell death in diabetes cardiomyopathy. Acta Pharm. Sin. B 2022, 12, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Soengas, M.S.; Alarcón, R.M.; Yoshida, H.; Giaccia, A.J.; Hakem, R.; Mak, T.W.; Lowe, S.W. Apaf-1 and caspase-9 in p53-dependent apoptosis and tumor inhibition. Science 1999, 284, 156–159. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Coy, S.; Petrova, B.; Dreishpoon, M.; Verma, A.; Abdusamad, M.; Rossen, J.; Joesch-Cohen, L.; Humeidi, R.; Spangler, R.D.; et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 2022, 375, 1254–1261, Erratum in Science 2022, 376, eabq4855. [Google Scholar] [CrossRef]
- Liu, R.; Duan, T.; Yu, L.; Tang, Y.; Liu, S.; Wang, C.; Fang, W.-J. Acid sphingomyelinase promotes diabetic cardiomyopathy via NADPH oxidase 4 mediated apoptosis. Cardiovasc. Diabetol. 2023, 22, 25. [Google Scholar] [CrossRef] [PubMed]
- Kuethe, F.; Sigusch, H.H.; Bornstein, S.R.; Hilbig, K.; Kamvissi, V.; Figulla, H.R. Apoptosis in patients with dilated cardiomyopathy and diabetes: A feature of diabetic cardiomyopathy? Horm. Metab. Res. 2007, 39, 672–676. [Google Scholar] [CrossRef]
- Li, X.; Li, Z.; Dong, X.; Wu, Y.; Li, B.; Kuang, B.; Chen, G.; Zhang, L. Astragaloside IV attenuates myocardial dysfunction in diabetic cardiomyopathy rats through downregulation of CD36-mediated ferroptosis. Phytother. Res. 2023, 37, 3042–3056. [Google Scholar] [CrossRef] [PubMed]
- Xue, F.; Cheng, J.; Liu, Y.; Cheng, C.; Zhang, M.; Sui, W.; Chen, W.; Hao, P.; Zhang, Y.; Zhang, C. Cardiomyocyte-specific knockout of ADAM17 ameliorates left ventricular remodeling and function in diabetic cardiomyopathy of mice. Signal Transduct. Target. Ther. 2022, 7, 259. [Google Scholar] [CrossRef] [PubMed]
- Zychlinsky, A.; Prevost, M.C.; Sansonetti, P.J. Shigella flexneri induces apoptosis in infected macrophages. Nature 1992, 358, 167–169. [Google Scholar] [CrossRef]
- Cookson, B.T.; Brennan, M.A. Pro-inflammatory programmed cell death. Trends Microbiol. 2001, 9, 113–114. [Google Scholar] [CrossRef]
- Broz, P.; Pelegrín, P.; Shao, F. The gasdermins, a protein family executing cell death and inflammation. Nat. Rev. Immunol. 2020, 20, 143–157. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, Z.; Li, H. Insights into innate immune signalling in controlling cardiac remodelling. Cardiovasc. Res. 2017, 113, 1538–1550. [Google Scholar] [CrossRef]
- Malireddi, R.K.S.; Kesavardhana, S.; Kanneganti, T.-D. ZBP1 and TAK1: Master Regulators of NLRP3 Inflammasome/Pyroptosis, Apoptosis, and Necroptosis (PAN-optosis). Front. Cell. Infect. Microbiol. 2019, 9, 406. [Google Scholar] [CrossRef]
- Hao, Y.; Yang, B.; Yang, J.; Shi, X.; Yang, X.; Zhang, D.; Zhao, D.; Yan, W.; Chen, L.; Zheng, H.; et al. ZBP1: A Powerful Innate Immune Sensor and Double-Edged Sword in Host Immunity. Int. J. Mol. Sci. 2022, 23, 224. [Google Scholar] [CrossRef]
- Kesavardhana, S.; Malireddi, R.K.S.; Burton, A.R.; Porter, S.N.; Vogel, P.; Pruett-Miller, S.M.; Kanneganti, T.-D. The Zα2 domain of ZBP1 is a molecular switch regulating influenza-induced PANoptosis and perinatal lethality during development. J. Biol. Chem. 2020, 295, 8325–8330. [Google Scholar] [CrossRef]
- Zheng, M.; Kanneganti, T.-D. The regulation of the ZBP1-NLRP3 inflammasome and its implications in pyroptosis, apoptosis, and necroptosis (PANoptosis). Immunol. Rev. 2020, 297, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Gitlin, A.D.; Heger, K.; Schubert, A.F.; Reja, R.; Yan, D.; Pham, V.C.; Suto, E.; Zhang, J.; Kwon, Y.C.; Freund, E.C.; et al. Integration of innate immune signalling by caspase-8 cleavage of N4BP1. Nature 2020, 587, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, M.; Günther, S.D.; Schwarzer, R.; Albert, M.-C.; Schorn, F.; Werthenbach, J.P.; Schiffmann, L.M.; Stair, N.; Stocks, H.; Seeger, J.M.; et al. Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature 2019, 575, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Bertheloot, D.; Latz, E.; Franklin, B.S. Necroptosis, pyroptosis and apoptosis: An intricate game of cell death. Cell. Mol. Immunol. 2021, 18, 1106–1121. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Orning, P.; Lien, E.; Fitzgerald, K.A. Gasdermins and their role in immunity and inflammation. J. Exp. Med. 2019, 216, 2453–2465. [Google Scholar] [CrossRef]
- Jorgensen, I.; Miao, E.A. Pyroptotic cell death defends against intracellular pathogens. Immunol. Rev. 2015, 265, 130–142. [Google Scholar] [CrossRef]
- Lee, B.L.; Stowe, I.B.; Gupta, A.; Kornfeld, O.S.; Roose-Girma, M.; Anderson, K.; Warming, S.; Zhang, J.; Lee, W.P.; Kayagaki, N. Caspase-11 auto-proteolysis is crucial for noncanonical inflammasome activation. J. Exp. Med. 2018, 215, 2279–2288. [Google Scholar] [CrossRef]
- Shao, R.; Lou, X.; Xue, J.; Ning, D.; Chen, G.; Jiang, L. Review: The role of GSDMD in sepsis. Inflamm. Res. 2022, 71, 1191–1202. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Zhou, Z.; He, H.; Wang, K.; Shi, X.; Wang, Y.; Su, Y.; Wang, Y.; Li, D.; Liu, W.; Zhang, Y.; et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science 2020, 368, eaaz7548. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Zhao, R.; Xia, W.; Chang, C.-W.; You, Y.; Hsu, J.-M.; Nie, L.; Chen, Y.; Wang, Y.-C.; Liu, C.; et al. PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat. Cell Biol. 2020, 22, 1264–1275. [Google Scholar] [CrossRef]
- Rogers, C.; Fernandes-Alnemri, T.; Mayes, L.; Alnemri, D.; Cingolani, G.; Alnemri, E.S. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat. Commun. 2017, 8, 14128. [Google Scholar] [CrossRef] [PubMed]
- Devant, P.; Kagan, J.C. Molecular mechanisms of gasdermin D pore-forming activity. Nat. Immunol. 2023, 24, 1064–1075. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Liu, W.-C.; Chen, X.-Y.; Wang, X.; Li, J.-L.; Zhang, X. Gasdermin D-mediated pyroptosis: Mechanisms, diseases, and inhibitors. Front. Immunol. 2023, 14, 1178662. [Google Scholar] [CrossRef]
- Xia, S.; Zhang, Z.; Magupalli, V.G.; Pablo, J.L.; Dong, Y.; Vora, S.M.; Wang, L.; Fu, T.-M.; Jacobson, M.P.; Greka, A.; et al. Gasdermin D pore structure reveals preferential release of mature interleukin-1. Nature 2021, 593, 607–611. [Google Scholar] [CrossRef]
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.-C.; Shao, F. Erratum: Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016, 540, 150. [Google Scholar] [CrossRef]
- Wang, J.; Deobald, K.; Re, F. Gasdermin D Protects from Melioidosis through Pyroptosis and Direct Killing of Bacteria. J. Immunol. 2019, 202, 3468–3473. [Google Scholar] [CrossRef]
- Rogers, C.; Alnemri, E.S. Gasdermins: Novel mitochondrial pore-forming proteins. Mol. Cell Oncol. 2019, 6, e1621501. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.S.; Hong, Z.; Wu, W.; Xiong, S.; Zhong, M.; Gao, X.; Rehman, J.; Malik, A.B. mtDNA Activates cGAS Signaling and Suppresses the YAP-Mediated Endothelial Cell Proliferation Program to Promote Inflammatory Injury. Immunity 2020, 52, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Weindel, C.G.; Martinez, E.L.; Zhao, X.; Mabry, C.J.; Bell, S.L.; Vail, K.J.; Coleman, A.K.; VanPortfliet, J.J.; Zhao, B.; Wagner, A.R.; et al. Mitochondrial ROS promotes susceptibility to infection via gasdermin D-mediated necroptosis. Cell 2022, 185, 3214–3231. [Google Scholar] [CrossRef] [PubMed]
- Miao, N.; Wang, Z.; Wang, Q.; Xie, H.; Yang, N.; Wang, Y.; Wang, J.; Kang, H.; Bai, W.; Wang, Y.; et al. Oxidized mitochondrial DNA induces gasdermin D oligomerization in systemic lupus erythematosus. Nat. Commun. 2023, 14, 872. [Google Scholar] [CrossRef]
- Chen, W.; Chen, S.; Yan, C.; Zhang, Y.; Zhang, R.; Chen, M.; Zhong, S.; Fan, W.; Zhu, S.; Zhang, D.; et al. Allergen protease-activated stress granule assembly and gasdermin D fragmentation control interleukin-33 secretion. Nat. Immunol. 2022, 23, 1021–1030. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, R.; Kamachi, F.; Nakamura, M.; Yamazaki, S.; Kamiya, T.; Takasugi, M.; Cheng, Y.; Nonaka, Y.; Yukawa-Muto, Y.; Thuy, L.T.T.; et al. Gasdermin D-mediated release of IL-33 from senescent hepatic stellate cells promotes obesity-associated hepatocellular carcinoma. Sci. Immunol. 2022, 7, eabl7209. [Google Scholar] [CrossRef] [PubMed]
- Du, G.; Healy, L.B.; David, L.; Walker, C.; Fontana, P.; Dong, Y.; Devant, P.; Puthenveetil, R.; Ficarro, S.B.; Banerjee, A.; et al. ROS-dependent palmitoylation is an obligate licensing modification for GSDMD pore formation. bioRxiv 2023, 3, 531538. [Google Scholar] [CrossRef]
- Balasubramanian, A.; Ghimire, L.; Hsu, A.Y.; Kambara, H.; Liu, X.; Hasegawa, T.; Xu, R.; Tahir, M.; Yu, H.; Lieberman, J.; et al. Palmitoylation of gasdermin D directs its membrane translocation and pore formation in pyroptosis. bioRxiv 2023, 1, 529402. [Google Scholar] [CrossRef]
- Wang, W.; Prokopec, J.S.; Zhang, Y.; Sukhoplyasova, M.; Shinglot, H.; Wang, M.-T.; Linkermann, A.; Stewart-Ornstein, J.; Gong, Y.-N. Sensing plasma membrane pore formation induces chemokine production in survivors of regulated necrosis. Dev. Cell 2022, 57, 228–245. [Google Scholar] [CrossRef]
- Rühl, S.; Shkarina, K.; Demarco, B.; Heilig, R.; Santos, J.C.; Broz, P. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science 2018, 362, 956–960. [Google Scholar] [CrossRef]
- Nozaki, K.; Maltez, V.I.; Rayamajhi, M.; Tubbs, A.L.; Mitchell, J.E.; Lacey, C.A.; Harvest, C.K.; Li, L.; Nash, W.T.; Larson, H.N.; et al. Caspase-7 activates ASM to repair gasdermin and perforin pores. Nature 2022, 606, 960–967. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yu, Q.; Jiang, D.; Yu, K.; Yu, W.; Chi, Z.; Chen, S.; Li, M.; Yang, D.; Wang, Z.; et al. Epithelial Gasdermin D shapes the host-microbial interface by driving mucus layer formation. Sci. Immunol. 2022, 7, eabk2092. [Google Scholar] [CrossRef] [PubMed]
- Santa Cruz Garcia, A.B.; Schnur, K.P.; Malik, A.B.; Mo, G.C.H. Gasdermin D pores are dynamically regulated by local phosphoinositide circuitry. Nat. Commun. 2022, 13, 52. [Google Scholar] [CrossRef] [PubMed]
- Bugger, H.; Abel, E.D. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia 2014, 57, 660–671. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Fox, D.; Man, S.M. Mechanisms of Gasdermin Family Members in Inflammasome Signaling and Cell Death. J. Mol. Biol. 2018, 430, 3068–3080. [Google Scholar] [CrossRef]
- Xia, S.; Hollingsworth, L.R.; Wu, H. Mechanism and Regulation of Gasdermin-Mediated Cell Death. Cold Spring Harb. Perspect. Biol. 2020, 12, a036400. [Google Scholar] [CrossRef]
- Chen, K.W.; Monteleone, M.; Boucher, D.; Sollberger, G.; Ramnath, D.; Condon, N.D.; von Pein, J.B.; Broz, P.; Sweet, M.J.; Schroder, K. Noncanonical inflammasome signaling elicits gasdermin D-dependent neutrophil extracellular traps. Sci. Immunol. 2018, 3, eaar6676. [Google Scholar] [CrossRef]
- Fender, A.C.; Kleeschulte, S.; Stolte, S.; Leineweber, K.; Kamler, M.; Bode, J.; Li, N.; Dobrev, D. Thrombin receptor PAR4 drives canonical NLRP3 inflammasome signaling in the heart. Basic Res. Cardiol. 2020, 115, 10. [Google Scholar] [CrossRef]
- Shi, H.; Gao, Y.; Dong, Z.; Yang, J.e.; Gao, R.; Li, X.; Zhang, S.; Ma, L.; Sun, X.; Wang, Z.; et al. GSDMD-Mediated Cardiomyocyte Pyroptosis Promotes Myocardial I/R Injury. Circ. Res. 2021, 129, 383–396. [Google Scholar] [CrossRef]
- Miao, N.; Yin, F.; Xie, H.; Wang, Y.; Xu, Y.; Shen, Y.; Xu, D.; Yin, J.; Wang, B.; Zhou, Z.; et al. The cleavage of gasdermin D by caspase-11 promotes tubular epithelial cell pyroptosis and urinary IL-18 excretion in acute kidney injury. Kidney Int. 2019, 96, 1105–1120. [Google Scholar] [CrossRef]
- Baker, P.J.; Boucher, D.; Bierschenk, D.; Tebartz, C.; Whitney, P.G.; D’Silva, D.B.; Tanzer, M.C.; Monteleone, M.; Robertson, A.A.B.; Cooper, M.A.; et al. NLRP3 inflammasome activation downstream of cytoplasmic LPS recognition by both caspase-4 and caspase-5. Eur. J. Immunol. 2015, 45, 2918–2926. [Google Scholar] [CrossRef] [PubMed]
- Rühl, S.; Broz, P. Caspase-11 activates a canonical NLRP3 inflammasome by promoting K+ efflux. Eur. J. Immunol. 2015, 45, 2927–2936. [Google Scholar] [CrossRef] [PubMed]
- Linder, A.; Hornung, V. Irgm2 and Gate-16 put a break on caspase-11 activation. EMBO Rep. 2020, 21, e51787. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gao, W.; Shi, X.; Ding, J.; Liu, W.; He, H.; Wang, K.; Shao, F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 2017, 547, 99–103. [Google Scholar] [CrossRef]
- Orning, P.; Weng, D.; Starheim, K.; Ratner, D.; Best, Z.; Lee, B.; Brooks, A.; Xia, S.; Wu, H.; Kelliher, M.A.; et al. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science 2018, 362, 1064–1069. [Google Scholar] [CrossRef]
- Kenny, H.C.; Abel, E.D. Heart Failure in Type 2 Diabetes Mellitus. Circ. Res. 2019, 124, 121–141. [Google Scholar] [CrossRef]
- Hölscher, M.E.; Bode, C.; Bugger, H. Diabetic Cardiomyopathy: Does the Type of Diabetes Matter? Int. J. Mol. Sci. 2016, 17, 2136. [Google Scholar] [CrossRef]
- van Heerebeek, L.; Hamdani, N.; Handoko, M.L.; Falcao-Pires, I.; Musters, R.J.; Kupreishvili, K.; Ijsselmuiden, A.J.J.; Schalkwijk, C.G.; Bronzwaer, J.G.F.; Diamant, M.; et al. Diastolic stiffness of the failing diabetic heart: Importance of fibrosis, advanced glycation end products, and myocyte resting tension. Circulation 2008, 117, 43–51. [Google Scholar] [CrossRef]
- Xue, M.; Li, T.; Wang, Y.; Chang, Y.; Cheng, Y.; Lu, Y.; Liu, X.; Xu, L.; Li, X.; Yu, X.; et al. Empagliflozin prevents cardiomyopathy via sGC-cGMP-PKG pathway in type 2 diabetes mice. Clin. Sci. 2019, 133, 1705–1720. [Google Scholar] [CrossRef]
- Yan, M.; Li, Y.; Luo, Q.; Zeng, W.; Shao, X.; Li, L.; Wang, Q.; Wang, D.; Zhang, Y.; Diao, H.; et al. Mitochondrial damage and activation of the cytosolic DNA sensor cGAS-STING pathway lead to cardiac pyroptosis and hypertrophy in diabetic cardiomyopathy mice. Cell Death Discov. 2022, 8, 258. [Google Scholar] [CrossRef]
- He, Y.; Zeng, M.Y.; Yang, D.; Motro, B.; Núñez, G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 2016, 530, 354–357. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Planillo, R.; Kuffa, P.; Martínez-Colón, G.; Smith, B.L.; Rajendiran, T.M.; Núñez, G. K⁺ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Lu, Y.; Meng, J.; Wang, Z. Pyroptosis and Its Regulation in Diabetic Cardiomyopathy. Front. Physiol. 2021, 12, 791848. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.-F.; Zhai, B.-Z.; Hu, W.-L.; Lou, M.-H.; Chen, Y.-H.; Liu, Y.-F.; Chen, J.-G.; Mei, S.; You, Z.-Q.; Liu, Z.; et al. Pyrroloquinoline quinone ameliorates diabetic cardiomyopathy by inhibiting the pyroptosis signaling pathway in C57BL/6 mice and AC16 cells. Eur. J. Nutr. 2022, 61, 1823–1836. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Yao, Q.; Hu, T.; Cai, Z.; Xie, Q.; Zhao, J.; Yuan, Y.; Ni, J.; Wu, Q.Q. Cathepsin B deteriorates diabetic cardiomyopathy induced by streptozotocin via promoting NLRP3-mediated pyroptosis. Mol. Ther. Nucleic Acids 2022, 30, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Lin, H.; Huang, X.; Weng, J.; Peng, F.; Wu, S. METTL14 suppresses pyroptosis and diabetic cardiomyopathy by downregulating TINCR lncRNA. Cell Death Dis. 2022, 13, 38. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Li, Y.; Jin, Y.; Chen, Y.; Tian, L.; He, W. Synergistic cardioptotection by tilianin and syringin in diabetic cardiomyopathy involves interaction of TLR4/NF-κB/NLRP3 and PGC1a/SIRT3 pathways. Int. Immunopharmacol. 2021, 96, 107728. [Google Scholar] [CrossRef]
- Wei, C.; Xu, J.; Liu, Y.; Qadir, J.; Zhang, S.; Yuan, H. Exogenous Spermidine Alleviates Diabetic Myocardial Fibrosis Via Suppressing Inflammation and Pyroptosis in db/db Mice. Balk. Med. J. 2023, 40, 333–343. [Google Scholar] [CrossRef]
- Begum, R.; Thota, S.; Abdulkadir, A.; Kaur, G.; Bagam, P.; Batra, S. NADPH oxidase family proteins: Signaling dynamics to disease management. Cell Mol. Immunol. 2022, 19, 660–686. [Google Scholar] [CrossRef]
- Devant, P.; Boršić, E.; Ngwa, E.M.; Xiao, H.; Chouchani, E.T.; Thiagarajah, J.R.; Hafner-Bratkovič, I.; Evavold, C.L.; Kagan, J.C. Gasdermin D pore-forming activity is redox-sensitive. Cell Rep. 2023, 42, 112008. [Google Scholar] [CrossRef]
- Wei, H.; Bu, R.; Yang, Q.; Jia, J.; Li, T.; Wang, Q.; Chen, Y. Exendin-4 Protects against Hyperglycemia-Induced Cardiomyocyte Pyroptosis via the AMPK-TXNIP Pathway. J. Diabetes Res. 2019, 2019, 8905917. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.-L.; Fu, Y.; Wu, C.-W.; Zhang, Y.; Ren, H.; Zhou, S.-S. Signaling Pathways Related to Oxidative Stress in Diabetic Cardiomyopathy. Front. Endocrinol. 2022, 13, 907757. [Google Scholar] [CrossRef] [PubMed]
- Evavold, C.L.; Hafner-Bratkovič, I.; Devant, P.; D’Andrea, J.M.; Ngwa, E.M.; Boršić, E.; Doench, J.G.; LaFleur, M.W.; Sharpe, A.H.; Thiagarajah, J.R.; et al. Control of gasdermin D oligomerization and pyroptosis by the Ragulator-Rag-mTORC1 pathway. Cell 2021, 184, 4495–4511. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Yin, Q. AIM2 inflammasome activation and regulation: A structural perspective. J. Struct. Biol. 2017, 200, 279–282. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Pan, J.; Liu, H.; Zhang, M.; Liu, D.; Lu, L.; Tian, J.; Liu, M.; Jin, T.; An, F. AIM2 gene silencing attenuates diabetic cardiomyopathy in type 2 diabetic rat model. Life Sci. 2019, 221, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, C.; Chen, J.; Sang, T.; Peng, H.; Lin, X.; Zhao, Q.; Chen, S.; Eling, T.; Wang, X. Overexpression of NAG-1/GDF15 prevents hepatic steatosis through inhibiting oxidative stress-mediated dsDNA release and AIM2 inflammasome activation. Redox Biol. 2022, 52, 102322. [Google Scholar] [CrossRef] [PubMed]
- Nie, L.; Zhao, P.; Yue, Z.; Zhang, P.; Ji, N.; Chen, Q.; Wang, Q. Diabetes induces macrophage dysfunction through cytoplasmic dsDNA/AIM2 associated pyroptosis. J. Leukoc. Biol. 2021, 110, 497–510. [Google Scholar] [CrossRef]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef]
- Hu, B.; Jin, C.; Li, H.-B.; Tong, J.; Ouyang, X.; Cetinbas, N.M.; Zhu, S.; Strowig, T.; Lam, F.C.; Zhao, C.; et al. The DNA-sensing AIM2 inflammasome controls radiation-induced cell death and tissue injury. Science 2016, 354, 765–768. [Google Scholar] [CrossRef]
- Fidler, T.P.; Xue, C.; Yalcinkaya, M.; Hardaway, B.; Abramowicz, S.; Xiao, T.; Liu, W.; Thomas, D.G.; Hajebrahimi, M.A.; Pircher, J.; et al. The AIM2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis. Nature 2021, 592, 296–301. [Google Scholar] [CrossRef]
- Yarovinsky, T.O.; Su, M.; Chen, C.; Xiang, Y.; Tang, W.H.; Hwa, J. Pyroptosis in cardiovascular diseases: Pumping gasdermin on the fire. Semin. Immunol. 2023, 69, 101809. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.M.; Geng, K.; Law, B.Y.-K.; Wang, P.; Pu, Y.L.; Chen, Q.; Xu, H.W.; Tan, X.Z.; Jiang, Z.Z.; Xu, Y. Lipotoxicity-induced mtDNA release promotes diabetic cardiomyopathy by activating the cGAS-STING pathway in obesity-related diabetes. Cell Biol. Toxicol. 2023, 39, 277–299. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Dai, S.; Zhong, L.; Shi, X.; Fan, X.; Zhong, X.; Lin, W.; Su, L.; Lin, S.; Han, B.; et al. GSDMD (Gasdermin D) Mediates Pathological Cardiac Hypertrophy and Generates a Feed-Forward Amplification Cascade via Mitochondria-STING (Stimulator of Interferon Genes) Axis. Hypertension 2022, 79, 2505–2518. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Li, X.; Dai, F.; Liu, J.; Zhang, Q.; Guo, W. GSDMD-mediated pyroptosis promotes cardiac remodeling in pressure overload. Clin. Exp. Hypertens. 2023, 45, 2189138. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-F.; Zhou, L.; Mao, H.-Q.; Yang, F.-H.; Chen, Z.; Zhang, L. Mitochondrial DNA leakage exacerbates odontoblast inflammation through gasdermin D-mediated pyroptosis. Cell Death Discov. 2021, 7, 381. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Zhang, J.; Lin, Z.; Li, Y.; Qin, G. CircularRNA circ_0071269 knockdown protects against from diabetic cardiomyopathy injury by microRNA-145/gasdermin A axis. Bioengineered 2022, 13, 2398–2411. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Sun, Y.; Yang, F.; Yan, D.; Shen, M.; Jin, Z.; Zhan, L.; Liu, G.; Yang, L.; Zhou, Q.; et al. CircRNA DICAR as a novel endogenous regulator for diabetic cardiomyopathy and diabetic pyroptosis of cardiomyocytes. Signal Transduct. Target. Ther. 2023, 8, 99. [Google Scholar] [CrossRef]
- Yang, F.; Qin, Y.; Lv, J.; Wang, Y.; Che, H.; Chen, X.; Jiang, Y.; Li, A.; Sun, X.; Yue, E.; et al. Silencing long non-coding RNA Kcnq1ot1 alleviates pyroptosis and fibrosis in diabetic cardiomyopathy. Cell Death Dis. 2018, 9, 1000. [Google Scholar] [CrossRef]
- Xiao, W.; Zheng, D.; Chen, X.; Yu, B.; Deng, K.; Ma, J.; Wen, X.; Hu, Y.; Hou, J. Long non-coding RNA MIAT is involved in the regulation of pyroptosis in diabetic cardiomyopathy via targeting miR-214-3p. iScience 2021, 24, 103518. [Google Scholar] [CrossRef]
- Wu, A.; Sun, W.; Mou, F. lncRNA-MALAT1 promotes high glucose-induced H9C2 cardiomyocyte pyroptosis by downregulating miR-141-3p expression. Mol. Med. Rep. 2021, 23, 259. [Google Scholar] [CrossRef]
- Abo-Saif, M.A.; Ragab, A.E.; Ibrahim, A.O.; Abdelzaher, O.F.; Mehanyd, A.B.M.; Saber-Ayad, M.; El-Feky, O.A. Pomegranate peel extract protects against the development of diabetic cardiomyopathy in rats by inhibiting pyroptosis and downregulating LncRNA-MALAT1. Front. Pharmacol. 2023, 14, 1166653. [Google Scholar] [CrossRef]
- Song, S.; Ding, Y.; Dai, G.-L.; Zhang, Y.; Xu, M.-T.; Shen, J.-R.; Chen, T.-T.; Chen, Y.; Meng, G.-L. Sirtuin 3 deficiency exacerbates diabetic cardiomyopathy via necroptosis enhancement and NLRP3 activation. Acta Pharmacol. Sin. 2021, 42, 230–241. [Google Scholar] [CrossRef]
- Xiong, J.; Zhou, Q. The lncRNA HOTAIR attenuates pyroptosis of diabetic cardiomyocytes by recruiting FUS to regulate SIRT3 expression. Kaohsiung J. Med. Sci. 2023, 39, 458–467. [Google Scholar] [CrossRef]
- Liu, J.; Sun, M.; Wang, J.; Sun, Z.; Wang, G. HOTAIR regulates SIRT3-mediated cardiomyocyte survival after myocardial ischemia/reperfusion by interacting with FUS. BMC Cardiovasc. Disord. 2023, 23, 171. [Google Scholar] [CrossRef]
- Tang, G.; Duan, F.; Li, W.; Wang, Y.; Zeng, C.; Hu, J.; Li, H.; Zhang, X.; Chen, Y.; Tan, H. Metformin inhibited Nod-like receptor protein 3 inflammasomes activation and suppressed diabetes-accelerated atherosclerosis in apoE−/− mice. Biomed. Pharmacother. 2019, 119, 109410. [Google Scholar] [CrossRef]
- Yang, F.; Qin, Y.; Wang, Y.; Meng, S.; Xian, H.; Che, H.; Lv, J.; Li, Y.; Yu, Y.; Bai, Y.; et al. Metformin Inhibits the NLRP3 Inflammasome via AMPK/mTOR-dependent Effects in Diabetic Cardiomyopathy. Int. J. Biol. Sci. 2019, 15, 1010–1019. [Google Scholar] [CrossRef]
- Zou, R.; Nie, C.; Pan, S.; Wang, B.; Hong, X.; Xi, S.; Bai, J.; Yu, M.; Liu, J.; Yang, W. Co-administration of hydrogen and metformin exerts cardioprotective effects by inhibiting pyroptosis and fibrosis in diabetic cardiomyopathy. Free Radic Biol. Med. 2022, 183, 35–50. [Google Scholar] [CrossRef]
- Liu, B.; Xu, J.; Lu, L.; Gao, L.; Zhu, S.; Sui, Y.; Cao, T.; Yang, T. Metformin induces pyroptosis in leptin receptor-defective hepatocytes via overactivation of the AMPK axis. Cell Death Dis. 2023, 14, 82. [Google Scholar] [CrossRef]
- Chen, X.; Ren, L.; Liu, X.; Sun, X.; Dong, C.; Jiang, Y.; Qin, Y.; Qu, H.; Jiao, J.; Wang, S.; et al. Ranolazine protects against diabetic cardiomyopathy by activating the NOTCH1/NRG1 pathway. Life Sci. 2020, 261, 118306. [Google Scholar] [CrossRef]
- Ren, L.; Chen, X.; Nie, B.; Qu, H.; Ju, J.; Bai, Y. Ranolazine Inhibits Pyroptosis via Regulation of miR-135b in the Treatment of Diabetic Cardiac Fibrosis. Front. Mol. Biosci. 2022, 9, 806966. [Google Scholar] [CrossRef]
- Kim, S.R.; Lee, S.-G.; Kim, S.H.; Kim, J.H.; Choi, E.; Cho, W.; Rim, J.H.; Hwang, I.; Lee, C.J.; Lee, M.; et al. SGLT2 inhibition modulates NLRP3 inflammasome activity via ketones and insulin in diabetes with cardiovascular disease. Nat. Commun. 2020, 11, 2127. [Google Scholar] [CrossRef]
- Biegus, J.; Voors, A.A.; Collins, S.P.; Kosiborod, M.N.; Teerlink, J.R.; Angermann, C.E.; Tromp, J.; Ferreira, J.P.; Nassif, M.E.; Psotka, M.A.; et al. Impact of empagliflozin on decongestion in acute heart failure: The EMPULSE trial. Eur. Heart J. 2023, 44, 41–50. [Google Scholar] [CrossRef]
- Voors, A.A.; Angermann, C.E.; Teerlink, J.R.; Collins, S.P.; Kosiborod, M.; Biegus, J.; Ferreira, J.P.; Nassif, M.E.; Psotka, M.A.; Tromp, J.; et al. The SGLT2 inhibitor empagliflozin in patients hospitalized for acute heart failure: A multinational randomized trial. Nat. Med. 2022, 28, 568–574. [Google Scholar] [CrossRef]
- Liu, P.; Zhang, Z.; Wang, J.; Zhang, X.; Yu, X.; Li, Y. Empagliflozin protects diabetic pancreatic tissue from damage by inhibiting the activation of the NLRP3/caspase-1/GSDMD pathway in pancreatic β cells: In vitro and in vivo studies. Bioengineered 2021, 12, 9356–9366. [Google Scholar] [CrossRef]
- Madonna, R.; Moscato, S.; Cufaro, M.C.; Pieragostino, D.; Mattii, L.; Del Boccio, P.; Ghelardoni, S.; Zucchi, R.; De Caterina, R. Empagliflozin inhibits excessive autophagy through the AMPK/GSK3β signalling pathway in diabetic cardiomyopathy. Cardiovasc. Res. 2023, 119, 1175–1189. [Google Scholar] [CrossRef]
- Martín-Sánchez, F.; Diamond, C.; Zeitler, M.; Gomez, A.I.; Baroja-Mazo, A.; Bagnall, J.; Spiller, D.; White, M.; Daniels, M.J.D.; Mortellaro, A.; et al. Inflammasome-dependent IL-1β release depends upon membrane permeabilisation. Cell Death Differ. 2016, 23, 1219–1231. [Google Scholar] [CrossRef]
- Rathkey, J.K.; Zhao, J.; Liu, Z.; Chen, Y.; Yang, J.; Kondolf, H.C.; Benson, B.L.; Chirieleison, S.M.; Huang, A.Y.; Dubyak, G.R.; et al. Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci. Immunol. 2018, 3, eaat2738. [Google Scholar] [CrossRef]
- Li, Y.; Tu, Z.; Chen, F.; Yang, X.; Deng, R.; Su, F.; Cheng, Z.; Li, S.; Ong, S.-B.; Wang, D.; et al. Anti-inflammatory effect of Danhong injection through inhibition of GSDMD-mediated pyroptosis. Phytomedicine 2023, 113, 154743. [Google Scholar] [CrossRef]
- Humphries, F.; Shmuel-Galia, L.; Ketelut-Carneiro, N.; Li, S.; Wang, B.; Nemmara, V.V.; Wilson, R.; Jiang, Z.; Khalighinejad, F.; Muneeruddin, K.; et al. Succination inactivates gasdermin D and blocks pyroptosis. Science 2020, 369, 1633–1637. [Google Scholar] [CrossRef]
- Hu, J.J.; Liu, X.; Xia, S.; Zhang, Z.; Zhang, Y.; Zhao, J.; Ruan, J.; Luo, X.; Lou, X.; Bai, Y.; et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat. Immunol. 2020, 21, 736–745. [Google Scholar] [CrossRef]
- Tang, G.; Li, S.; Zhang, C.; Chen, H.; Wang, N.; Feng, Y. Clinical efficacies, underlying mechanisms and molecular targets of Chinese medicines for diabetic nephropathy treatment and management. Acta Pharm. Sin. B 2021, 11, 2749–2767. [Google Scholar] [CrossRef]
- Zhou, J.; Yan, S.; Guo, X.; Gao, Y.; Chen, S.; Li, X.; Zhang, Y.; Wang, Q.; Zheng, T.; Chen, L. Salidroside protects pancreatic β-cells against pyroptosis by regulating the NLRP3/GSDMD pathway in diabetic conditions. Int. Immunopharmacol. 2023, 114, 109543. [Google Scholar] [CrossRef]
- Han, J.; Zuo, Z.; Shi, X.; Zhang, Y.; Peng, Z.; Xing, Y.; Pang, X. Hirudin ameliorates diabetic nephropathy by inhibiting Gsdmd-mediated pyroptosis. Cell Biol. Toxicol. 2023, 39, 573–589. [Google Scholar] [CrossRef]
- Zhu, J.; Chen, H.; Le, Y.; Guo, J.; Liu, Z.; Dou, X.; Lu, D. Salvianolic acid A regulates pyroptosis of endothelial cells via directly targeting PKM2 and ameliorates diabetic atherosclerosis. Front. Pharmacol. 2022, 13, 1009229. [Google Scholar] [CrossRef]
- Sun, S.; Dawuti, A.; Gong, D.; Wang, R.; Yuan, T.; Wang, S.; Xing, C.; Lu, Y.; Du, G.; Fang, L. Puerarin-V Improve Mitochondrial Respiration and Cardiac Function in a Rat Model of Diabetic Cardiomyopathy via Inhibiting Pyroptosis Pathway through P2X7 Receptors. Int. J. Mol. Sci. 2022, 23, 13015. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, X.; Zong, B.; Yuan, H.; Wang, Z.; Wei, Y.; Wang, X.; Liu, G.; Zhang, J.; Li, S.; et al. Gypenosides improve diabetic cardiomyopathy by inhibiting ROS-mediated NLRP3 inflammasome activation. J. Cell Mol. Med. 2018, 22, 4437–4448. [Google Scholar] [CrossRef]
- Yang, L.; Cheng, C.-F.; Li, Z.-F.; Huang, X.-J.; Cai, S.-Q.; Ye, S.-Y.; Zhao, L.-J.; Xiong, Y.; Chen, D.-F.; Liu, H.-L.; et al. Berberine blocks inflammasome activation and alleviates diabetic cardiomyopathy via the miR-18a-3p/Gsdmd pathway. Int. J. Mol. Med. 2023, 51, 49. [Google Scholar] [CrossRef]
- Gao, G.; Fu, L.; Xu, Y.; Tao, L.; Guo, T.; Fang, G.; Zhang, G.; Wang, S.; Qin, T.; Luo, P.; et al. Cyclovirobuxine D Ameliorates Experimental Diabetic Cardiomyopathy by Inhibiting Cardiomyocyte Pyroptosis via NLRP3 in vivo and in vitro. Front. Pharmacol. 2022, 13, 906548. [Google Scholar] [CrossRef]
- Jiang, K.; Tu, Z.; Chen, K.; Xu, Y.; Chen, F.; Xu, S.; Shi, T.; Qian, J.; Shen, L.; Hwa, J.; et al. Gasdermin D inhibition confers antineutrophil-mediated cardioprotection in acute myocardial infarction. J. Clin. Investig. 2022, 132. [Google Scholar] [CrossRef]
- Sun, S.; Gong, D.; Liu, R.; Wang, R.; Chen, D.; Yuan, T.; Wang, S.; Xing, C.; Lv, Y.; Du, G.; et al. Puerarin Inhibits NLRP3-Caspase-1-GSDMD-Mediated Pyroptosis via P2X7 Receptor in Cardiomyocytes and Macrophages. Int. J. Mol. Sci. 2023, 24, 13169. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Signaling Pathway | Conditions | Treatments | Model | Pyroptotic Marker | Inflammatory Marker/ROS | Other Cell Death Markers/Relevant Findings | Ref | |||
|---|---|---|---|---|---|---|---|---|---|---|
| GSDMD NT | GSDMD | caspase-1 | Others | |||||||
| PAR4/NLRP3 | HFD for 8 weeks | PAR4−/− | C57BL/6J mice | ↓ | ↓ | IL-1β ↓ | [58] | |||
| HFD for 8 weeks | Control | C57BL/6J mice | ↑ | - | ↑ | ASC- NLRP3- | IL-1β and IL-18 ↑ | P10, p20, p37 caspase-1 ↑ | ||
| Patients with type 2 diabetes mellitus | Atria of patients with type 2 diabetes | ↑ | - | ↑ | IL-1β ↑ | |||||
| CTSB/NLRP3 | HG (33.3 mM) | Si-CTSB | NRCMs | ↓ | ↓ | ↓ | NLRP3 ASC ↓ | TUNEL, LDH ↓ | [75] | |
| STZ (50 mg/kg) for 5 days | CTSB−/+ heterozygous | C57BL/6J mice | ↓ | ↓ | ↓ | NLRP3 ASC ↓ | IL-1β and IL-18 ↓ | EF, FS ↑ LVEDd, LVESd, TUNEL, CD45, CD68 ↓ | ||
| METTL14/TINCR | HG (50 mM) for 24 h | MCC950 (5 μg/mL) for 24 h | H9c2 and NRVMs | ↓ | ↓ | NLRP3 ↓ | IL-1β and IL-18 ↓ | [76] | ||
| HG (50 mM) for 24 h | Overexpression METTL14 + overexpression TINCR | H9c2 and NRVMs | ↑ | ↑ | NLRP3 ↑ | IL-1β and IL-18 ↑ | TUNEL ↓ | |||
| STZ (65 mg/kg) | METTL14 overexpression | SD rats | ↓ | ↓ | NLRP3 ↓ | |||||
| STZ (65 mg/kg) | MCC950 (10 mg/kg) | SD rats | ↓ | NLRP3 ↓ | EF, FS ↑ CKMB, LDH, and AST ↓ | |||||
| cGAS-STING/p-TBK1/p-IRF3 | STZ (50 mg/kg) for 5 days with HFD | STING-shRNA | Male C57BL/6J mice | ↓ | - | ↓ | NLRP3 ↓ | TNF-α, IFN-β, IL-1β and IL-18 ↓ | E/A, EF, FS ↑WGA, LDH, ANP, BNP, and β-MHC ↓ | [70] |
| PA (400 μM) for 24 h | PA + EtBr | H9c2 and NMCMs | ↓ | - | ↓ | NLRP3 ↓ | TNF-α, IFN-β, IL-1β, and IL-18 ↓ | mtDNA, LDH, TUNEL, α-actinin, ANP, BNP, and β-MHC ↓ | ||
| AIM2 | HG 25 (mM) for 24 h | 5 mmol/L NAC | H9c2 | ↓ | ↓ | ASC ↓ | ROS ↓ | EthD-III, TUNEL ↓ | [85] | |
| HFD for 4 weeks + STZ (40 mg/kg) | AIM2-shRNA | SD rats | ↓ | ↓ | ASC ↓ | IL-1β ↓ | E/A, EF, FS ↑ TUNEL, collagen I, and collagen III ↓ | |||
| CircRNA DICAR/VCP/Med12 | AGEs (200 μg/mL) for 48 h | Overexpressed DICAR | HL-1 cardiomyocytes | ↓ | ↓ | ASC NLRP3 ↓ | [97] | |||
| db/db mice | DICAR Tg | C57BL/KsJ mice | ↓ | ↓ | ASC NLRP3 ↓ | EF, FS ↑ WGA, LVEDd, LVEDV ↓ | ||||
| DICAR+/− | C57BL/KsJ mice | ↑ | ↑ | ASC NLRP3 ↑ | EF, FS ↓ LVEDD, LVEDV, WGA ↑ | |||||
| lncRNA Kcnq1ot1/miR-214-3p | STZ (50 mg/kg) for 5 days | Kcnq1ot1-shRNA | C57BL/6 mice | ↓ | ↓ | NLRP3 ↓ | IL-1β ↓ | EF, FS ↑ TGF-β1, p-smad2, p-smad3, collagen I, and collagen III ↓ | [98] | |
| HG (30 mM) for 24 h | Si-Kcnq1ot1 | Cardiac fibroblasts | ↓ | ↓ | NLRP3 ↓ | IL-1β ↓ | TGF-β1, p-smad2, p-smad3, collagen I, and collagen III ↓ | |||
| lncRNA MIAT/miR-214-3p | HG (25 mM) for 24 h | Si-MIAT | Cardiac fibroblasts | ↓ | ↓ | IL-1β and IL-18 ↓ | [99] | |||
| STZ (150 mg/kg) | LV-siMIAT | C57BL/6 mice | ↓ | ↓ | IL-1β and IL-18 ↓ | EF, FS ↑ | ||||
| lncRNA MALAT1/miR-141-3p | HG (30 mM) for 24 h | Control | H9C2 | ↑ | ↓ | ↑ | ASC ↑ NLRP3 ↑ | TUNEL ↑ | [100] | |
| Overexpression MALAT1 | ↑↑ | ↓↓ | ↑↑ | ASC ↑↑ NLRP3 ↑↑ | TUNEL ↑↑ | |||||
| Si-MALAT1 | ↓ | ↑ | ↓ | ASC ↓ NLRP3 ↓ | TUNEL ↓ | |||||
| Overexpression MALAT1 + overexpression-miR-141-3p | ↓ | ↑ | ↓ | ASC ↓ NLRP3 ↓ | TUNEL ↓ | |||||
| lncRNA HOTAIR/FUS/SIRT3 | HG (30 mM) for 24 h | Overexpression HOTAIR | H9C2 | ↓ | ↓ | NLRP3 ↓ | IL-1β and IL-18 ↓ | [103] | ||
| HG (30 mM) for 24 h | Overexpression SIRT3 | ↓ | ↓ | NLRP3 ↓ | IL-1β and IL-18 ↓ | |||||
| HG (30 mM) for 24 h | Overexpression HOTAIR + sh- SIRT3 | ↑ | ↑ | NLRP3 ↑ | IL-1β and IL-18 ↑ | |||||
| Drug Therapies | Dosage | Conditions | Model | Pyroptotic Marker | Inflammatory Marker/ROS | Other Cell Death Markers/Relevant Findings | Ref. | |||
|---|---|---|---|---|---|---|---|---|---|---|
| GSDMDNT | GSDMD | Caspase-1 | Others | |||||||
| Metformin | 2 mM metformin | HG (30 mM) for 24 h | Primary cardiomyocytes | ↓ | ↓ | NLRP3 ↓ | IL-1β ↓ | [106] | ||
| Ranolazine | 30 mg/kg for 12 weeks | 8 weeks of HFD + STZ (35 mg/kg) | SD rats | ↓ | ↓ | - | IL-1β ↓ | EF, FS ↑ TGF-β1, collagen I and collagen III ↓ | [110] | |
| Empagliflozin | 10 mg/kg/day for 8 weeks | db/db mice | ↓ | ↑ | ↓ | NLRP3 ↓ | IL-1β ↓ | TUNEL, caspase-3, Bax, SOD2, NOX2/4, ANP, BNP, β-MHC, TGF-β, collagen I and collagen III ↓ | [69] | |
| H2 | Inhaled 2% hydrogen for 3 h per day | STZ (50 mg/kg) for 5 days | C57BL/6 mice | ↓ | ↑ | ↓ | NLRP3 ↓ | IL-18, IL-1β ↓ | TGF-β1, p-smad3, p-smad2, COL-I, COL-III, α-SMA ↓ | [107] |
| Puerarin-V | 50, 100, or 150 mg/kg/day for 6 weeks | HFD for 6 weeks + STZ (30 mg/kg) + ISO (5 mg/kg) | SD rats | ↓ | ↓ | P2X7R, NLRP3,ASC ↓ | IL-18, IL-1β ↓ | cTn-T, NT-proBNP, AST, LDH, CK-MB, MDA ↓SOD, GSH ↑ | [125] | |
| Berberine | 200 mg/kg/day for 4 weeks | HFD for 4 weeks + STZ (30 mg/kg) for 5 days | SD rats | ↓ | ↓ | - | NLRP3- | IL-1β ↓ | cTn-I, CK-MB ↓ | [127] |
| MiR-18a-3p overexpres- sion | HFD for 4 weeks + STZ (30 mg/kg) for 5 days | SD rats | ↓ | ↓ | - | NLRP3- | IL-1β ↓ | |||
| MiR-18a-3p mimics | HG (33 mM) for 2 days | H9C2 cells | ↓ | ↓ | IL-1β ↓ | PI ↓ | ||||
| Cyclovirobuxine D | 0.5 mg/kg/day or 1 mg/kg/day for 2 months | HFD + STZ (30 mg/kg) for 4 days | C57BL/6 mice | ↓ | ↓ | ↓ | NLRP3 ↓ | IL-18, IL-1β ↓ | TUNEL, LDH ↓EF, FS ↑ | [128] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Z.; Chen, Y.; Mei, Y.; Yan, M.; Liang, H. Gasdermin D-Mediated Pyroptosis in Diabetic Cardiomyopathy: Molecular Mechanisms and Pharmacological Implications. Molecules 2023, 28, 7813. https://doi.org/10.3390/molecules28237813
Liu Z, Chen Y, Mei Y, Yan M, Liang H. Gasdermin D-Mediated Pyroptosis in Diabetic Cardiomyopathy: Molecular Mechanisms and Pharmacological Implications. Molecules. 2023; 28(23):7813. https://doi.org/10.3390/molecules28237813
Chicago/Turabian StyleLiu, Zhou, Yifan Chen, Yu Mei, Meiling Yan, and Haihai Liang. 2023. "Gasdermin D-Mediated Pyroptosis in Diabetic Cardiomyopathy: Molecular Mechanisms and Pharmacological Implications" Molecules 28, no. 23: 7813. https://doi.org/10.3390/molecules28237813
APA StyleLiu, Z., Chen, Y., Mei, Y., Yan, M., & Liang, H. (2023). Gasdermin D-Mediated Pyroptosis in Diabetic Cardiomyopathy: Molecular Mechanisms and Pharmacological Implications. Molecules, 28(23), 7813. https://doi.org/10.3390/molecules28237813